
Structures and Spectroscopic Properties of Polysulfide Radical Anions: A Theoretical Perspective

Abstract
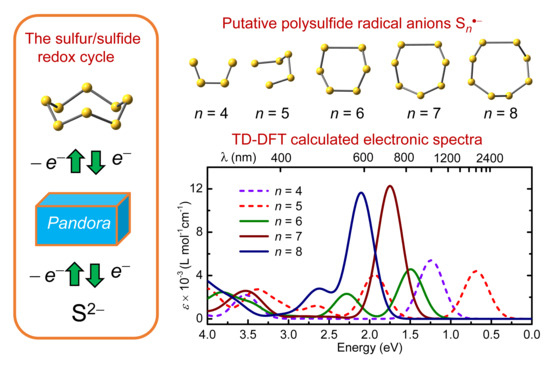
1. Introduction
2. Results
2.1. Structural Trends
2.1.1. S2•− and S3•−
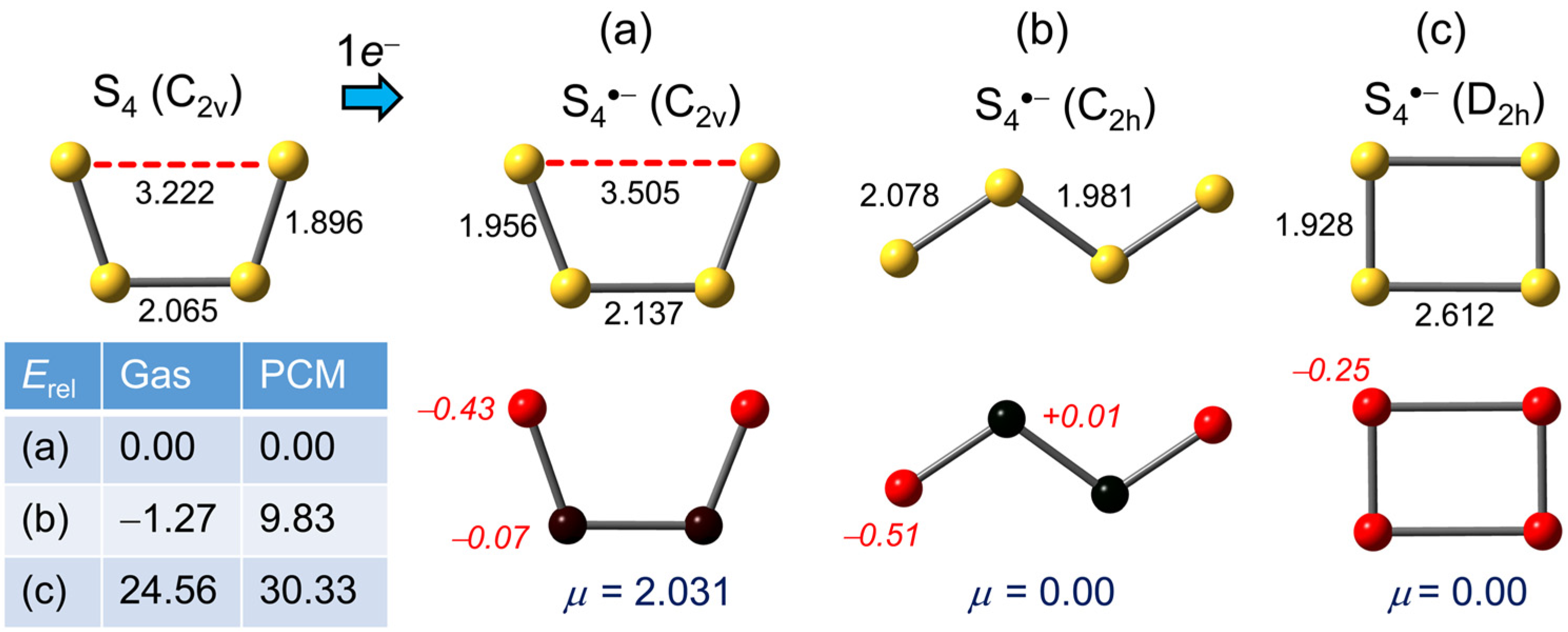
2.1.2. S4•−
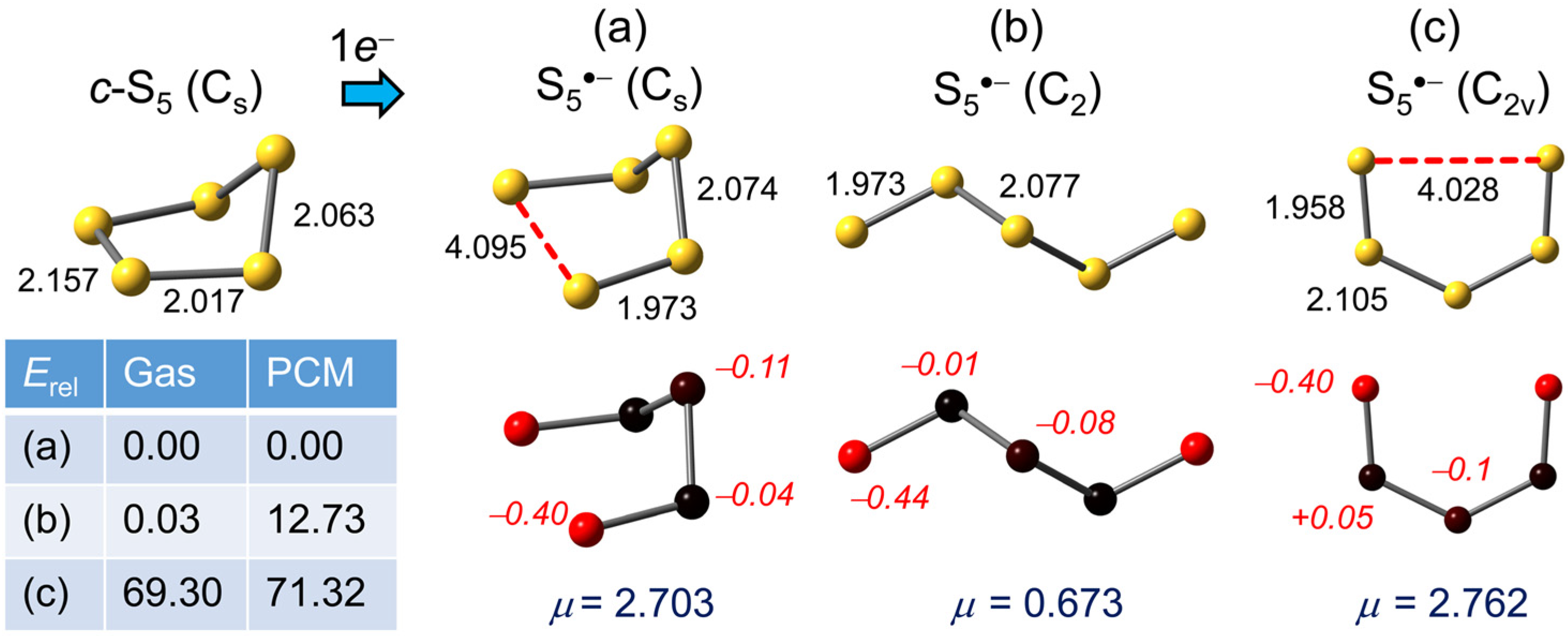
2.1.3. S5•−
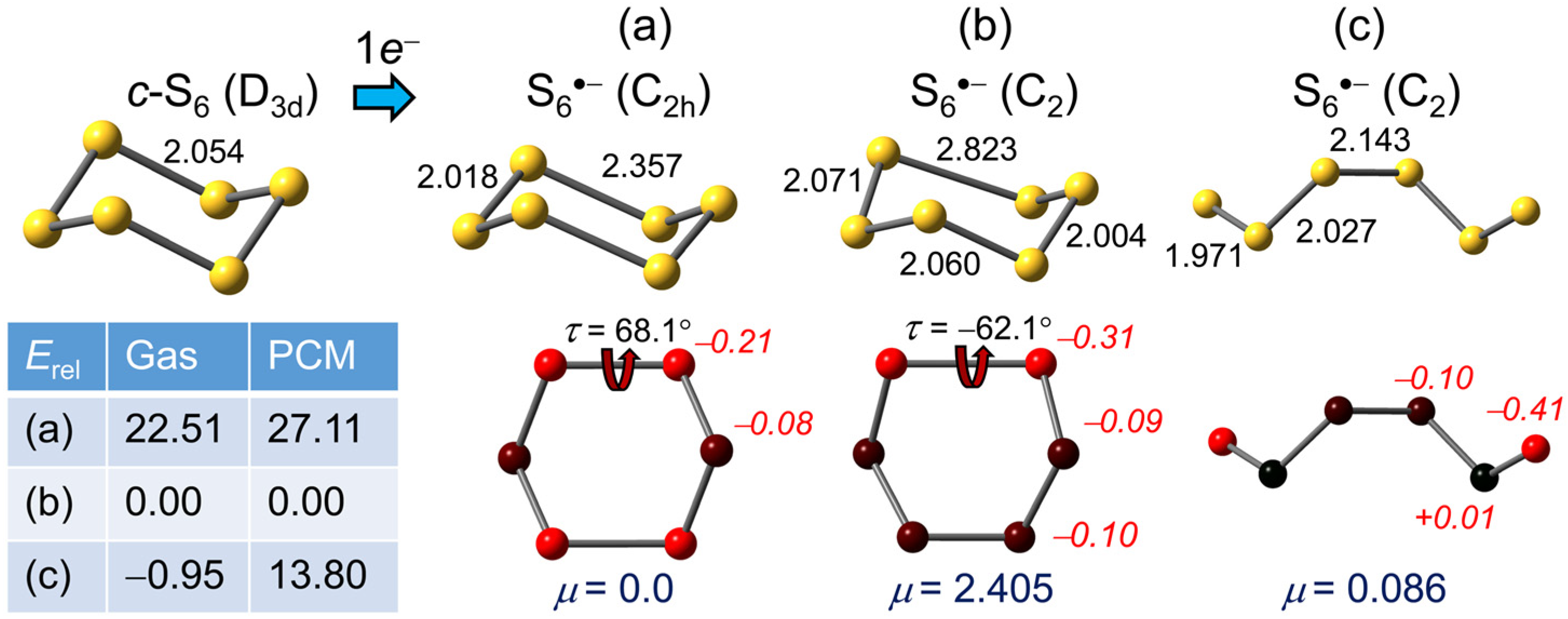
2.1.4. S6•−
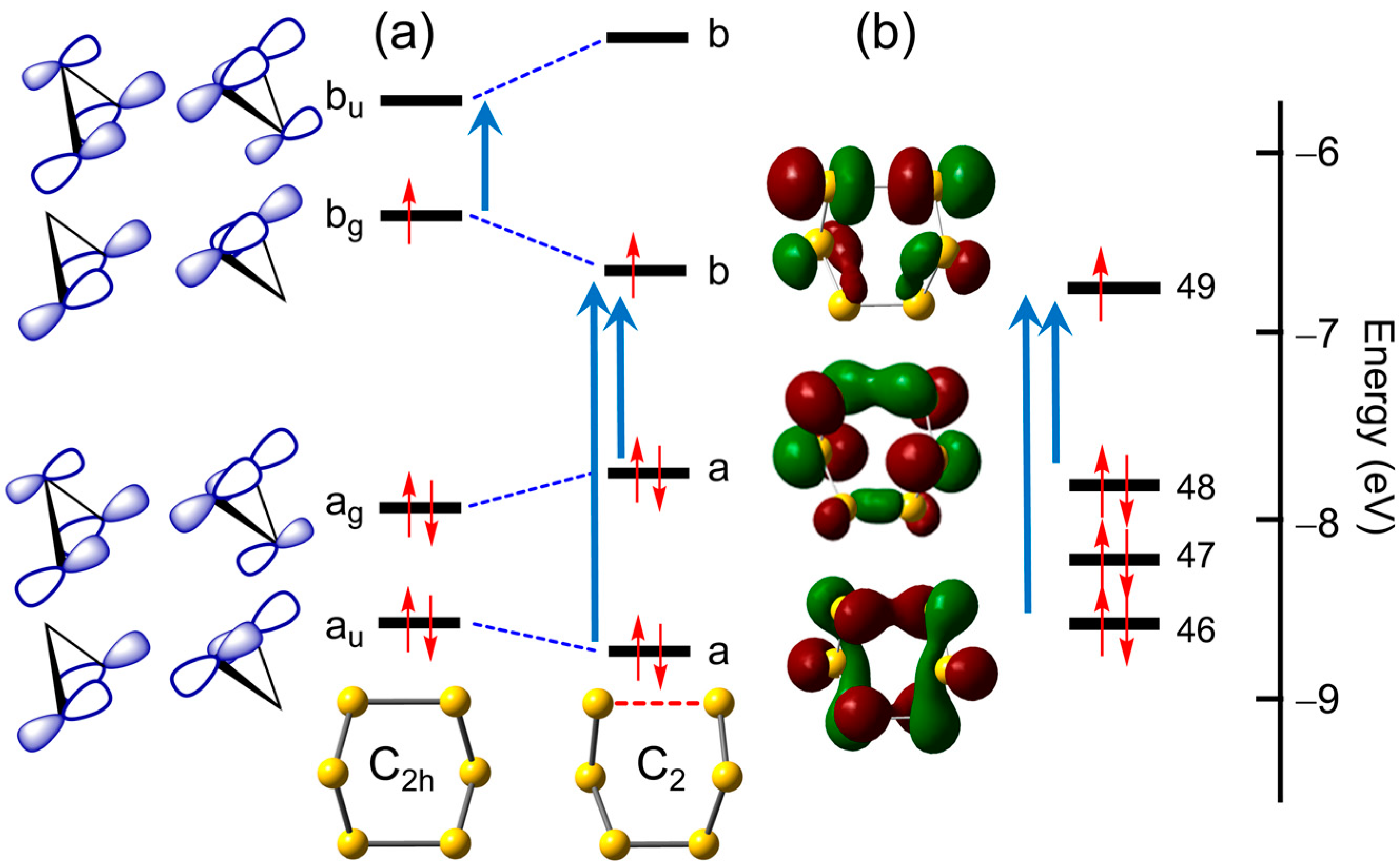
2.1.5. S7•−
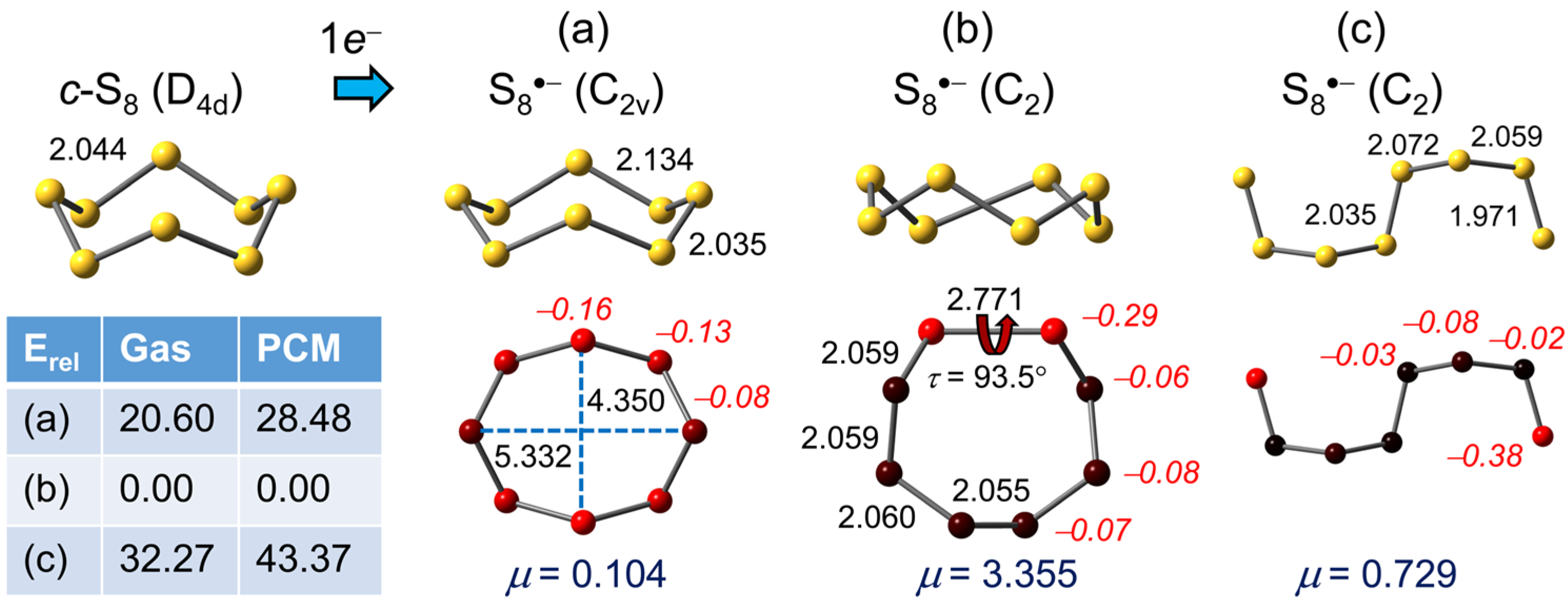
2.1.6. S8•−
2.2. Electronic Spectra
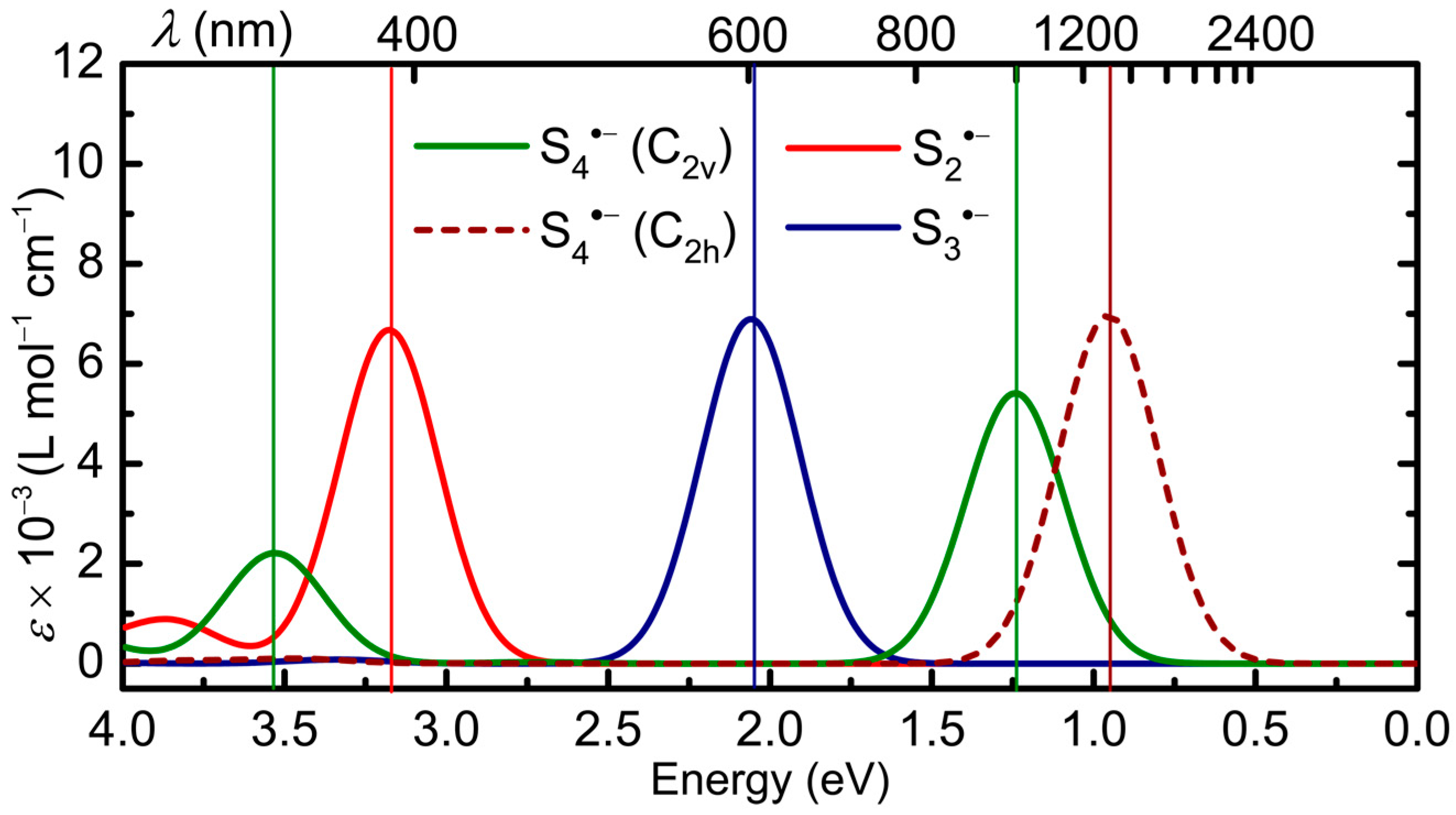
2.2.1. Sn•− (n = 2–4)
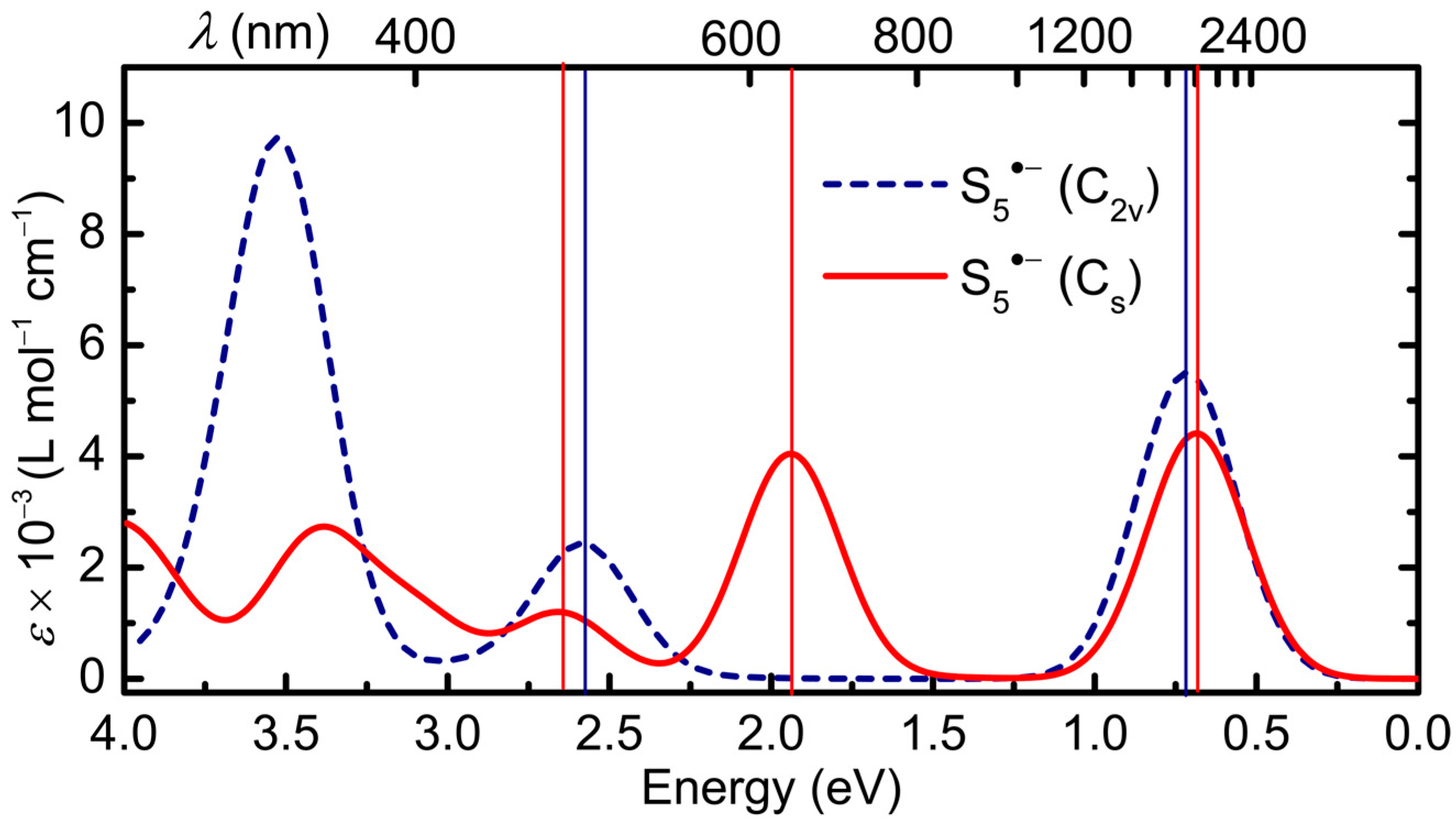
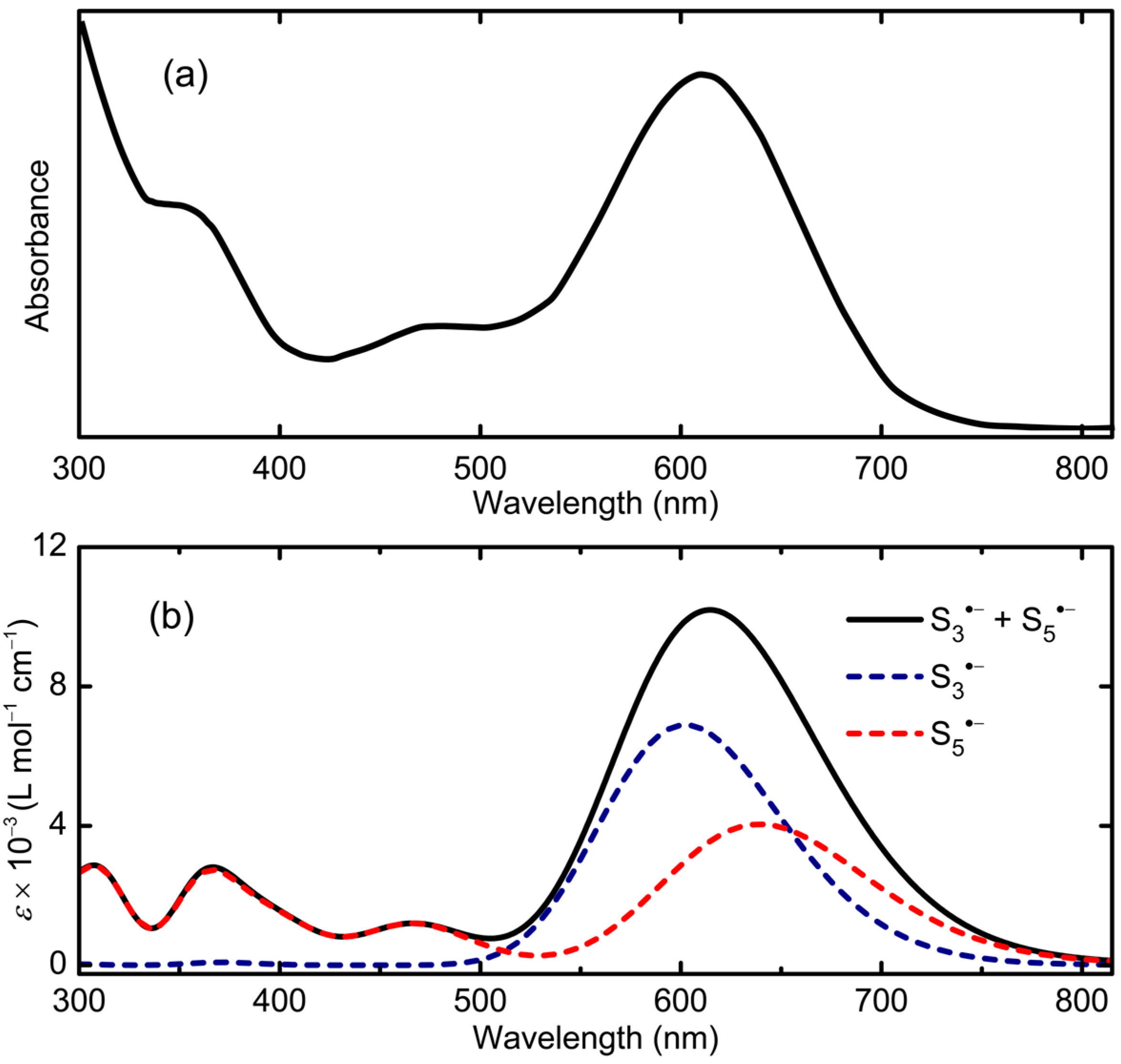
2.2.2. S5•−
2.2.3. S6•−
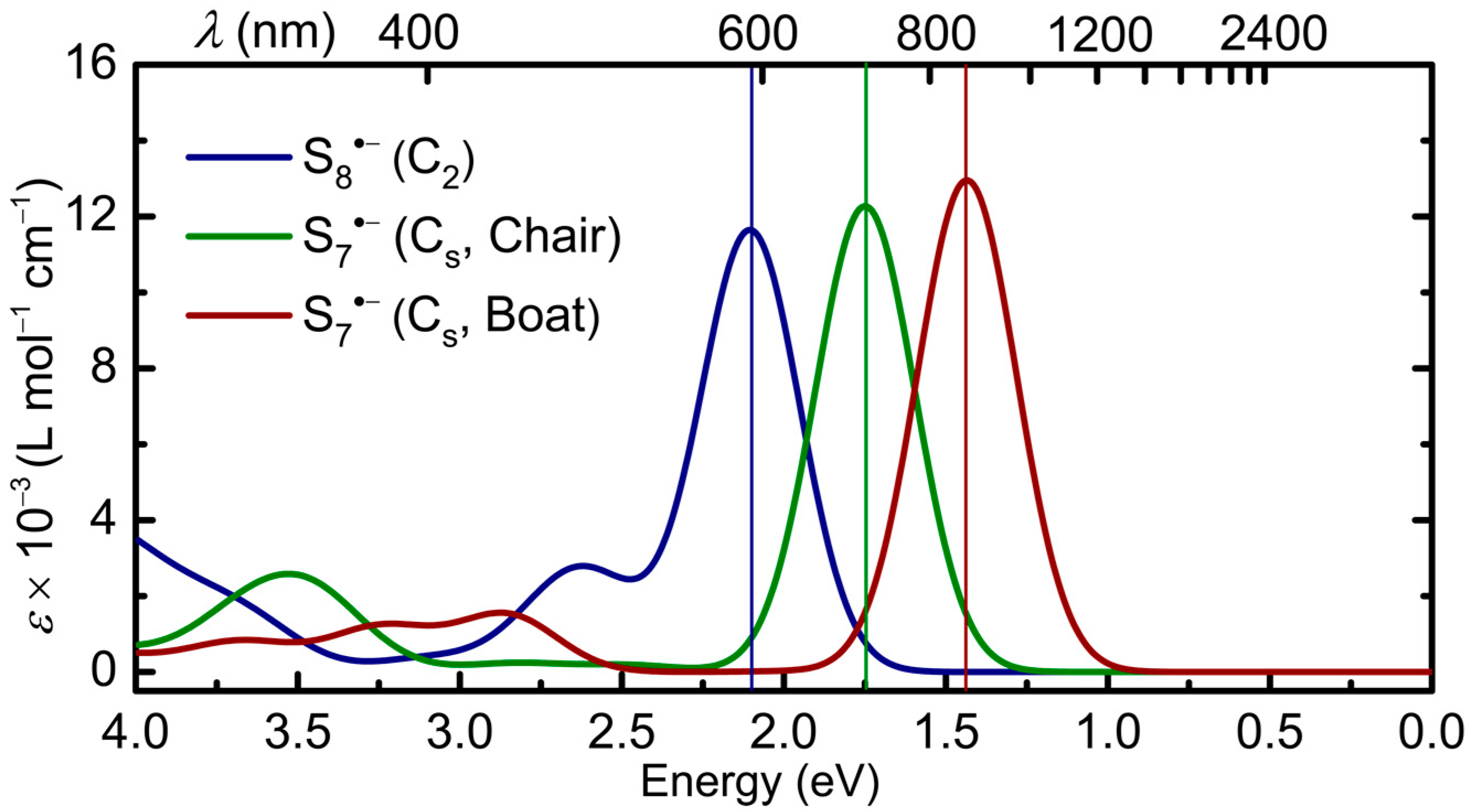
2.2.4. S7•− and S8•−
3. Discussion
4. Computational Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chivers, T. Ubiquitous trisulphur radical ion S3•−. Nature 1974, 252, 32–33. [Google Scholar] [CrossRef]
- Steudel, R. Inorganic polysulfides Sn2− and radical anions Sn•−. In Elemental Sulfur and Sulfur-Rich compounds II. Topics in Current Chemistry; Steudel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 231, pp. 127–152. [Google Scholar]
- Steudel, R.; Chivers, T. The role of polysulfide dianions and radical anions in the chemical, physical and biological sciences, including sulfur-based batteries. Chem. Soc. Rev. 2019, 48, 3279–3319. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.S.; Oilunkaniemi, R.; Chivers, T.; McGeachie, L.; Kelly, P.F.; King, R.S.P. Polychalcogen molecules, ligands, and ions Part 2: Catenated acyclic molecules, ions, and p-block element derivatives. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2023; pp. 970–1020. [Google Scholar]
- Zheng, D.; Wang, G.; Liu, D.; Si, J.; Ding, T.; Qu, D.; Yang, X.; Qu, D. The progress of Li−S batteries—Understanding of the sulfur redox mechanism: Dissolved polysulfide ions in the electrolytes. Adv. Mater. Technol. 2018, 3, 1700233. [Google Scholar] [CrossRef]
- Zhao, E.; Nie, K.; Yu, X.; Hu, Y.-S.; Wang, F.; Xiao, J.; Li, H.; Huang, X. Advanced characterization techniques in promoting mechanism understanding for lithium-sulfur batteries. Adv. Func. Mater. 2018, 28, 1707543. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Z.-W.; Peng, H.-J.; Huang, J.-Q.; Zhang, Q. A toolbox for lithium-sulfur research: Methods and protocols. Small Methods 2017, 1, 1700134. [Google Scholar] [CrossRef]
- Song, P.; Rao, W.; Chivers, T.; Wang, S.-Y. Applications of trisulfide radical anion S3•− in organic chemistry. Org. Chem. Front. 2023, 10, 3378–3401. [Google Scholar] [CrossRef]
