
Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes

 and
and
Abstract
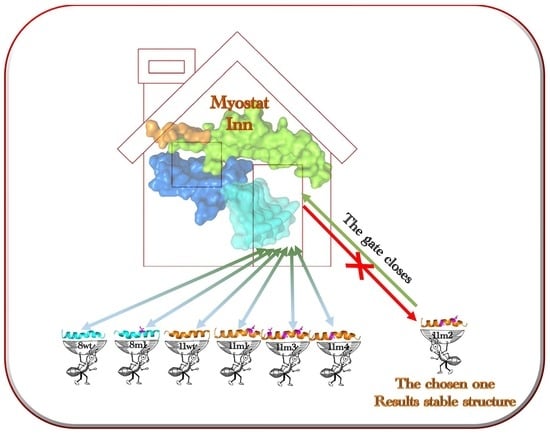
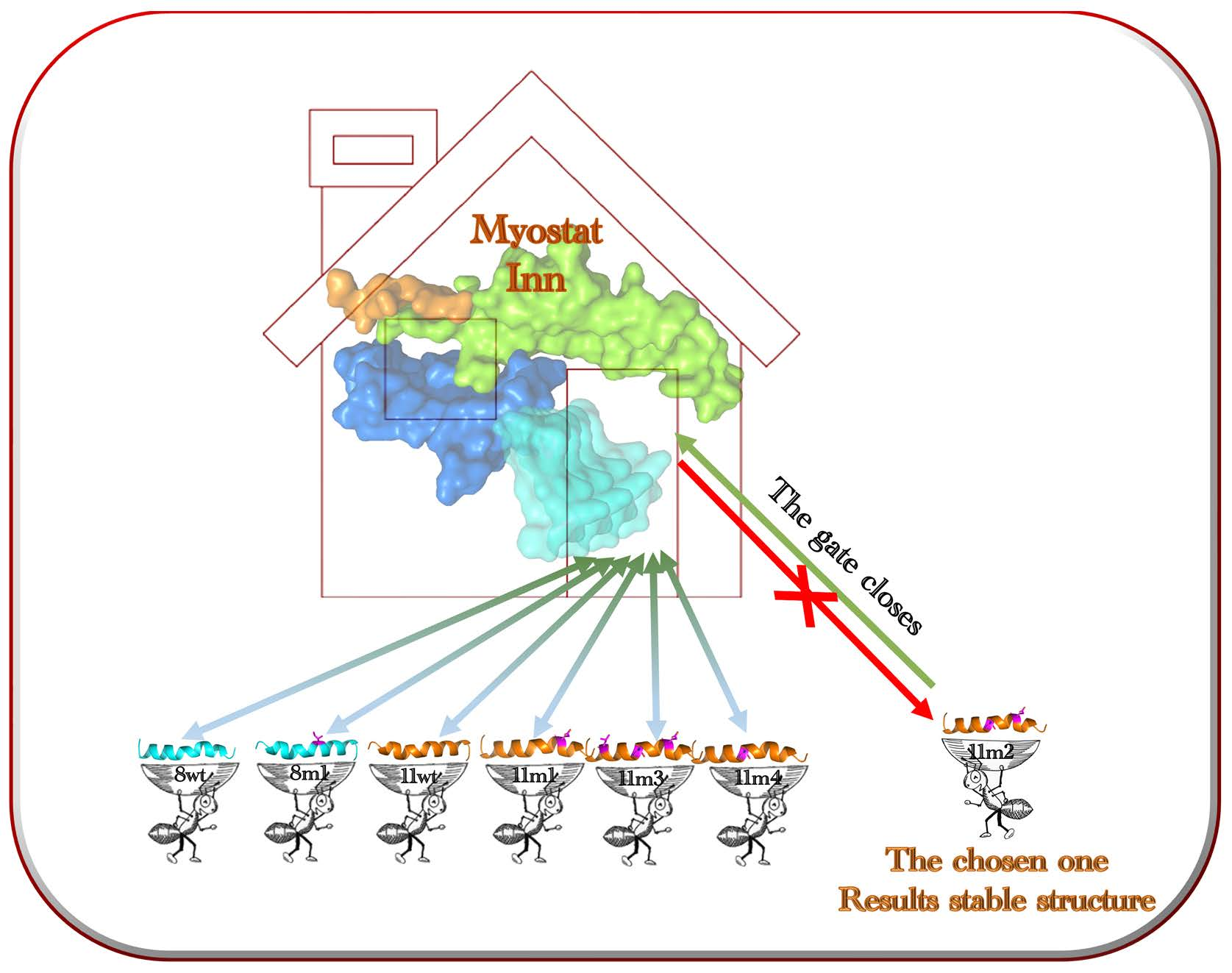
1. Introduction
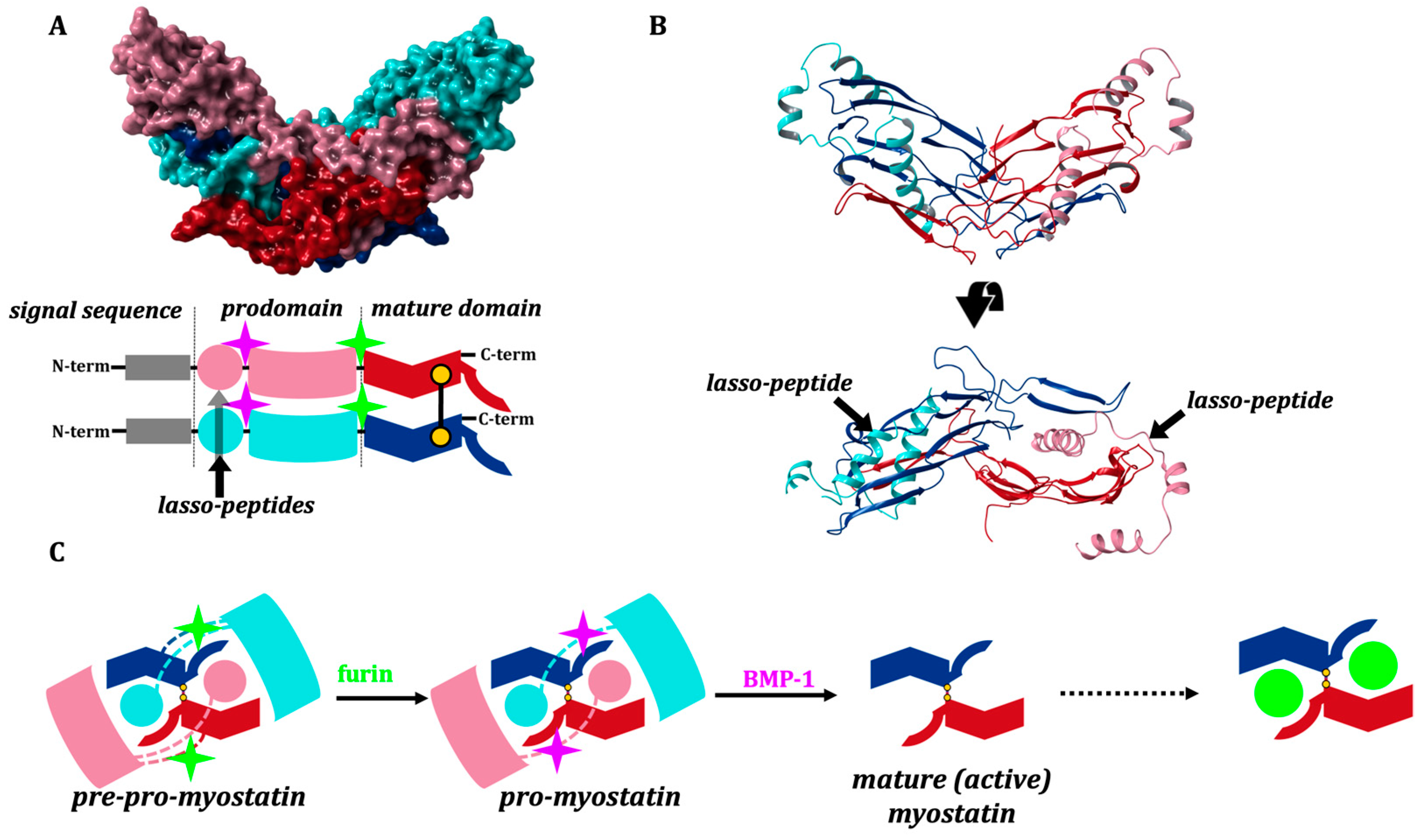
2. Results and Discussion
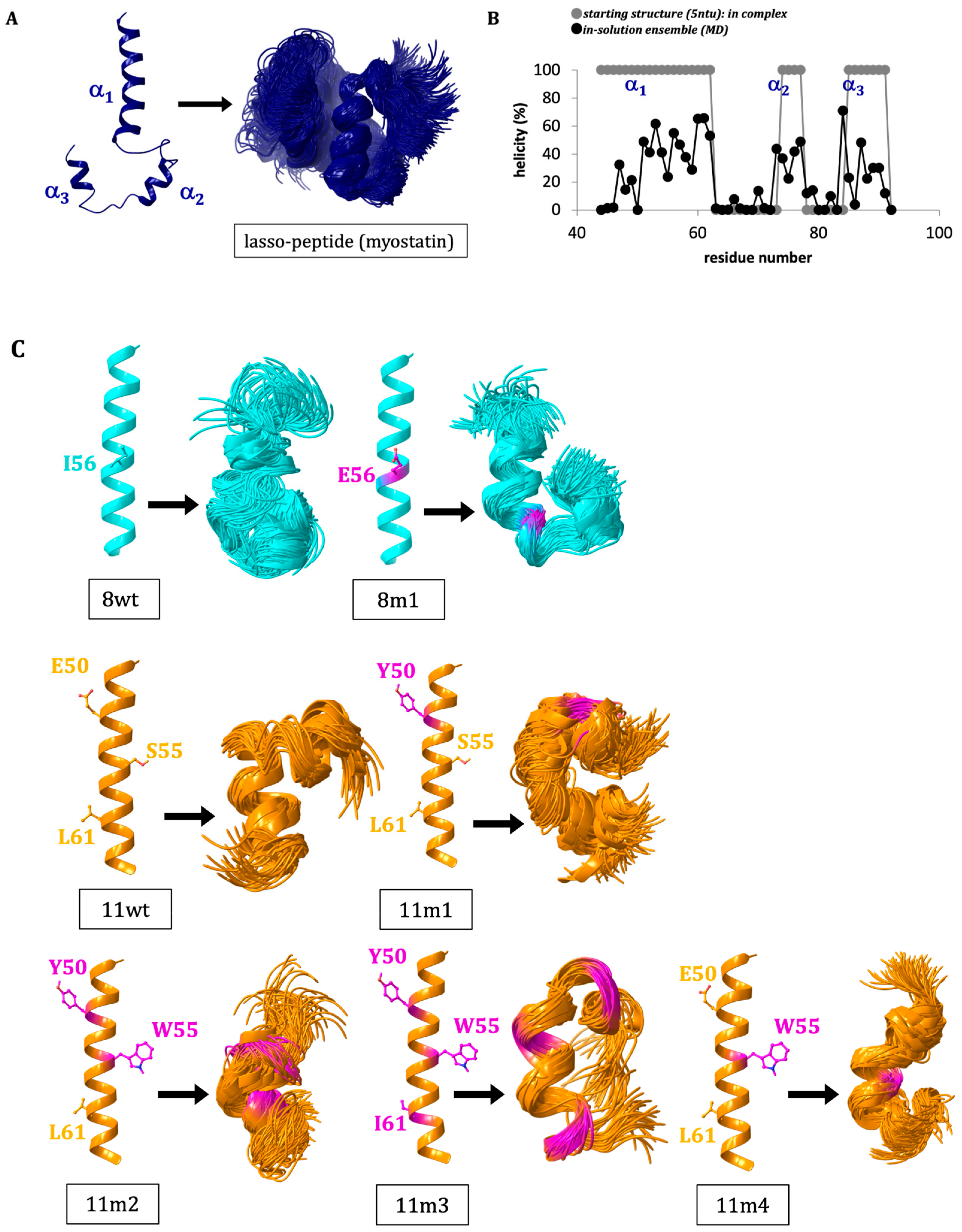
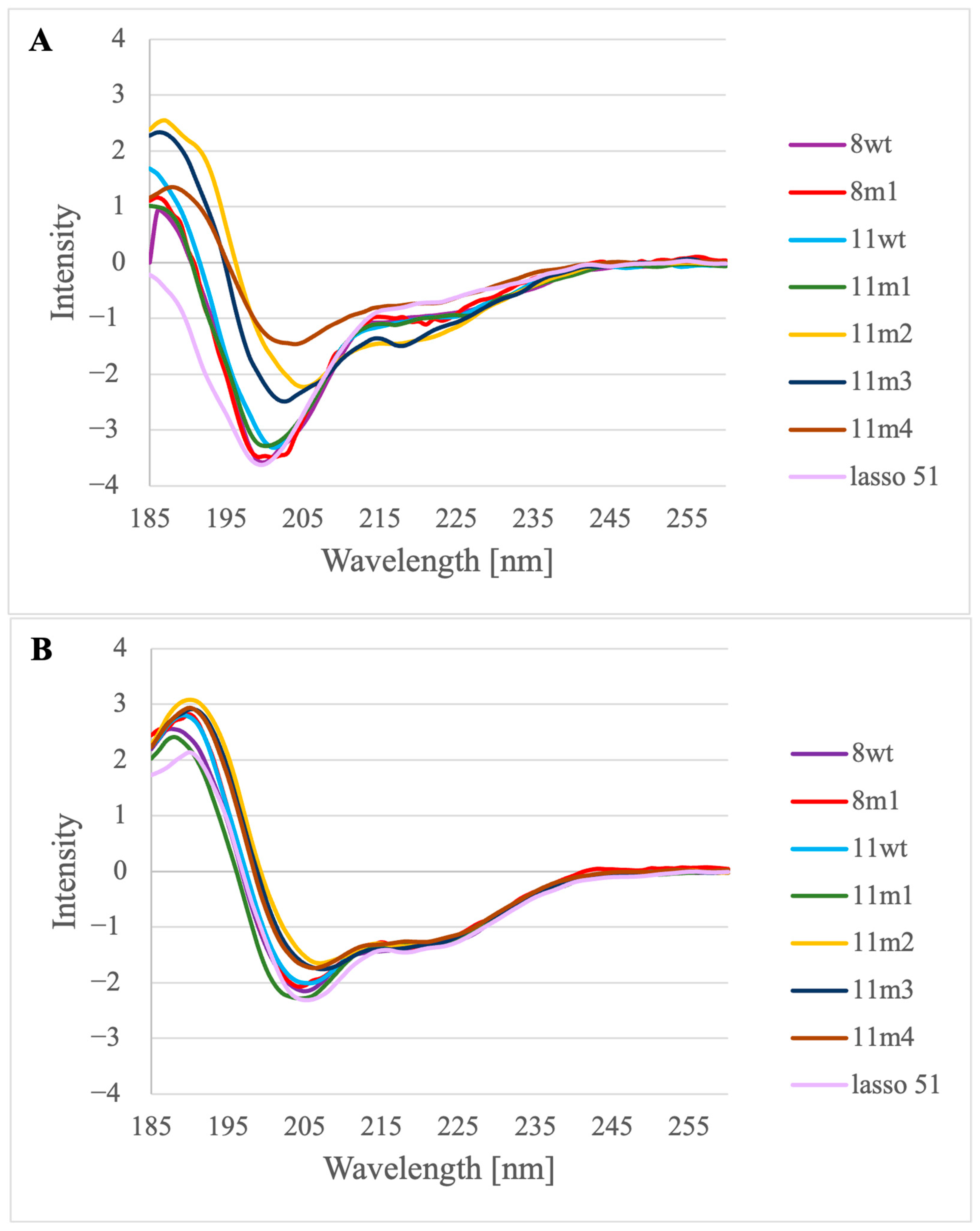
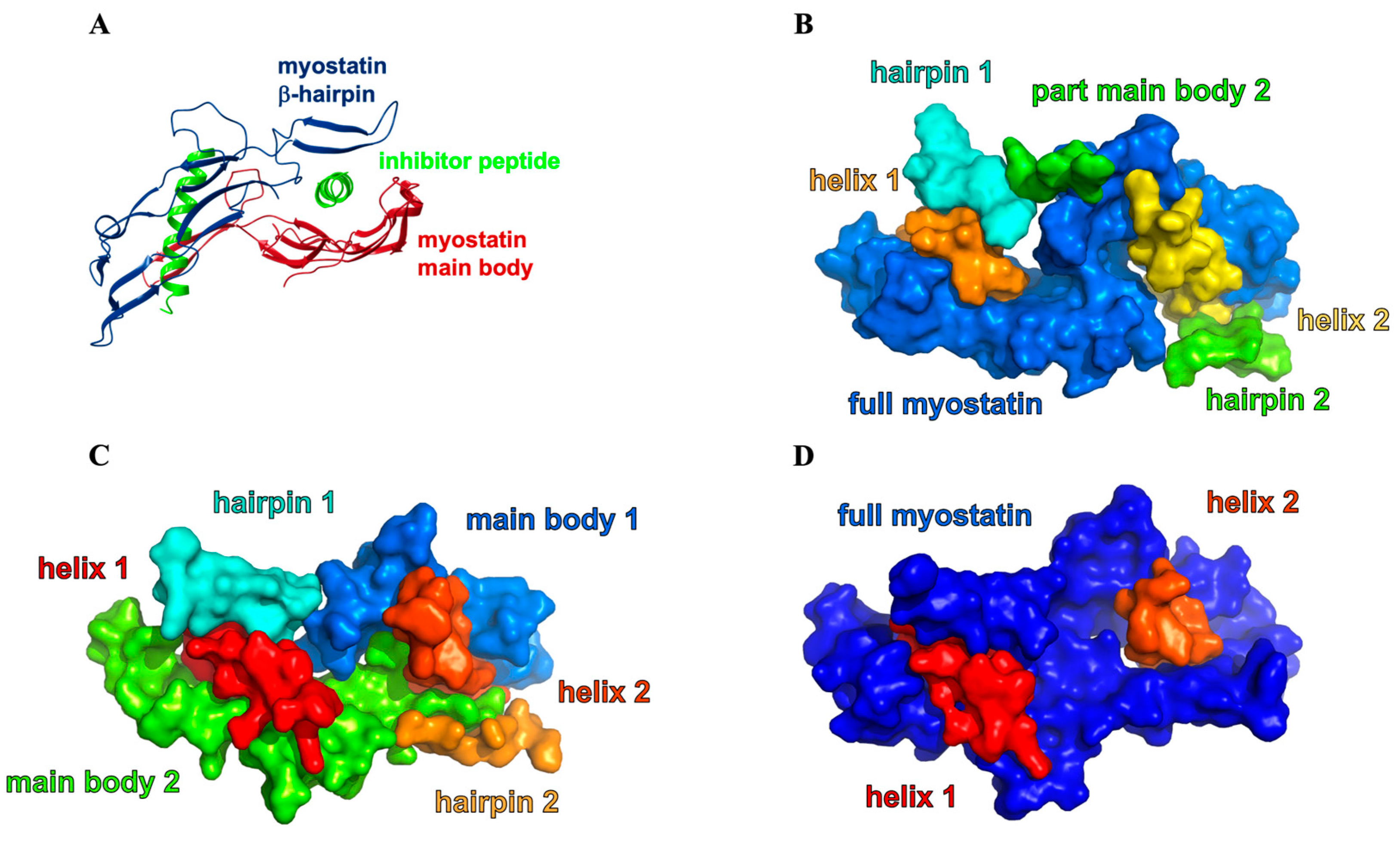
2.1. Solution Structure of the Lasso-Peptides of Myostatin (GDF8), GDF11 and Their Variants
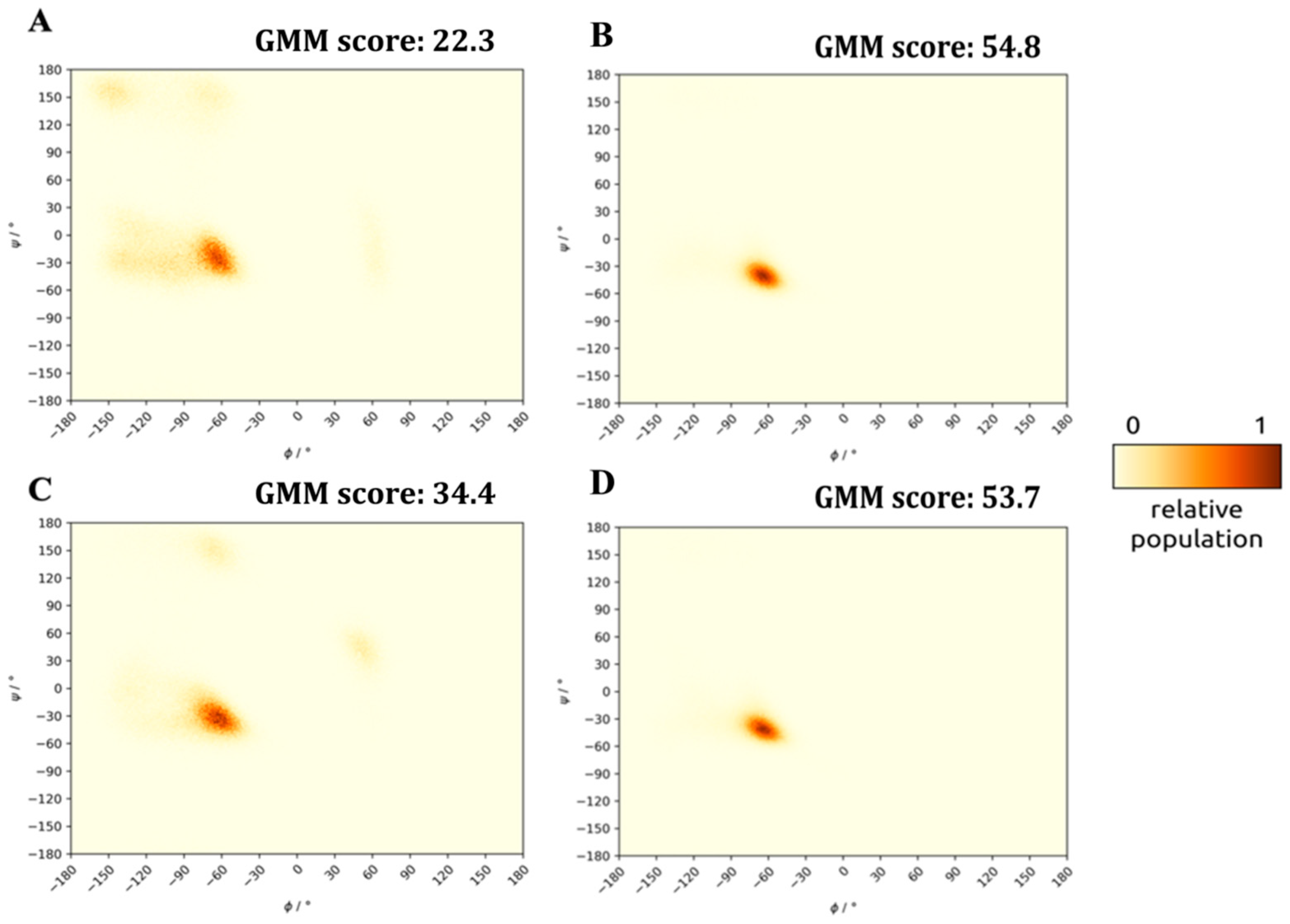
2.2. Structure of the Inhibited Complex: Helicity
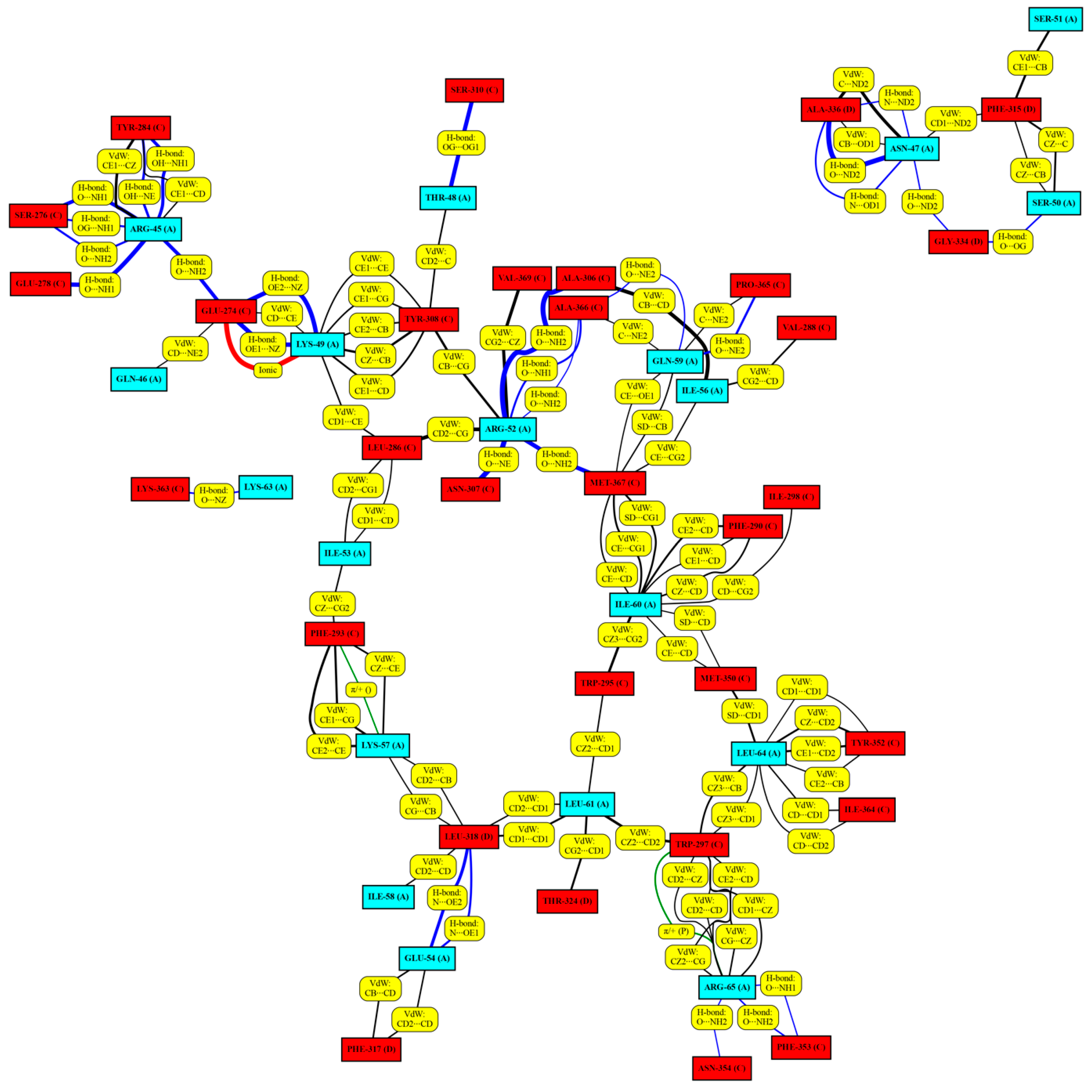
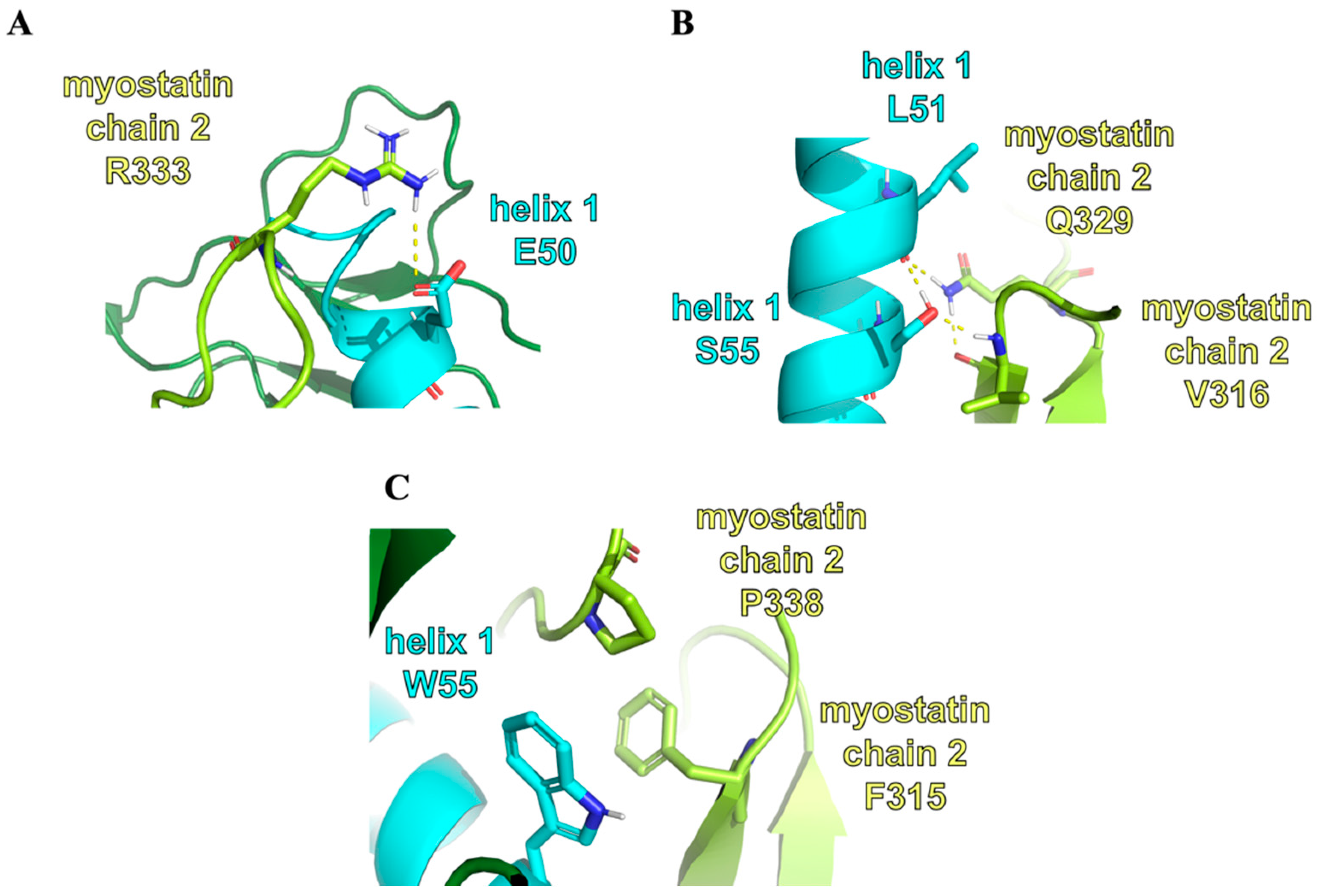
2.3. Structure of the Inhibited Complex: Interaction Networks
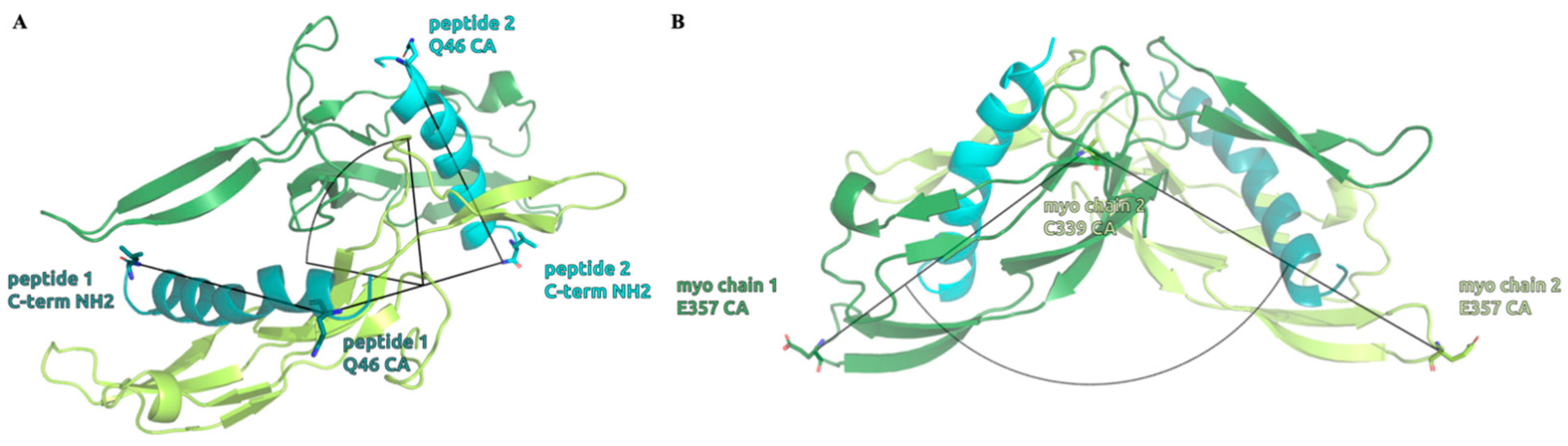
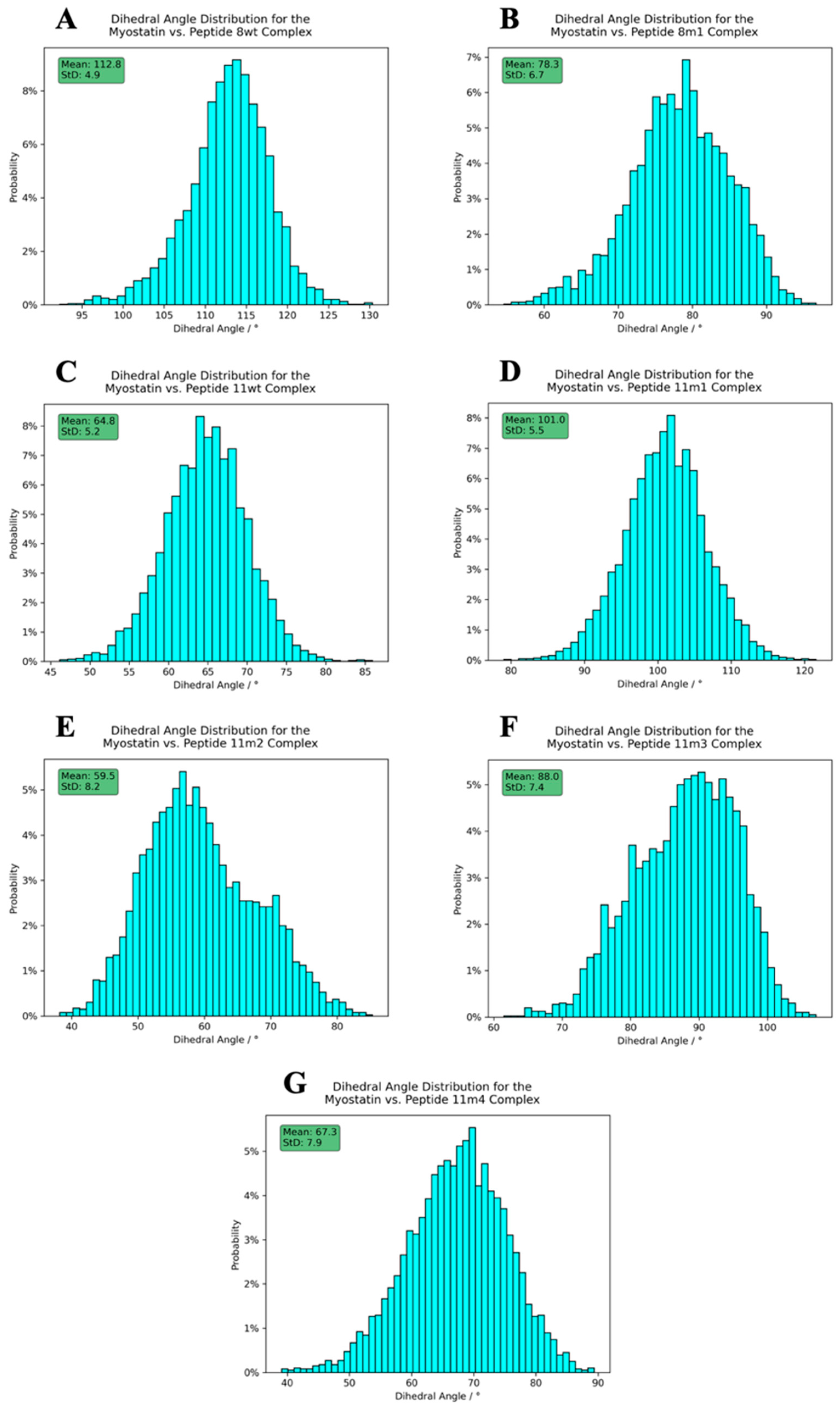
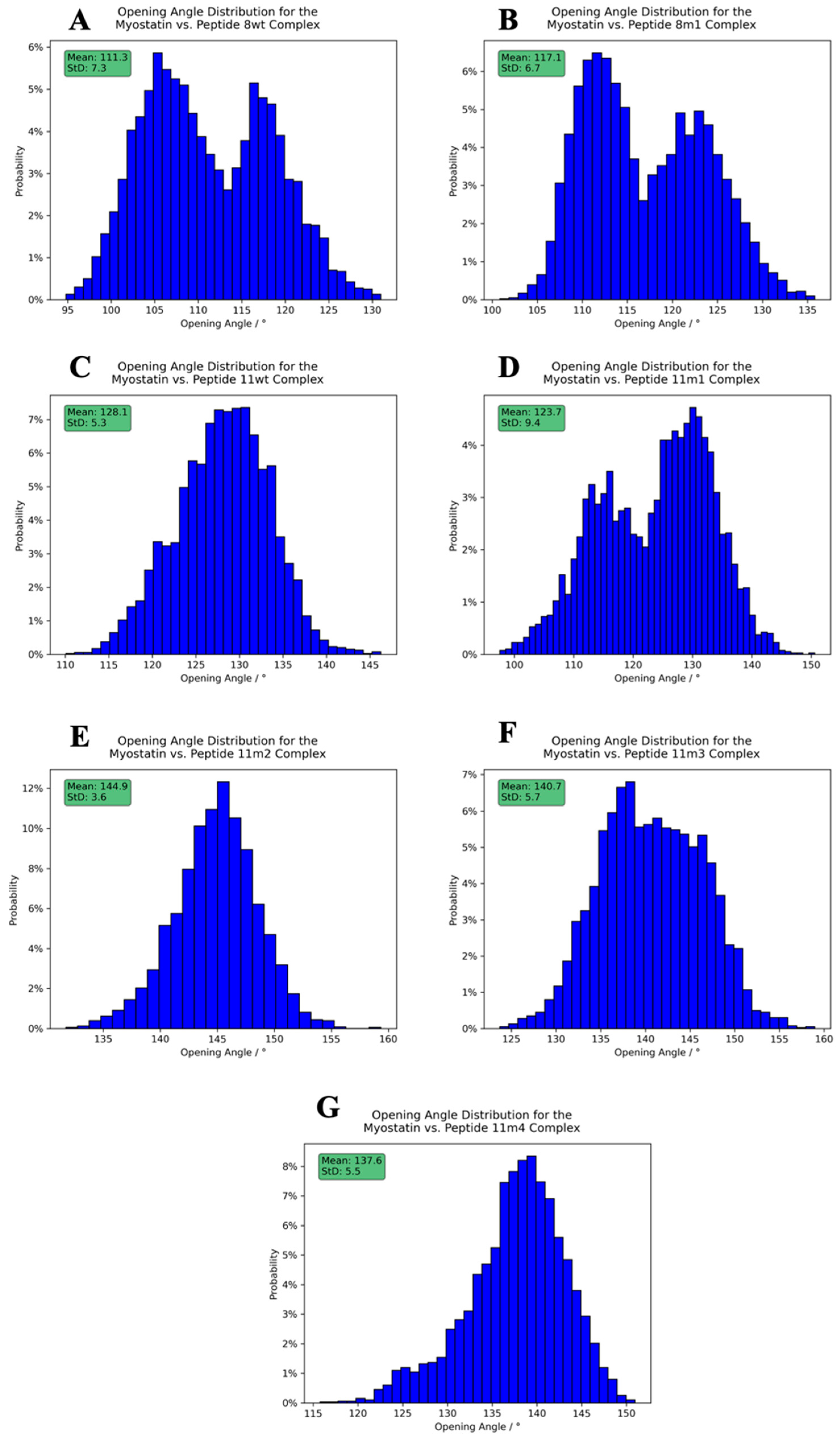
2.4. Structure of the Inhibited Complex: Dynamics
3. Materials and Methods
3.1. Molecular Modeling, Dynamics Calculations and Trajectory Analysis
3.2. Solid Phase Peptide Synthesis
3.3. Purification by High Liquid Performance Chromatography
3.4. Mass Spectrometry Analysis
3.5. Circular Dichroism Measurements
3.6. CCA+ Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ALK4/5/7 | Activin-like receptor Kinase receptor 4/5/7 |
