
Determination of the Bioactive Compounds from Echinacea purpurea (L.) Moench Leaves Extracts in Correlation with the Antimicrobial Activity and the In Vitro Wound Healing Potential

 ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Total Polyphenols Content
2.2. Total Flavonoids Content
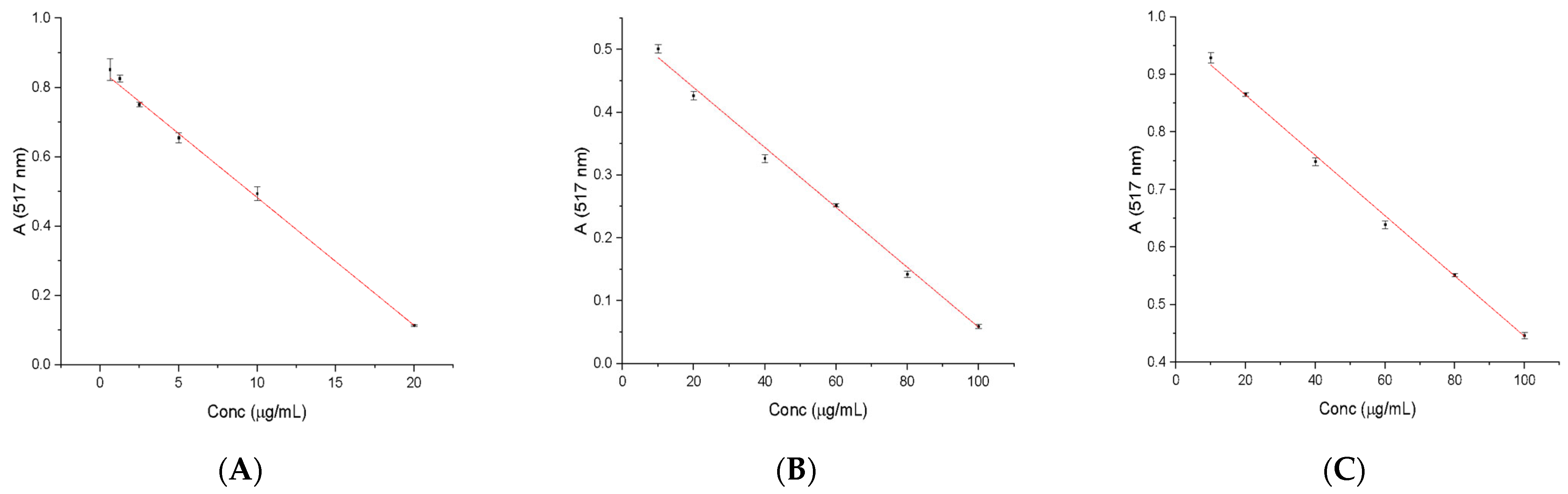
2.3. Determination of Free Radical Scavenging Activity
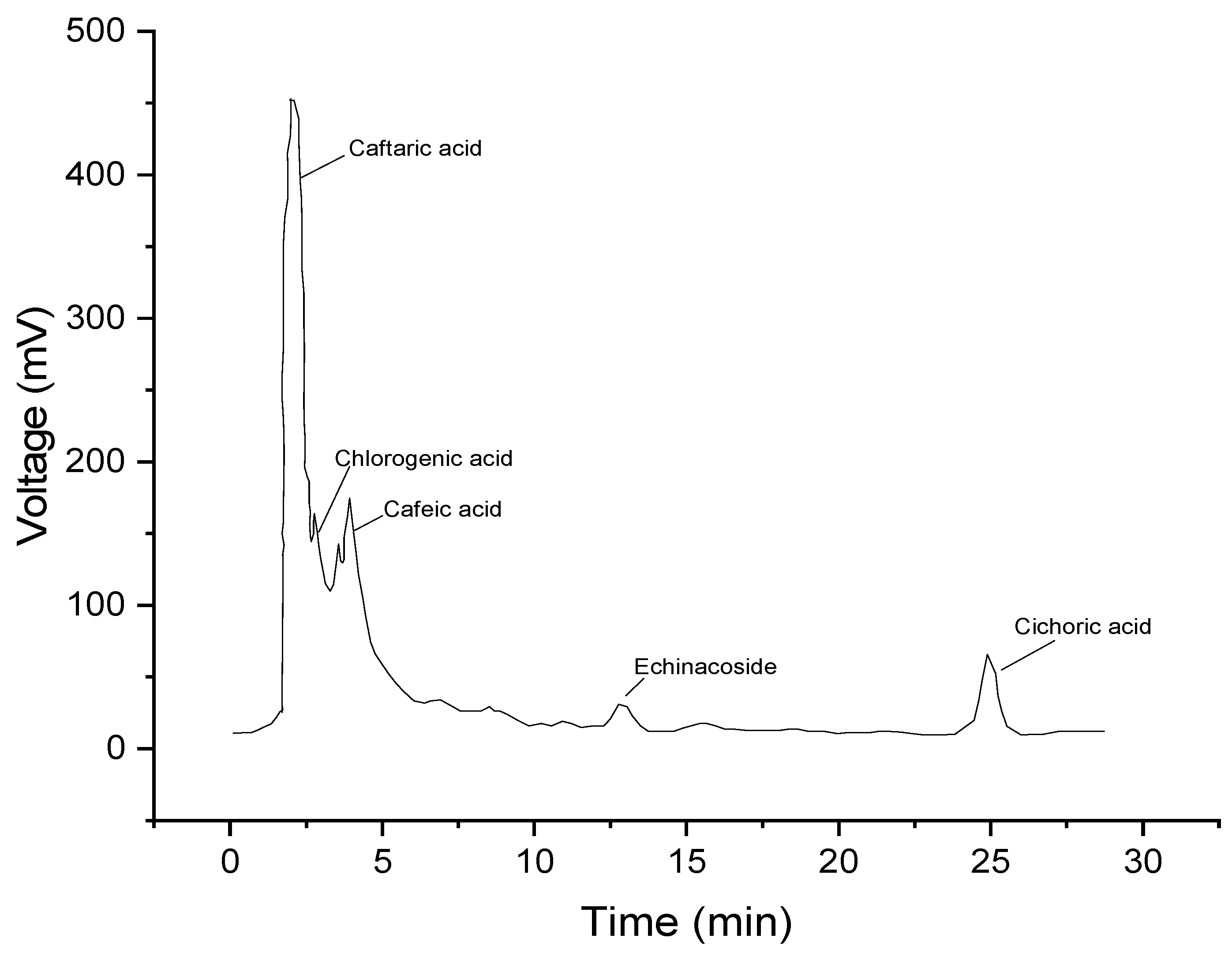
2.4. The Results of the HPLC Analysis
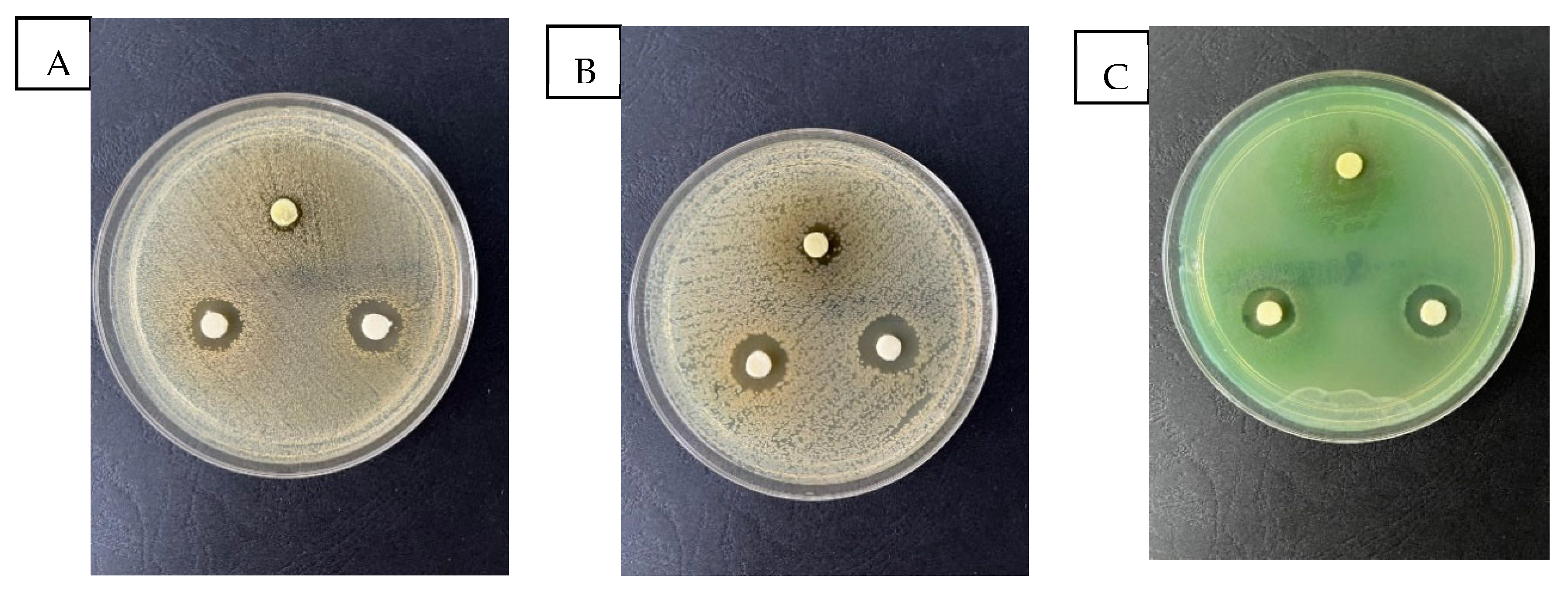
2.5. Evaluation of the Antimicrobial Activity
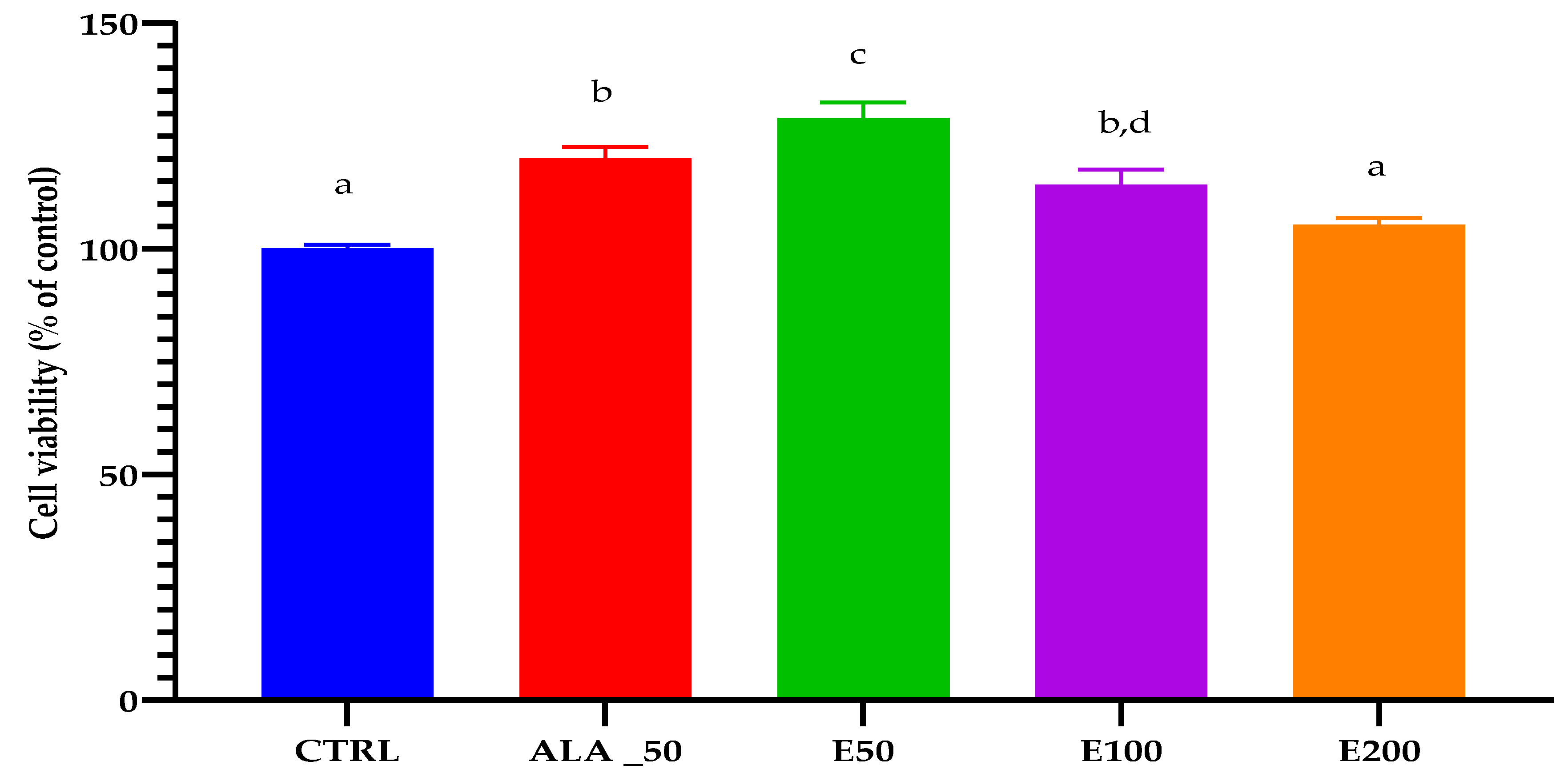
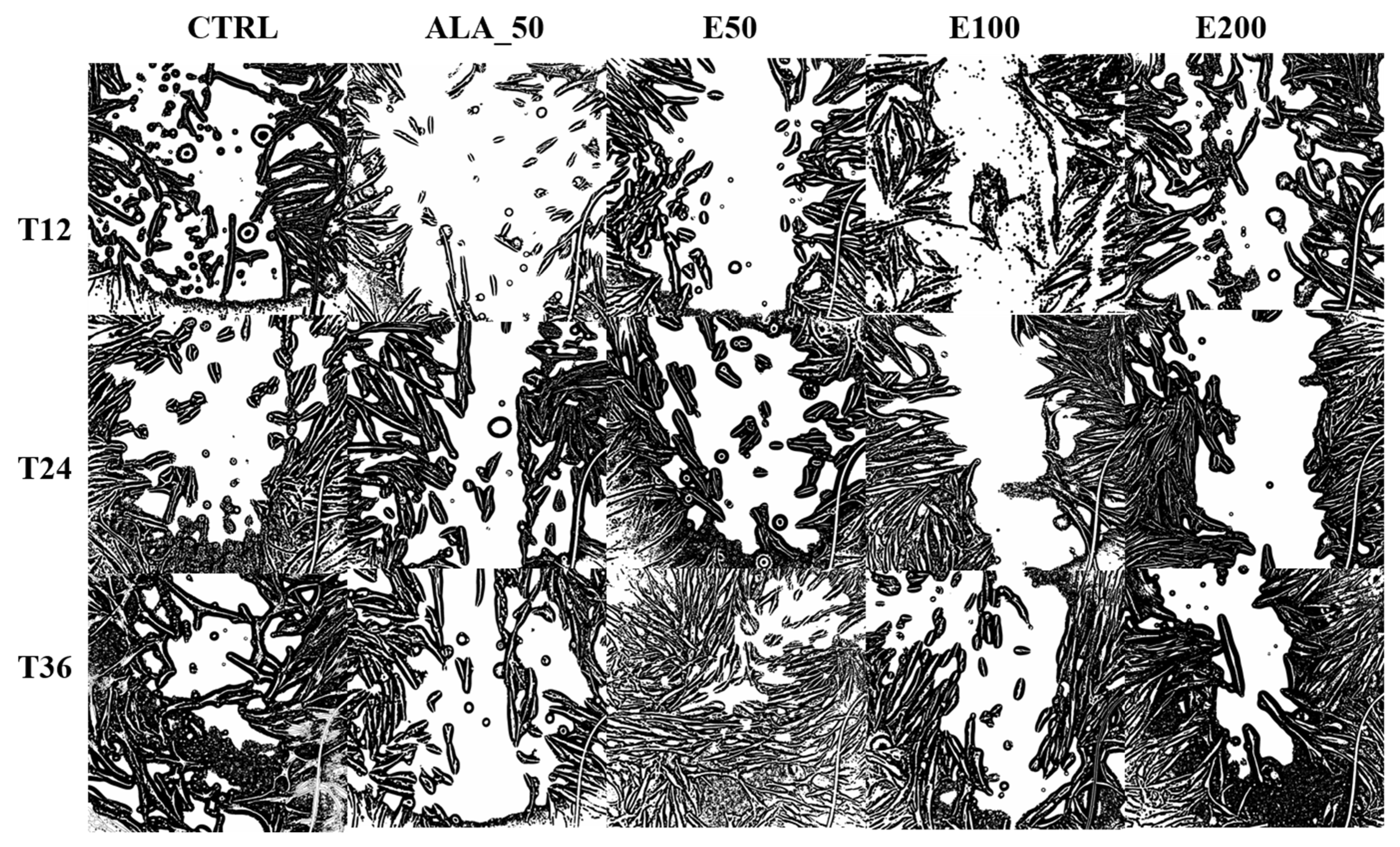
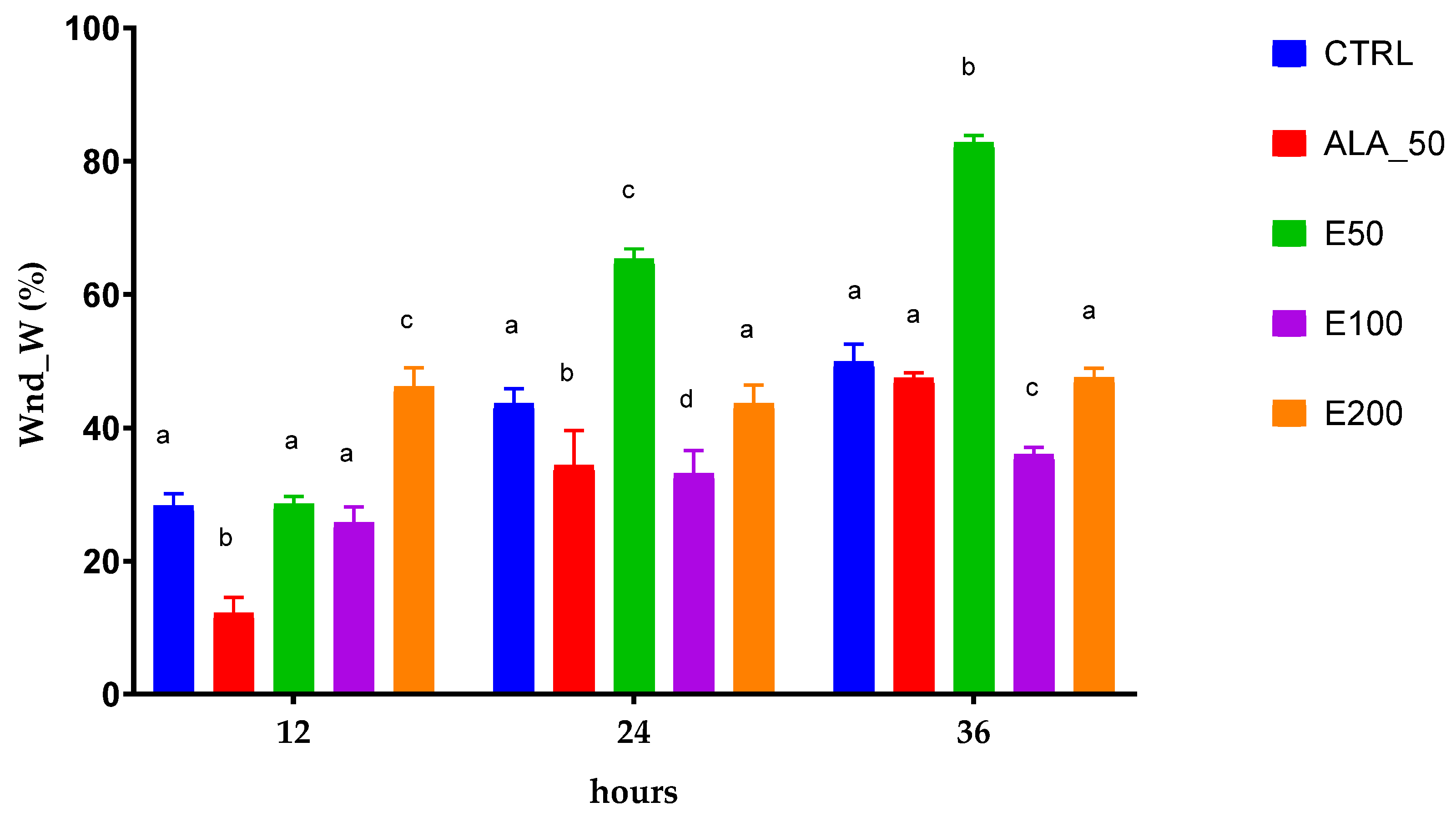
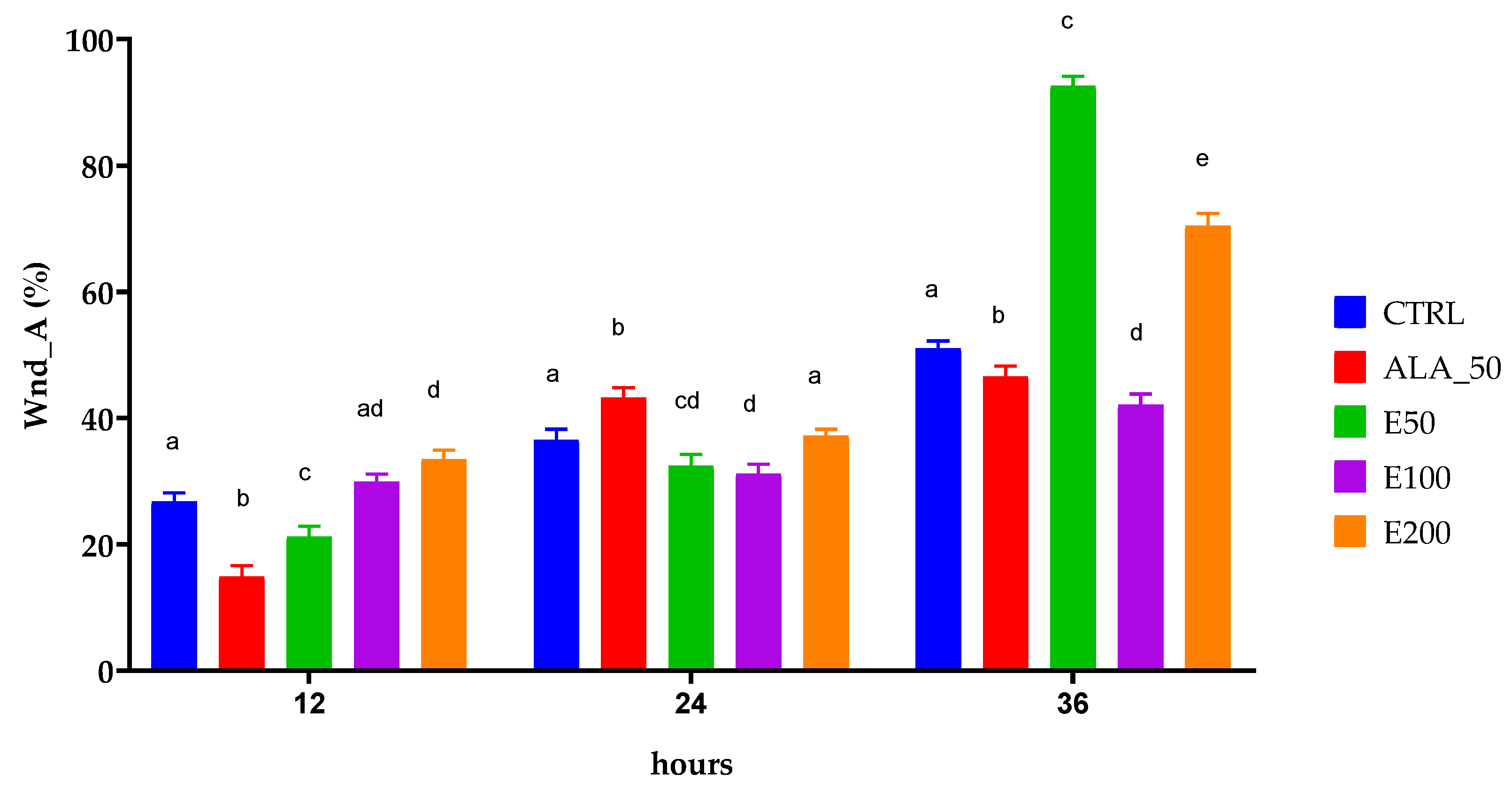
2.6. Evaluation of the Wound Healing Effect of EPF Extract Using In Vitro Scratch Assay
3. Materials and Methods
3.1. Plant Material
3.2. Chemicals and Equipments
3.3. Determination of Total Phenols
3.4. Determination of Total Flavonoids
3.5. Determination of Free Radical Scavenging Activity
- (a)
- Ascorbic acid
- (b)
- Gallic and caffeic acid
3.6. HPLC Analysis
3.7. Antimicrobial Activity
3.8. In Vitro Evaluation of the Healing Effect of the Extract Obtained from EPF
3.8.1. Cell Culture Formation
3.8.2. Cell Viability Assay
3.8.3. Cytotoxicity Testing of EPF Extract by the MTS Method
3.8.4. In Vitro Testing of the Biological Healing Effect of EPF Extract Using the Scratch Method
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Glevitzky, I.; Dumitrel, G.A.; Glevitzky, M.; Pasca, B.; Otrisal, P.; Bungau, S.; Cioca, G.; Pantis, C.; Popa, M. Statistical Analysis of the Relationship between Antioxidant Activity and the Structure of Flavonoid Compounds. Rev. Chim. 2019, 70, 3103–3107. [Google Scholar] [CrossRef]
- Hegedűs, C.S.; Muresan, M.; Badale, A.; Bombicz, M.; Varga, B.; Szilágyi, A.; Sinka, D.; Bácskay, I.; Popoviciu, M.; Magyar, I.; et al. SIRT1 Activation by Equisetum arvense L. (Horsetail) Modulates Insulin Sensitivity in Streptozotocin Induced Diabetic Rats. Molecules 2020, 25, 2541. [Google Scholar] [CrossRef] [PubMed]
- Pallag, A.; Bungau, S.G.; Tit, D.M.; Jurca, T.; Sirbu, V.; Honiges, A.; Horhogea, C. Comparative study of polyphenols, flavonoids, and chlorophylls in Equisetum arvense L. populations. Rev. Chim. 2016, 67, 530–533. [Google Scholar]
- Stanisavljevic, I.; Stojiþevic, S.; Veliþkovic, D.; Veljkovic, V.; Lazic, M. Antioxidant and Antimicrobial Activities of Echinacea (Echinacea purpurea L.) Extracts Obtained by Classical and Ultrasound Extraction. Chin. J. Chem. Eng. 2009, 17, 478–483. [Google Scholar] [CrossRef]
- Kayaa, M.; Merdivanb, M.; Tashakkoria, P.; Erdemb, P.; Andersonc, J.L. Analysis of Echinacea flower volatile constituents by HS-SPME-GC/MS using laboratory-prepared and commercial SPME fibers. J. Essent. Oil Res. 2018, 31, 91–98. [Google Scholar] [CrossRef]