- Bogdándi, V.; Ida, T.; Sutton, T.R.; Bianco, C.; Ditrói, T.; Koster, G.; Henthorn, H.A.; Minnion, M.; Toscano, J.P.; van der Vliet, A.; et al. Speciation of reactive sulfur species and their reactions with alkylating agents: Do we have any clue about what is present in the cell? Br. J. Pharmacol. 2019, 176, 646–670. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Kuhnle, G.G.C.; Dyson, A.; Fernandez, B.O.; Grman, M.; DuMond, J.F.; Barrow, M.P.; McLeod, G.; Nakagawa, H.; Ondrias, K.; et al. Key bioactive reaction products of the NO/H2S interaction are S/N hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. USA 2015, 112, E4651–E4660. [Google Scholar] [CrossRef] [PubMed]
- Pokrovski, G.S.; Dubrovinsky, L.S. The S3•− ion is stable in geological fluids at elevated temperatures and pressures. Science 2011, 331, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Pokrovski, G.S.; Kokh, M.A.; Guillaume, D.; Borisova, A.Y.; Gisquet, P.; Hazemann, J.-L.; Lahera, E.; Del Net, W.; Proux, O.; Testemale, D.; et al. Sulfur radical species form gold deposits on earth. Proc. Nat. Acad. Sci. USA 2015, 112, 13484–13489. [Google Scholar] [CrossRef]
- Pokrovski, G.S.; Kokh, M.A.; Desmaele, E.; Laskar, C.; Bazarkina, E.F.; Borisova, A.Y.; Testemale, D.; Hazemann, J.-L.; Vuilleumier, R.; Ferlat, G.; et al. The trisulfur radical ion S3•− controls platinum transport by hydrothermal fluids. Proc. Nat. Acad. Sci. USA 2021, 118, e2109768118. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; McNaughter, P.D.; O’Brien, P.; Minamimoto, H.; Murakoshi, K. Photoelectrochemical formation of polysulfide at PbS QD-sensitized plasmonic electrodes. J. Phys. Chem. Lett. 2019, 10, 5357–5363. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, V.; Baker, D.; Kamat, P.V. Understanding the role of the sulfide redox couple (S2−/Sn2−) in quantum dot-sensitized solar cells. J. Am. Chem. Soc. 2011, 133, 9607–9615. [Google Scholar] [CrossRef]
- Reinen, D.; Lindner, G.-G. The nature of the chalcogen colour centres in ultramarine type solids. Chem. Soc. Rev. 1999, 28, 75–84. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Sapozhnikov, A.N.; Shendrik, R.Y.; Vigasina, M.F.; Steudel, R. Spectroscopic and crystal-chemical features of sodalite-group minerals from gem lazurite deposits. Minerals 2020, 10, 1042. [Google Scholar] [CrossRef]
- Chivers, T.; Elder, P.J.W. Ubiquitous trisulfur radical anion: Fundamentals and applications in materials science, electrochemistry, analytical chemistry, and geochemistry. Chem. Soc. Rev. 2013, 42, 5996–6005. [Google Scholar] [CrossRef]
- Seel, F. Polysulfide radical anions. Angew. Chem. Int. Ed. 1973, 12, 420–421. [Google Scholar] [CrossRef]
- Seel, F.; Guttler, H.-J.; Simon, G.; Wieckowski, A. Colored sulfur species. Pure Appl. Chem. 1977, 49, 45–54. [Google Scholar] [CrossRef]