| BMP-1 | Bone Morphogenetic Protein-1 |
| CD | Circular Dichroism |
| DIC | N,N-Diisopropylcarbodiimide |
| DMF | Dimethylformamide |
| ECD | Electronic Circular Dichroism |
| ESI-ITMS | Electrospray Ionization Ion Trap Mass Spectrometry |
| FUV-ECD | Far-UV Electronic Circular Dichroism |
| GDF11 | Growth Differentiation Factor 11 |
| GDF8 | Growth Differentiation Factor 8 |
| GMM | Gaussian Mahalanobis Mean |
| GMX | GROMACS |
| MD | Molecular Dynamics |
| MS | Mass Spectrometry |
| NMP | N-Methyl-Pyrrolidon |
| PDB | Protein Data Bank |
| RP-HPLC | Reverse Phase—High Performance Liquid Chromatography |
| StD | Standard Deviation |
| TFA | Trifluoroacetic Acid |
| TFE | 2,2,2-Trifluoroethanol |
| TGF-β | Transforming Growth Factor β |
References
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of Skeletal Muscle in Mice by a New TGF-Fl Member. Nature 1997, 3887, 83–90. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Bondy, C. Growth Factors and Cytokines in Health and Disease. In A Multi-Volume Treatise; JAI Press: Greenwich, UK, 1996; Volume 1, ISBN 0762300914. [Google Scholar]