- Lekar, A.V.; Borisenko, S.N.; Filonova, O.V.; Vetrova, E.V.; Maksimenko, E.V.; Borisenko, N.I.; Minkin, V.-I. Extraction of caftaric and cichoric acids from Echniacea purpurea L. in subcritical water. Russ. J. Phys. Chem. 2013, 7, 968–975. [Google Scholar] [CrossRef]
- Ponoran, C.; Olah, N. Care este adevårul despre Echinacea Truth about Echinacea. Rom. J. Med. Pract. 2008, 3, 144–148. [Google Scholar]
- Senica, M.; Mlinsek, G.; Veberic, R.; Mikulic-Petkovsek, M. Which Plant Part of Purple Coneflower (Echinacea purpurea (L.) Moench) Should be Used for Tea and Which for Tincture? J. Med. Food 2019, 22, 102–108. [Google Scholar] [CrossRef]
- Aarland, R.C.; Bañuelos-Hernández, A.E.; Fragoso-Serrano, M.; Del Carmen Sierra-Palacios, E.; Díaz de León-Sánchez, F.; Pérez-Flores, L.J.; Rivera-Cabrera, F.; Mendoza-Espinoza, J.A. Studies on phytochemical, antioxidant, anti-inflammatory, hypoglycaemic and antiproliferative activities of Echinacea purpurea and Echinacea angustifolia extracts. Pharm. Biol. 2017, 55, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.; Barros, L.; Dias, M.I.; Finimundy, T.C.; Amaral, J.S.; José Alves, M.; Calhelha, R.C.; Santos, P.F.; Ferreira, I.C.F.R. Echinacea purpurea (L.) Moench: Chemical Characterization and Bioactivity of Its Extracts and Fractions. Pharmaceuticals 2020, 13, 125. [Google Scholar] [CrossRef]
- Burlou-Nagy, C.; Bănică, F.; Jurca, T.; Vicaș, L.G.; Marian, E.; Muresan, M.E.; Bácskay, I.; Kiss, R.; Fehér, P.; Pallag, A. Echinacea purpurea (L.) Moench: Biological and Pharmacological Properties. A Review. Plants 2022, 11, 1244. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; Brighenti, V.; Caesar, L.K.; Bertelli, D.; Cech, N.B.; Pellati, F. Analytical methods for the study of bioactive compounds from medicinally used Echinacea species. J. Pharm. Biomed. Anal. 2018, 160, 443–477. [Google Scholar] [CrossRef]
- Banica, F.; Bungau, S.; Țit, D.M.; Behl, T.; Otrisal, P.; Nechifor, A.C.; Gitea, D.; Pavel, F.M.; Nemeth, S. Determination of the Total Polyphenols Content and Antioxidant Activity of Echinacea purpurea Extracts Using Newly Manufactured Glassy Carbon Electrodes Modified with Carbon Nanotubes. Processes 2020, 8, 833. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Cimpeanu, C.; Predoi, G. Antioxidant Capacity Determination in Plants and Plant-Derived Products: A Review. Oxid. Med. Cell. Longev. 2016, 2016, 9130976. [Google Scholar] [CrossRef] [PubMed]
- Meeran, M.F.N.; Javed, H.; Sharma, C.; Goyald, S.N.; Kumar, S.; Kumar, J.N.; Ojha, S. Can Echinacea be a potential candidate to target immunity, inflammation, and infection—The trinity of coronavirus disease. Heliyon 2021, 7, e05990. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.-Y.; Sung, J.-M.; Huang, P.-W.; Lin, S.-D. Antioxidant, Antidiabetic, and Antihypertensive Properties of Echinacea purpurea Flower Extract and Caffeic Acid Derivatives Using In Vitro Models. J. Med. Food 2017, 20, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, R.; Bächler, A.; Feldhaus, S.; Lang, G.; Klein, P.; Schoop, R. Safety and Dose-Dependent Effects of Echinacea for the Treatment of Acute Cold Episodes in Children: A Multicenter, Randomized, Open-Label Clinical Trial. Children 2020, 7, 292. [Google Scholar] [CrossRef]