- Schwamm, R.J.; Lein, M.; Coles, M.P.; Fitchett, C.M. Bismuth (III) complex of the S4•− radical anion: Dimer formation via pancake bonds. J. Am. Chem. Soc. 2017, 139, 16490–16493. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.H.; Dines, T.J.; Kurmoo, M. The nature of the sulfur chromophore in ultramarine blue, green, violent, and pink and of the selenium chromophore in ultramarine selenium. Characterization of radical anions by electronic and resonance Raman spectroscopy and the determination of their excited-state geometries. Inorg. Chem. 1983, 22, 2766–2772. [Google Scholar]
- Rauh, R.D.; Shuker, F.S.; Marston, J.M.; Brummer, S.B. Formation of lithium polysulfides in aprotic media. J. Inorg. Nucl. Chem. 1977, 39, 1761–1766. [Google Scholar] [CrossRef]
- Li, H.; Tang, X.; Pang, J.H.; Wu, X.; Yeow, E.K.L.; Wu, J.; Chiba, S. Polysulfide anions as visible light photoredox catalysts for aryl cross-couplings. J. Am. Chem. Soc. 2021, 143, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Chiba, S. Leveraging of sulfur anions in photoinduced molecular transformations. JACS Au 2021, 1, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Chiba, S. Anti-Markovnikov hydroarylation of alkenes via polysulfide anion photocatalysis. Chem. Commun 2021, 57, 6264–6267. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chiba, S. Synthesis of α-tertiary amines by polysulfide anions photocatalysis via single-electron transfer and hydrogen atom transfer in relays. Chem. Catal. 2022, 2, 1128–1142. [Google Scholar] [CrossRef]
- Hagen, M.; Schiffels, P.; Hammer, M.; Dörfler, S.; Tübke, J.; Hoffmann, M.; Althues, H.; Kaskel, S. In-situ Raman investigation of polysulfide formation in Li-S cells. J. Electrochem. Soc. 2013, 160, A1205–A1215. [Google Scholar] [CrossRef]
- Clark, R.J.H.; Cobbold, D.G. Characterization of sulfur radical anions in solutions of alkali polysulfides in DMF and HMPA and in the solid state in ultramarine blue, green, and red. Inorg. Chem. 1978, 17, 3169–3174. [Google Scholar] [CrossRef]
- Wu, H.-L.; Huff, L.A.; Gewirth, A.A. In-situ Raman spectroscopy of sulfur speciation in Li-S batteries. Appl. Mater. Interfaces 2015, 7, 1709–1719. [Google Scholar] [CrossRef]
- Levillain, E.; Leghié, P.; Gobeltz, N.; Lelieur, J.-P. Identification of the S4•− radical anion in solution. New J. Chem. 1997, 21, 335–341. [Google Scholar]
- Wujcik, K.H.; Wang, D.R.; Raghunathan, A.; Drake, M.; Pascal, T.A.; Prendergast, D.; Balsara, N.P. Lithium polysulfide radical anions in ether-based solvents. J. Phys. Chem. C 2016, 120, 18403–18410, and references cited therein. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Shendrik, R.Y.; Vigasina, M.F.; Pekov, I.V.; Sapozhnikov, A.N.; Shcherbakov, V.D.; Varlamov, D.A. Crystal chemistry, isomorphism, and thermal conversions of extra-framework components in sodalite-group minerals. Minerals 2022, 12, 887. [Google Scholar] [CrossRef]