- Walker, R.G.; McCoy, J.C.; Czepnik, M.; Mills, M.J.; Hagg, A.; Walton, K.L.; Cotton, T.R.; Hyvönen, M.; Lee, R.T.; Gregorevic, P.; et al. Molecular Characterization of Latent GDF8 Reveals Mechanisms of Activation. Proc. Natl. Acad. Sci. USA 2018, 115, E866–E875. [Google Scholar] [CrossRef]
- Takayama, K.; Noguchi, Y.; Aoki, S.; Takayama, S.; Yoshida, M.; Asari, T.; Yakushiji, F.; Nishimatsu, S.I.; Ohsawa, Y.; Itoh, F.; et al. Identification of the Minimum Peptide from Mouse Myostatin Prodomain for Human Myostatin Inhibition. J. Med. Chem. 2015, 58, 1544–1549. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.J. Induction of Cachexia in Mice by Systemically Administered Myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming Growth Factor-β Superfamily Ligand Trap ACE-536 Corrects Anemia by Promoting Late-Stage Erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, Y.; Qin, D.; Yao, Y.; Zhang, X.; Zhao, Y.; Zheng, C. Regulatory Role and Potential Importance of GDF-8 in Ovarian Reproductive Activity. Front. Endocrinol. 2022, 13, 878069. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Z.; Wu, Z.; Yan, Y.; Gao, Y.; Li, Y.; Cheng, J.-C.; Sun, Y.-P. GDF-8 Stimulates Trophoblast Cell Invasion by Inducing ALK5-SMAD2/3-Mediated MMP2 Expression. Reproduction 2021, 162, 331–338. [Google Scholar] [CrossRef]