- Yao, L.; Bai, L.; Tan, Y.; Sun, J.; Qu, Q.; Shi, D.; Guo, S.; Liu, C. The immunoregulatory effect of sulfated Echinacea purpurea polysaccharide on chicken bone marrow-derived dendritic cells. Int. J. Biol. Macromol. 2019, 139, 1123–1132. [Google Scholar] [CrossRef]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef]
- Chiellini, C.; Maida, I.; Maggini, V.; Bosi, E.; Mocali, S.; Emiliani, G.; Perrin, E.; Firenzuoli, F.; Mengoni, A.; Fani, R. Preliminary data on antibacterial activity of Echinacea purpurea-associated bacterial communities against Burkholderia cepacia complex strains, opportunistic pathogens of Cystic Fibrosis patients. Microbiol. Res. 2017, 196, 34–43. [Google Scholar] [CrossRef]
- Raduner, S.; Majewska, A.; Chen, J.Z.; Xie, X.Q.; Hamon, J.; Faller, B.; Altmann, K.H.; Gertsch, J. Alkylamides from Echinacea are a new class of cannabinomimetics. Cannabinoid type 2 receptor-dependent and -independent immunomodulatory effects. J. Biol. Chem. 2006, 281, 14192–14206. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Premoli, M.; Aria, F.; Bonini, S.A.; Maccarinelli, G.; Gianoncelli, A.; Memo, M.; Mastinu, A. Cannabimimetic plants: Are they new cannabinoidergic modulators? Planta 2019, 249, 1681–1694. [Google Scholar] [CrossRef]
- Ren, W.; Ban, J.; Xia, Y.; Zhou, F.; Yuan, C.; Jia, H.; Huang, H.; Jiang, M.; Liang, M.; Li, Z.; et al. Echinacea purpurea-derived homogeneous polysaccharide exerts anti-tumor efficacy via facilitating M1 macrophage polarization. Innovation 2023, 4, 100391. [Google Scholar] [PubMed]
- Park, S.J.; Lee, M.; Kim, D.; Oh, D.H.; Prasad, K.S.; Eun, S.; Lee, J. Echinacea purpurea Extract Enhances Natural Killer Cell Activity In Vivo by Upregulating MHC II and Th1-type CD4+ T Cell Responses. J. Med. Food 2021, 24, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.E.; Cohen, S.N.; Archer, C.B. On behalf of British Association of Dermatologists and Royal College of General Practitioners. Guidance on the diagnosis and clinical management of atopic eczema. Clin. Exp. Dermatol. 2012, 37, 7–12. [Google Scholar] [CrossRef]
- Kapur, S.; Watson, W.; Carr, S. Atopic dermatitis. Allergy Asthma Clin. Immunol. 2018, 14, 52. [Google Scholar] [CrossRef]
- Kilic, A.; Harder, A.; Reich, H.; Knie, U.; Masur, C.; Abels, C. Efficacy of hydrophilic or lipophilic emulsions containing Echinacea purpurea extract in treatment of different types of pruritus. Clin. Cosmet. Investig. Dermatol. 2018, 11, 591–602. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Wysocki, A.; Warburton, D.; Tuan, T.