- Merritt, M.V.; Sawyer, D.T. Electrochemical reduction of elemental sulfur in aprotic solvents. Formation of a stable S8•− species. Inorg. Chem. 1970, 9, 211–215. [Google Scholar] [CrossRef]
- Martin, R.P.; Doub, W.H.; Roberts, J.L.; Sawyer, D.T. Further studies of the electrochemical reduction of sulfur in aprotic solvents. Inorg. Chem. 1973, 12, 1921–1924. [Google Scholar] [CrossRef]
- Bonnaterre, R.; Cauquis, G. Spectrophotometric study of the electrochemical reduction of sulphur in organic media. J. Chem. Soc. Chem. Commun. 1972, 293–294. [Google Scholar] [CrossRef]
- Manan, N.S.A.; Aldous, L.; Alias, Y.; Murray, P.; Yellowlees, L.J.; Lagunas, M.C.; Hardacre, C. Electrochemistry of sulfur and polysulfides in ionic liquids. J. Phys. Chem. B 2011, 115, 13873–13879. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, S.; Kim, B.S.; Han, D.H.; Park, S.M.; Kwak, J. Effect of organic solvents and electrode materials on electrochemical reduction of sulfur. J. Electrochem. Soc. 2008, 3, 566–577. [Google Scholar] [CrossRef]
- Gaillard, F.; Levillain, E.; Lelieur, J.-P. Polysulfides in DMF: Only the radical anions S3•− and S4•− are reducible. J. Electroanal. Chem. 1997, 432, 129–138. [Google Scholar] [CrossRef]
- Evans, A.; Montenegro, M.I.; Pletcher, D. The mechanism for the cathodic reduction of sulphur in DMF: Low temperature voltammetry. Electrochem. Commun. 2001, 3, 514–518. [Google Scholar] [CrossRef]
- Levillain, E.; Gaillard, F.; Leghié, P.; Demortier, A.; Lelieur, J.-P. On the understanding of the reduction of sulfur (S8) in dimethylformamide (DMF). J. Electroanal. Chem. 1997, 420, 167–177. [Google Scholar] [CrossRef]
- Leghié, P.; Lelieur, J.-P.; Levillain, E. Final comment on Reply to “Comments on the mechanism of the electrochemical reduction of sulphur in DMF”. Electrochem. Commun. 2002, 4, 406–411. [Google Scholar] [CrossRef]
- Steudel, R.; Steudel, Y. Polysulfide chemistry in sodium-sulfur batteries and related systems: A computational study by G3X(MP2) and PCM calculations. Chem. Eur. J. 2013, 19, 3162–3176. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-S.; Park, S.-M. In situ spectroelectrochemical studies on the reduction of sulfur in dimethyl sulfoxide solutions. J. Electrochem. Soc. 1993, 140, 115–122. [Google Scholar] [CrossRef]
- Han, D.-H.; Kim, B.-S.; Choi, S.-J.; Jung, Y.; Kwak, J.; Park, S.-M. Time-resolved in situ spectroelectrochemical study on reduction of sulfur in DMF. J. Electrochem. Soc. 2004, 151, E283–E290. [Google Scholar] [CrossRef]
- Cuisinier, M.; Hart, C.; Balusubramanian, M.; Garsuch, A.; Nazar, L.F. Radical or not radical: Revisiting lithium-sulfur electrochemistry in non-aqueous solvents. Adv. Energy Mater. 2015, 5, 1401801. [Google Scholar] [CrossRef]
- Schliephke, A.; Falius, H.; Buchkremer-Hermanns, H.; Bottcher, P. Preparation and crystal structure of the bis(triethylammonium) octasulfide, [HN(C2H5)3]2S8. Z. Naturforsch. 1988, 43b, 21–24. [Google Scholar]