- Lee, S.-J.; McPherron, A.C. Regulation of Myostatin Activity and Muscle Growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The Role of Myostatin in Muscle Wasting: An Overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef]
- Wolfman, N.M.; Mcpherron, A.C.; Pappano, W.N.; Davies, M.V.; Song, K.; Tomkinson, K.N.; Wright, J.F.; Zhao, L.; Sebald, S.M.; Greenspan, D.S.; et al. Activation of Latent Myostatin by the BMP-1tolloid Family of Metalloproteinases. Proc. Natl. Acad. Sci. USA 2003, 100, 15842–15846. [Google Scholar] [CrossRef]
- Cotton, T.R.; Fischer, G.; Wang, X.; McCoy, J.C.; Czepnik, M.; Thompson, T.B.; Hyvönen, M. Structure of the Human Myostatin Precursor and Determinants of Growth Factor Latency. EMBO J. 2018, 37, 367–383. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Cash, J.N.; Angerman, E.B.; Kattamuri, C.; Nolan, K.; Zhao, H.; Sidis, Y.; Keutmann, H.T.; Thompson, T.B. Structure of Myostatin·follistatin-like 3: N-Terminal Domains of Follistatin-Type Molecules Exhibit Alternate Modes of Binding. J. Biol. Chem. 2012, 287, 1043–1053. [Google Scholar] [CrossRef]
- Kondás, K.; Szlaḿa, G.; Nagy, A.; Trexler, M.; Ṕatthy, L. Biological Functions of the WAP Domain-Containing Multidomain Proteins WFIKKN1 and WFIKKN2. Biochem. Soc. Trans. 2011, 39, 1416–1420. [Google Scholar] [CrossRef]