-L. Wound healing in development. Birth Defects Res. Part C Embryo Today Rev. 2012, 96, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Endres, L.; Tit, D.M.; Bungau, S.; Pascalau, N.A.; Maghiar Todan, L.; Bimbo-Szuhai, E.; Iancu, G.M.; Negrut, N. Incidence and clinical implications of autoimmune thyroiditis in the development of acne in young patients. Diagnostics 2021, 11, 794. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Kvasnička, F.; Čopíková, J.; Ševčík, R.; Krátká, J.; Syntytsia, A.; Voldřich, M. Determination of phenolic acids by capillary zone electrophoresis and HPLC. Open Chem. 2008, 6, 410–418. [Google Scholar] [CrossRef]
- Vicas, L.; Teusdea, A.; Vicas, S.; Marian, E.; Jurca, T.; Muresan, M.; Gligor, F. Assessment of antioxidant capacity of some extracts for further use in therapy. Farmacia 2015, 63, 267–274. [Google Scholar]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef]
- Tabar, R.S.; Moieni, A.; Monfared, S.R. Improving biomass and chicoric acid content in hairy roots of Echinacea purpurea L. Biologia 2019, 74, 941–951. [Google Scholar] [CrossRef]
- Maggini, V.; De Leo, M.; Granchi, C.; Tuccinardi, T.; Mengoni, A.; Gallo, E.R.; Biffi, S.; Fani, R.; Pistelli, L.; Firenzuoli, F.; et al. The influence of Echinacea purpurea leaf microbiota on chicoric acid level. Sci. Rep. 2019, 9, 10897. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.G.; Santos, L.F.F.; Pianetti, G.A.; César, I.C. A Rapid UPLC Method for the Simultaneous Quantitation of Caffeic Acid Derivatives in Dried Extracts of Echinacea Purpurea. J. Chromatogr. Sci. 2021, 59, 439–444. [Google Scholar] [CrossRef]
- Chen, X.-L.; Zhang, J.-J.; Chen, R.; Li, Q.-L.; Yang, Y.-S.; Wu, H. An Uncommon Plant Growth Regulator, Diethyl Aminoethyl Hexanoate, Is Highly Effective in Tissue Cultures of the Important Medicinal Plant Purple Coneflower (Echinacea purpurea L.). BioMed Res. Int. 2013, 2013, 540316. [Google Scholar] [CrossRef][Green Version]
- Lv, X.; Feng, S.; Zhang, J.; Sun, S.; Geng, Y.; Yang, M.; Liu, Y.; Qin, L.; Zhao, T.; Wang, C.; et al. Application of HPLC Fingerprint Combined with Chemical Pattern Recognition and Multi-Component Determination in Quality Evaluation of Echinacea purpurea (L.) Moench. Molecules 2022, 27, 6463. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hsia, H.C. The Impact of Microbial Communities on Wound Healing: A Review. Ann. Plast. Surg. 2018, 81, 113–123. [Google Scholar] [CrossRef]
- Miere, F.; Teușdea, A.C.; Laslo, V.; Cavalu, S.; Fritea, L.; Dobjanschi, L.; Zdrinca, M.; Zdrinca, M.; Ganea, M.; Pașc, P.; et al. Evaluation of In Vitro Wound-Healing Potential, Antioxidant Capacity, and Antimicrobial Activity of Stellaria media (L.) Vill. Appl. Sci. 2021, 11, 11526. [Google Scholar] [CrossRef]