- Dev, S.; Ramli, E.; Rauchfuss, T.B.; Wilson, S.R. Synthesis and structure of [M(N-methylimidazole)6]S8 (M = Mn, Fe, Ni, Mg). Polysulfide salts prepared by the reaction of N-methylimidazole + metal powder + sulfur. Inorg. Chem. 1991, 30, 2514–2519. [Google Scholar] [CrossRef]
- Fujinaga, T.; Kuwamoto, T.; Okazaki, S.; Hojo, M. Electrochemical Reduction of Elemental Sulfur in Acetonitrile. Bull. Chem. Soc. Jpn. 1980, 53, 2851–2855. [Google Scholar] [CrossRef]
- Mondal, M.K.; Zhang, L.; Feng, Z.; Tang, S.; Feng, R.; Zhao, Y.; Tan, G.; Ruan, H.; Wang, X. Tricoordinate pnictogen-centered radical anions: Isolation, characterization, and reactivity. Angew. Chem. Int. Ed. 2019, 58, 15829–15833. [Google Scholar] [CrossRef]
- Liebing, P.; Kühling, M.; Swanson, C.; Feneberg, M.; Hilfert, L.; Goldhahn, R.; Chivers, T.; Edelmann, F.T. Catenated and spirocyclic polychalcogenides from potassium carbonate and elemental chalcogens. Chem. Commun. 2019, 55, 14965–14967. [Google Scholar] [CrossRef] [PubMed]
- Neumüller, B.; Schmock, F.; Kirmse, R.; Voigt, A.; Diefenbach, A.; Bickelhaupt, F.M.; Dehnicke, K. (Ph4P)S6−A compound containing the cyclic radical anion S6•−. Angew. Chem. Int. Ed. 2002, 39, 4580–4582. [Google Scholar] [CrossRef]
- Fabian, J.; Komiha, N.; Linguerri, R.; Rosmus, P. The absorption wavelengths of sulfur chromophores of ultramarines calculated by TD-DFT. J. Mol. Struct. THEOCHEM 2006, 801, 63. [Google Scholar] [CrossRef]
- Rejmak, P. Computational refinement of the puzzling red tetrasulfur chromophore in ultramarine pigments. PhysChemChemPhys 2020, 22, 22684–22698. [Google Scholar] [CrossRef]
- Hunsicker, S.; Jones, R.O.; Ganteför, G. Rings and chains in sulfur cluster anions S− to S9−: Theory (simulated annealing) and experiment (photoelectron detachment). J. Chem. Phys. 1995, 102, 5917–5936. [Google Scholar] [CrossRef][Green Version]
- Wong, M.W. Quantum-chemical calculations of sulfur-rich compounds. Elemental Sulfur and Sulfur-Rich compounds II. Topics in Current Chemistry; Steudel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 231, pp. 1–29. [Google Scholar]
- Swope, W.C.; Lee, Y.-P.; Schaefer, H.F. Diatomic sulfur: Low lying bound molecular electronic states of S2. J. Chem. Phys. 1979, 70, 947–953. [Google Scholar] [CrossRef]
- Zakrzewski, V.G.; von Niessen, W. Structures, stabilities and adiabatic ionization and electron affinity energies of small sulfur clusters. Theor. Chim. Acta 1994, 88, 75–96. [Google Scholar] [CrossRef]
- Koch, W.; Natterer, J.; Heinemann, C. Quantum chemical study on the equilibrium geometries of S3 and S3−, The electron affinity of S3 and the low lying electronic states of S3−. J. Chem. Phys. 1995, 102, 6159–6167. [Google Scholar] [CrossRef]