- Kondás, K.; Szláma, G.; Trexler, M.; Patthy, L. Both WFIKKN1 and WFIKKN2 Have High Affinity for Growth and Differentiation Factors 8 and 11. J. Biol. Chem. 2008, 283, 23677–23684. [Google Scholar] [CrossRef]
- McCoy, J.C.; Walker, R.G.; Murray, N.H.; Thompson, T.B. Crystal Structure of the WFIKKN2 Follistatin Domain Reveals Insight into How It Inhibits Growth Differentiation Factor 8 (GDF8) and GDF11. J. Biol. Chem. 2019, 294, 6333–6343. [Google Scholar] [CrossRef]
- Padyana, A.K.; Vaidialingam, B.; Hayes, D.B.; Gupta, P.; Franti, M.; Farrow, N.A. Crystal Structure of Human GDF11. Acta Crystallogr. Sect. Struct. Biol. Commun. 2016, 72, 160–164. [Google Scholar] [CrossRef]
- Walker, R.G.; Czepnik, M.; Goebel, E.J.; McCoy, J.C.; Vujic, A.; Cho, M.; Oh, J.; Aykul, S.; Walton, K.L.; Schang, G.; et al. Structural Basis for Potency Differences between GDF8 and GDF11. BMC Biol. 2017, 15, 19. [Google Scholar] [CrossRef]
- Hayashi, Y.; Mikawa, S.; Masumoto, K.; Katou, F.; Sato, K. GDF11 Expression in the Adult Rat Central Nervous System. J. Chem. Neuroanat. 2018, 89, 21–36. [Google Scholar] [CrossRef]