- Givol, O.; Kornhaber, R.; Visentin, D.; Cleary, M.; Haik, J.; Harats, M. A Systematic Review of Calendula Officinalis Extract for Wound Healing. Wound Repair Regen. 2019, 27, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Song, N.; Liu, W.; Dong, A.; Wang, Y.-J.; Yang, Y.-W. Construction of Antibacterial N-Halamine Polymer Nanomaterials Capable of Bacterial Membrane Disruption for Efficient Anti-Infective Wound Therapy. Macromol. Biosci. 2019, 19, 1970010. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, W.; Tang, F.; Chen, X.; Ren, L. Antibacterial Activities of Flavonoids: Structure-Activity Relationship and Mechanism. Curr. Med. Chem. 2014, 22, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Górniak, I.; Bartoszewski, R.; Króliczewski, J. Comprehensive review of antimicrobial activities of plant flavonoids. Phytochem. Rev. 2019, 18, 241–272. [Google Scholar] [CrossRef]
- Shamsudin, N.F.; Ahmed, Q.U.; Mahmood, S.; Ali Shah, S.A.; Khatib, A.; Mukhtar, S.; Alsharif, M.A.; Parveen, H.; Zakaria, Z.A. Antibacterial Effects of Flavonoids and Their Structure-Activity Relationship Study: A Comparative Interpretation. Molecules 2022, 27, 1149. [Google Scholar] [CrossRef]
- Miere (Groza), F.; Vicas, S.I.; Timar, A.V.; Ganea, M.; Zdrinca, M.; Cavalu, S.; Fritea, L.; Vicas, L.; Muresan, M.; Pallag, A.; et al. Preparation and Characterization of Two Different Liposomal Formulations with Bioactive Natural Extract for Multiple Applications. Processes 2021, 9, 432. [Google Scholar] [CrossRef]
- Veloso, D.J.; Abrão, F.; Martins, C.H.G.; Bronzato, J.D.; Gomes, B.P.F.A.; Higino, J.S.; Sampaio, F.C. Potential Antibacterial and Anti-Halitosis Activity of Medicinal Plants against Oral Bacteria. Arch. Oral Biol. 2020, 110, 104585. [Google Scholar] [CrossRef]
- Stoll, A.; Renz, J.; Brack, A. Isolierung und Konstitution des Echinacosids, eines Glykosids aus den Wurzeln von Echinacea angustifolia D. C. 6. Mitteilung über antibakterielle Stoffe. Helv. Chim. Acta 1950, 33, 1877–1893. [Google Scholar] [CrossRef]
- Attarzadeh, M.; Balouchi, H.; Rajaie, M.; Dehnavi, M.M.; Salehi, A. Improving growth and phenolic compounds of Echinacea purpurea root by integrating biological and chemical resources of phosphorus under water deficit stress. Ind. Crop Prod. 2020, 154, 112763. [Google Scholar] [CrossRef]
- Garzoli, S.; Maggio, F.; Vinciguerra, V.; Rossi, C.; Donadu, M.G.; Serio, A. Chemical Characterization and Antimicrobial Properties of the Hydroalcoholic Solution of Echinacea purpurea (L.) Moench. and Propolis from Northern Italy. Molecules 2023, 28, 1380. [Google Scholar] [CrossRef]
- Vang Mouritzen, M.; Jenssen, H. Optimized Scratch Assay for In Vitro Testing of Cell Migration with an Automated Optical Camera. J. Vis. Exp. 2018, 138, 57691. [Google Scholar] [CrossRef]
- Ávila-Salas, F.; Marican, A.; Pinochet, S.; Carreño, G.; Valdés, O.; Venegas, B.; Donoso, W.; Cabrera-Barjas, G.; Vijayakumar, S.; Durán-Lara, E.F. Film Dressings Based on Hydrogels: Simultaneous and Sustained-Release of Bioactive Compounds with Wound Healing Properties. Pharmaceutics 2019, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Valle, K.Z.M.; Saucedo Acuña, R.A.; Ríos Arana, J.V.; Lobo, N.; Rodriguez, C.; Cuevas-Gonzalez, J.C.; Tovar-Carrillo, K.L. Natural Film Based on Pectin and Allantoin for Wound Healing: Obtaining, Characterization, and Rat Model. BioMed Res. Int. 2020, 2020, 6897497. [Google Scholar] [CrossRef] [PubMed]