- Wong, M.W.; Steudel, R. Structure and spectra of tetrasulfur S4 − an ab initio MO study. Chem. Phys. Lett. 2003, 379, 162–169. [Google Scholar] [CrossRef]
- Pearson, R.G. Symmetry Rules for Chemical Reactions: Orbital Topology and Elementary Processes; John Wiley and Sons: New York, NY, USA, 1976; pp. 78–79. [Google Scholar]
- Jones, R.O.; Ballone, P. Density functional and Monte Carlo studies of sulfur. I. Structure and bonding in Sn rings and chains (n = 2–18). J. Chem. Phys. 2003, 118, 9257–9265. [Google Scholar] [CrossRef]
- Chivers, T.; Laidlaw, W.G.; Oakley, R.T.; Trsic, M. Synthesis, crystal and molecular structure of [(Ph3P)2N+][S4N−], and the electronic structure of the acyclic anion, S4N−. J. Am. Chem. Soc. 1980, 102, 5773–5781. [Google Scholar] [CrossRef]
- Burford, N.; Chivers, T.; Cordes, A.W.; Oakley, R.T.; Pennington, W.T.; Swepston, P.N. Variable geometry of the S4N− anion: Crystal and molecular structure of Ph4As+S4N− and a refinement of the structure of PPN+ S4N− (PPN = [Ph3P]2N+). Inorg. Chem. 1981, 20, 4430–4432. [Google Scholar] [CrossRef]
- Donohue, J.; Caron, A.; Goldish, E. The crystal and molecular structure of S6 (sulfur-6). J. Am. Chem. Soc. 1961, 83, 3748–3751. [Google Scholar] [CrossRef]
- Steudel, R.; Steidel, J.; Pickardt, J.; Schuster, F.; Reinhardt, R. X-ray structural analyses of two allotropes of cycloheptasulfur (γ and δ-S7). Z. Naturforsch. 1980, 35b, 1378–1383. [Google Scholar] [CrossRef][Green Version]
- Schmidt, M.; Block, B.; Block, H.H.; Köpf, H.; Wilhelm, E. Cycloheptasulfur, S7, and cyclododecasulfur, S12: Two new sulfur rings. Angew. Chem. Int. Ed. 1968, 7, 632–633. [Google Scholar] [CrossRef]
- Rettig, S.R.; Trotter, J. Refinement of the structure of orthorhombic sulfur, α-S8. Acta Crystallogr. 1987, C43, 2260–2262. [Google Scholar] [CrossRef]
- Bonini, M.G.; Augusto, O. Carbon dioxide stimulates the production of thiyl, sulfinyl, and disulfide radical anion from thiol oxidation by peroxynitrite. J. Biol. Chem. 2001, 276, 9749–9754. [Google Scholar] [CrossRef]
- Yamaji, M.; Tojo, S.; Takehira, K.; Tobita, S.; Fujitsuka, M.; Majima, T. S-S bond mesolysis in α,α′-dinaphthyl disulfide radical anion generated during γ-radiolysis and pulse radiolysis in organic solution. J. Phys. Chem. A 2006, 110, 13487–13491. [Google Scholar] [CrossRef]
- Albright, T.A.; Burdett, J.K.; Whangbo, M.-H. Orbital Interactions in Chemistry; John Wiley and Sons: New York, NY, USA, 1985; pp. 212–213. [Google Scholar]
- Heilbronner, E.; Bock, H. The HMO Model and its Application. 1. Basis and Manipulation; John Wiley and Sons: New York, NY, USA, 1976; pp. 131–132. [Google Scholar]