- Jiang, M.S.; Liang, L.F.; Wang, S.; Ratovitski, T.; Holmstrom, J.; Barker, C.; Stotish, R. Characterization and Identification of the Inhibitory Domain of GDF-8 Propeptide. Biochem. Biophys. Res. Commun. 2004, 315, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Nakamura, A.; Rentier, C.; Mino, Y.; Asari, T.; Saga, Y.; Taguchi, A.; Yakushiji, F.; Hayashi, Y. Effect of N-Terminal Acylation on the Activity of Myostatin Inhibitory Peptides. ChemMedChem 2016, 11, 845–849. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Takayama, K.; Nishimatsu, S.I.; Okada, T.; Fujino, M.; Fukai, Y.; Murakami, T.; Hagiwara, H.; Itoh, F.; Tsuchida, K.; et al. The Inhibitory Core of the Myostatin Prodomain: Its Interaction with Both Type I and II Membrane Receptors, and Potential to Treat Muscle Atrophy. PLoS ONE 2015, 10, e0133713. [Google Scholar] [CrossRef] [PubMed]
- Asari, T.; Takayama, K.; Nakamura, A.; Shimada, T.; Taguchi, A.; Hayashi, Y. Structural Basis for the Effective Myostatin Inhibition of the Mouse Myostatin Prodomain-Derived Minimum Peptide. ACS Med. Chem. Lett. 2017, 8, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Rentier, C.; Asari, T.; Nakamura, A.; Saga, Y.; Shimada, T.; Nirasawa, K.; Sasaki, E.; Muguruma, K.; Taguchi, A.; et