- Ciganović, P.; Jakupović, L.; Momchev, P.; Nižić Nodilo, L.; Hafner, A.; Zovko Končić, M. Extraction Optimization, Antioxidant, Cosmeceutical and Wound Healing Potential of Echinacea purpurea Glycerolic Extracts. Molecules 2023, 28, 1177. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A. Folin-Ciocalteau Micro Method for Phenol Total in Wine; UCDavis: Davis, CA, USA, 2019. [Google Scholar]
- Jaradat, N.; Mohammed Hussen, F.; Al Ali, A. Preliminary Phytochemical Screening, Quantitative Estimation of Total Flavonoids, Total Phenols and Antioxidant Activity of Ephedra Alata Decne. J. Mater. Environ. Sci. 2015, 6, 1771–1778. [Google Scholar]
- Bukhari, S.B.; Memonb, S.; Mahroof-Tahir, M.; Bhanger Synthesis, M.I. Characterization and antioxidant activity copper–quercetin complex. Spectrochim. Acta 2009, 71, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Khan, S.; Avula, B.; Lata, H.; Hye Yang, M.; ElSohly, M.A.; Khan, I.A. Assessment of Total Phenolic and Flavonoid Content, Antioxidant Properties, and Yield of Aeroponically and Conventionally Grown Leafy Vegetables and Fruit Crops: A Comparative Study. Hindawi Publ. 2014, 2014, 253875. [Google Scholar] [CrossRef]
- Olah, N.K.; Petrescu, S.; Marian, E.; Jurca, T.; Marc, F.; Dobjanschi, L.; Honiges, A.; Kiss, R.; Bechir, E.S.; Bechir, F.; et al. The study of antioxidant capacity in extracts from vegetal sources with hypoglycaemic action. Rev. Chim. 2019, 70, 102–106. [Google Scholar] [CrossRef]
- Tufikul, I.; Mohammad, R.H.; Aumit, R.; Shafigul, I.; Afaz, U.; Ariful, I.; Nuruzzaman, N.; Sohel, R. Screening of in-vitro antioxidant, brine shrimp lethality bioassay and antimicrobial activities of extracts of Bridelia retusa (L.) Spreng. Fruit. Int. J. Pharm. 2015, 5, 1058–1067. [Google Scholar]
- Shirazi, O.U.; Muhammad Muzaffar, A.K.K.; Nor Azwani, M.S.; Mohd Nur, N.A. Determination of total phenolic, flavonoid content and free radical scavenging activities of common herbs and spices. J. Pharmacogn. Phytochem. 2014, 3, 104–108. [Google Scholar]
- Criste, A.; Urcan, A.C.; Bunea, A.; Pripon Furtuna, F.R.; Olah, N.K.; Madden, R.H.; Corcionivoschi, N. Phytochemical Composition and Biological Activity of Berries and Leaves from Four Romanian Sea Buckthorn (Hippophae rhamnoides L.) Varieties. Molecules 2020, 25, 1170. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Hernández, E.; Regalado-González, C.; Vázquez-Landaverde, P.; Guerrero-Legarreta, I.; García-Almendárez, B.E. Microencapsulation, Chemical Characterization, and Antimicrobial Activity of Mexican (Lippia graveolens H.B.K.) and European (Origanum vulgare L.) Oregano Essential Oils. Sci. World J. 2014, 2014, 641814. [Google Scholar] [CrossRef] [PubMed]