- Chivers, T.; Drummond, I. Characterization of the trisulfur radical anion S3•− in blue solutions of alkali polysulfides in hexamethylphosphoramide. Inorg. Chem. 1972, 11, 2525–2527. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Berry, D.E.; Fawkes, K.L.; Chivers, T. Student-designed experiment: Preparation and mass spectrum of cyclohexasulfur. Chem. Educ. 2001, 6, 109–111. [Google Scholar] [CrossRef]
- Boeré, R.T.; Chivers, T.; Roemmele, T.L.; Tuononen, H.M. An electrochemical and electronic structure investigation of the [S3N3]∙ radical and kinetic modeling of the [S4N4]n/[S3N3]n (n = 0, −1) interconversion. Inorg. Chem. 2009, 48, 7294–7306. [Google Scholar] [CrossRef]
- Derendorf, J.; Jenne, C.; Keßler, M. The first step of the oxidation of elemental sulfur: Crystal structure of the homopolyatomic sulfur radical cation [S8]∙+. Angew. Chem. Int. Ed. 2017, 56, 8281–8284. [Google Scholar] [CrossRef]
- Jagg, P.N.; Kelly, P.F.; Rzepa, H.S.; Williams, D.J.; Woollins, J.D.; Wylie, W. The preparation, x-ray crystal structure and theoretical study of [CoCp2][S3N3], (Cp = cyclopentadienyl), a novel stacking compound incorporating multiple C-H—N(pπ) interactions. J. Chem. Soc. Chem. Commun. 1991, 942–944. [Google Scholar] [CrossRef]
- Konchenko, S.N.; Gritsan, N.P.; Lonchakov, A.V.; Irtegova, I.G.; Mews, R.; Ovcharenko, V.I.; Radius, U.; Zibarev, A.V. Cobaltocenium [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl: Synthesis, structure, and magnetic properties. Eur. J. Inorg. Chem. 2008, 2008, 3833–3838. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Chivers, T.; Edelmann, F.; Richardson, J.F.; Schmidt, K.J. A convenient synthesis, X-ray crystal structure and Raman spectrum of the heptasulfide ion, S72−, in [PPN]2S7.2EtOH. Can. J. Chem. 1986, 64, 145–151. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V.J. Toward reliable density functional methods without adjustable parameters: The PBE0 model. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Treitel, N.; Shenhar, R.; Aprahamian, I.; Sheradsky, T.; Rabinovitz, M. Calculations of PAH anions: When are diffuse functions necessary? Phys. Chem. Chem. Phys. 2004, 6, 1113–1121. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. Eur. J. 2012, 18, 9955–9965. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Wang, C.-W.; Hui, K.; Chai, J.-D. Short- and long-range corrected hybrid density functionals with the D3 dispersion corrections. J. Chem. Phys. 2016, 145, 204101. [Google Scholar] [CrossRef]
- Li, S.L.; Truhlar, D.G. Improving Rydberg excitations within time-dependent density functional theory with generalized gradient approximations: The exchange-enhancement-for-large-gradient scheme. J. Chem. Theory Comput. 2015, 11, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, W. Critical assessment of TD-DFT for excited states of open-shell systems: I. doublet–doublet transitions. J. Chem. Theory Comput. 2016, 12, 238–260. [Google Scholar] [CrossRef]