al. Development of Potent Myostatin Inhibitory Peptides through Hydrophobic Residue-Directed Structural Modification. ACS Med. Chem. Lett. 2017, 8, 751–756. [Google Scholar] [CrossRef]
- Rentier, C.; Takayama, K.; Saitoh, M.; Nakamura, A.; Ikeyama, H.; Taguchi, A.; Taniguchi, A.; Hayashi, Y. Design and Synthesis of Potent Myostatin Inhibitory Cyclic Peptides. Bioorg. Med. Chem. 2019, 27, 1437–1443. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J.; et al. Myostatin Blockade with a Fully Human Monoclonal Antibody Induces Muscle Hypertrophy and Reverses Muscle Atrophy in Young and Aged Mice. Skelet. Muscle 2015, 5, 34. [Google Scholar] [CrossRef]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin Antibody (LY2495655) in Older Weak Fallers: A Proof-of-Concept, Randomised, Phase 2 Trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Farkas, V.; Ferentzi, K.; Horváti, K.; Perczel, A. Cost-Effective Flow Peptide Synthesis: Metamorphosis of HPLC. Org. Process Res. Dev. 2021, 25, 182–191. [Google Scholar] [CrossRef]
- Stráner, P.; Taricska, N.; Szabó, M.; Tóth, G.K.; Perczel, A. Bacterial Expression and/or Solid Phase Peptide Synthesis of 20-40 Amino Acid Long Polypeptides and Miniproteins, the Case