- Ghitea, T.C.; El-Kharoubi, A.; Ganea, M.; Bimbo-Szuhai, E.; Nemeth, T.S.; Ciavoi, G.; Foghis, M.; Dobjanschi, L.; Pallag, A.; Micle, O. The Antimicrobial Activity of Origanum vulgare L. Correlated with the Gastrointestinal Perturbation in Patients with Metabolic Syndrome. Molecules 2021, 26, 283. [Google Scholar] [CrossRef] [PubMed]
- Pallag, A.; Filip, G.A.; Olteanu, D.; Clichici, S.; Baldea, I.; Jurca, T.; Micle, O.; Vicaş, L.; Marian, E.; Soriţău, O.; et al. Equisetum arvense L. Extract Induces Antibacterial Activity and Modulates Oxidative Stress, Inflammation, and Apoptosis in Endothelial Vascular Cells Exposed to Hyperosmotic Stress. Hindawi 2018, 2018, 3060525. [Google Scholar] [CrossRef]
- Kabakov, A.E.; Gabai, V.L. Cell Death and Survival Assays. In Methods in Molecular Biology; Calderwood, S., Prince, T., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1709. [Google Scholar] [CrossRef]
- Malich, G.; Markovic, B.; Winder, C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology 1997, 124, 179–192. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard Substance | Equation of the Standard Curve | Coefficients of Determination (R2) |
|---|---|---|
| Gallic acid | A = 0.06209 + 9.06699 × 10−4 × C (μg/mL) | 0.99671 |
| Caffeic acid | A = 0.13564 + 6.47403 × 10−4 × C (μg/mL) | 0.99851 |
| Gallic Acid | Caffeic Acid | |
|---|---|---|
| Echinaceae purpureae folium | 1.41 ± 0.07 mg/g dry plant material | 1.22 ± 0.06 mg/g dry plant material |
| Standard Substance | Equation of the Standard Curve | Regression Coefficients (R2) |
|---|---|---|
| Rutin | A = 0.04786 + 0.00162 × C (μg/mL) | 0.99867 |
| Quercetin | A = −0.03537 + 0.02565 × C (μg/mL) | 0.99807 |
| Rutin | Quercetin | |
|---|---|---|
| Echinaceae purpureae folium | 1.32 ± 0.06 mg RuE/g dry plant material | 0.39 ± 0.02 mg QrE/g dry plant material |
| Standard Substance | Equation of the Standard Curve | Regression Coefficients (R2) |
|---|---|---|
| Ascorbic acid | A = 0.86962 − 0.03804 × C (μg/mL) | 0.99769 |
| Gallic acid | A = 0.53474 − 0.00477 × C (μg/mL) | 0.99573 |
| Caffeic acid | A = 0.96875 − 0.00524 × C (μg/mL) | 0.99871 |
| Ascorbic Acid | Gallic Acid | Caffeic Acid | |
|---|---|---|---|
| Echinaceae purpureae folium | 120.79 ± 0.0961 μg/g dry plant material | 660.71 ± 0.1182 μg/g dry plant material | 1153.63 ± 0.14 μg/g dry plant material |
| Extract | UM/disc | Staphylococcus aureus ATCC 25923 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 |
|---|---|---|---|---|
| Echinaceae purpureae folium 1:1 | 40 μL | 1.13 ± 0.21 | 2.16 ± 0.20 | 2.28 ± 0.17 |
| Echinaceae purpureae folium 1:4 | 40 μL | 15.56 ± 0.36 | 16.09 ± 0.25 | 14.40 ± 0.39 |
| Echinaceae purpureae folium 1:8 | 40 μL | 15.58 ± 0.34 | 18.60 ± 0.38 | 15.31 ± 0.17 |
| Penicillin | 10 mits | 33 | Not tested | Not tested |
| Neomicine | 10 µg | 17 | 17 | Not tested |