- Fedunov, R.G.; Pozdnyakov, I.P.; Isaeva, E.A.; Zherin, I.I.; Egorov, N.B.; Glebov, E.M. Sulfur-containing radical anions formed by photolysis of thiosulfate: Quantum-chemical analysis. J. Phys. Chem. A 2023, 127, 4704–4714. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Liang, J.; Feng, X.; Hait, D.; Head-Gordon, M. Revisiting the performance of time-dependent density functional theory for electronic excitations: Assessment of 43 popular and recently developed functionals from rungs one to four. J. Chem. Theory Comput. 2022, 18, 3460–3473. [Google Scholar] [CrossRef] [PubMed]
- GaussView, Version 6.0.16; Dennington, R., Keith, T.A., Millam, J.M., Eds.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n in Sn•− | State | Excitation a | State | ν, eV | λ, nm | f |
|---|---|---|---|---|---|---|
| 2 (D∞h) | 2Σg | 15β → 17β | 2Σu | 3.177 | 390.3 | 0.0891 |
| 3 (C2v) | 2B1 | 24β → 25β | 2B2 | 2.059 | 602.1 | 0.0920 |
| 4 (C2v) | 2A2 | 32β → 33β | 2B2 | 1.242 | 998.3 | 0.0722 |
| 2A2 | 29β → 33β | 2A1 | 3.533 | 351.0 | 0.0286 | |
| 4 (C2h) | 2Bg | 32β → 33β | 2Bu | 0.954 | 1299.5 | 0.0930 |
| 2Bg | 29β → 33β | 2Ag | 3.578 | 346.6 | 0.0000 b | |
| 5 (C2v) | 2B1 | 40β → 41β | 2B2 | 0.716 | 1732.3 | 0.0734 |
| 2B1 | 38β → 41β | 2A1 | 2.583 | 479.9 | 0.0332 | |
| 2B1 | 39β → 42β | 2A1 | 3.519 | 352.4 | 0.1258 | |
| 5 (Cs) | 2A″ | 40β → 41β | 2A″ | 0.685 | 1809.6 | 0.0589 |
| 2A″ | 38β → 41β | 2A″ | 1.940 | 638.2 | 0.0540 | |
| 2A″ | 37β → 41β | 2A″ | 2.653 | 467.4 | 0.0158 | |
| 6 (C2h) | Bg | 49α → 50α | Au | 1.231 | 1007.3 | 0.0445 |
| Bg | 48β → 49β | Bg | 1.978 | 627.5 | 0.0000 b | |
| 6 (C2) | 2B | 48β → 49β | 2B | 1.495 | 829.6 | 0.0611 |
| 2B | 46β → 49β | 2B | 2.283 | 543.1 | 0.0308 | |
| 7 (Cs chair) | 2A″ | 56β → 57β | 2A″ | 1.749 | 708.7 | 0.1637 |
| 7 (Cs boat) | 2A″ | 56β → 57β | 2A″ | 1.435 | 863.9 | 0.1729 |
| 8 (C2 crown) | 2B | 64β → 65β | 2B | 2.104 | 589.3 | 0.1551 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chivers, T.; Oakley, R.T. Structures and Spectroscopic Properties of Polysulfide Radical Anions: A Theoretical Perspective. Molecules 2023, 28, 5654. https://doi.org/10.3390/molecules28155654
Chivers T, Oakley RT. Structures and Spectroscopic Properties of Polysulfide Radical Anions: A Theoretical Perspective. Molecules. 2023; 28(15):5654. https://doi.org/10.3390/molecules28155654
Chicago/Turabian StyleChivers, Tristram, and Richard T. Oakley. 2023. "Structures and Spectroscopic Properties of Polysulfide Radical Anions: A Theoretical Perspective" Molecules 28, no. 15: 5654. https://doi.org/10.3390/molecules28155654
APA StyleChivers, T., & Oakley, R. T. (2023). Structures and Spectroscopic Properties of Polysulfide Radical Anions: A Theoretical Perspective. Molecules, 28(15), 5654. https://doi.org/10.3390/molecules28155654