| Tetracycline | 30 µg | 27 | 19 | Not tested |
| Gentamicin | 10 µg | 21 | 21 | 18 |
| Ampicillin | 10 µg | 16 | 17 | Not tested |
| Norfloxacin | 10 µg | 21 | 29 | 24 |
| Ciprofloxacin | 5 µg | 26 | 33 | 28 |
| Cefadroxil | 30 µg | Not tested | 17 | Not tested |
| Distilled water | - | 5 | 5 | 5 |
| Sample | % Cell Viability |
|---|---|
| CTRL | 94.25 ± 7.21 |
| ALA_50 | 94.687 ± 5.71 |
| E200 | 93.15 ± 6.80 |
| E100 | 94.55 ± 7.90 |
| E50 | 95.41 ± 4.11 |
| Sample | CTRL | ALA_50 | E50 | E100 | E200 |
|---|---|---|---|---|---|
| Time (h) | Wound closure by width (Wnd_W) | ||||
| 12 | 28.3175 ± 1.7873 | 12.1901 ± 2.4587 | 28.5831 ± 1.1171 | 25.7399 ± 2.3791 | 46.1769 ± 2.8161 |
| 24 | 43.59547 ± 2.2742 | 34.3750 ± 2.3513 | 65.3115 ± 1.5581 | 33.1601 ± 3.4085 | 43.6698 ± 2.7696 |
| 36 | 49.89126 ± 2.6339 | 47.4235 ± 0.8606 | 82.7769 ± 1.0817 | 35.9606 ± 1.0599 | 47.4837 ± 1.4903 |
| Wound closure by area or surface (Wnd_A) | |||||
| 12 | 26.7455 ± 1.4607 | 14.8338 ± 1.7761 | 21.2369 ± 1.6377 | 29.8948 ± 1.194 | 33.4771 ± 1.4980 |
| 24 | 36.5411 ± 1.6776 | 43.1676 ± 1.6365 | 32.3970 ± 1.8371 | 31.1618 ± 1.4967 | 37.160 ± 1.0570 |
| 36 | 51.06132 ± 1.1224 | 46.5938 ± 1.6265 | 92.5526 ± 1.5616 | 42.0614 ± 1.7360 | 70.4225 ± 1.9587 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlou-Nagy, C.; Bănică, F.; Negrean, R.A.; Jurca, T.; Vicaș, L.G.; Marian, E.; Bácskay, I.; Kiss, R.; Fehér, P.; Vicaș, S.I.; et al. Determination of the Bioactive Compounds from Echinacea purpurea (L.) Moench Leaves Extracts in Correlation with the Antimicrobial Activity and the In Vitro Wound Healing Potential. Molecules 2023, 28, 5711. https://doi.org/10.3390/molecules28155711
Burlou-Nagy C, Bănică F, Negrean RA, Jurca T, Vicaș LG, Marian E, Bácskay I, Kiss R, Fehér P, Vicaș SI, et al. Determination of the Bioactive Compounds from Echinacea purpurea (L.) Moench Leaves Extracts in Correlation with the Antimicrobial Activity and the In Vitro Wound Healing Potential. Molecules. 2023; 28(15):5711. https://doi.org/10.3390/molecules28155711
Chicago/Turabian StyleBurlou-Nagy, Cristina, Florin Bănică, Rodica Anamaria Negrean, Tünde Jurca, Laura Grațiela Vicaș, Eleonora Marian, Ildikó Bácskay, Rita Kiss, Pálma Fehér, Simona Ioana Vicaș, and et al. 2023. "Determination of the Bioactive Compounds from Echinacea purpurea (L.) Moench Leaves Extracts in Correlation with the Antimicrobial Activity and the In Vitro Wound Healing Potential" Molecules 28, no. 15: 5711. https://doi.org/10.3390/molecules28155711
APA StyleBurlou-Nagy, C., Bănică, F., Negrean, R. A., Jurca, T., Vicaș, L. G., Marian, E., Bácskay, I., Kiss, R., Fehér, P., Vicaș, S. I., Miere, F., Memete, A. R., & Pallag, A. (2023). Determination of the Bioactive Compounds from Echinacea purpurea (L.) Moench Leaves Extracts in Correlation with the Antimicrobial Activity and the In Vitro Wound Healing Potential. Molecules, 28(15), 5711. https://doi.org/10.3390/molecules28155711