Study of Class B GPCR Ligands. Curr. Protein Pept. Sci. 2016, 17, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Perczel, A.; Hollosi, M.; Tusnady, G.; Fasman, G.D. Convex Constraint Analysis: A Natural Deconvolution of Circular Dichroism Curves of Proteins. Protein Eng. Des. Sel. 1991, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Percze1, A.; Hollbsi, M.; Foxman, B.M.; Fasman, G.D. Conformational Analysis of Pseudocyclic Hexapeptides Based on Quantitative Circular Dichroism (CD), NOE, and X-Ray Data. The Pure CD Spectra of Type I and Type I1@-Turns. J. Am. Chem. Soc. 1991, 113, 9772–9784. [Google Scholar] [CrossRef]
- Oberg, K.A.; Ruysschaert, J.M.; Goormaghtigh, E. The Optimization of Protein Secondary Structure Determination with Infrared and Circular Dichroism Spectra. Eur. J. Biochem. 2004, 271, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- ChirasCan Series AppliCation Note. Available online: https://technoinfo.ru/technoinfo/files/012/000/0000012/AppNote%20ACD%20Biosimilarity%20Studies%20of%20Serum%20Albumins.pdf (accessed on 25 June 2023).
- Jákli, I.; Perczel, A. The Inherent Flexibility of Peptides and Protein Fragments Quantitized by CD in Conjunction with CCA+. J. Pept. Sci. 2009, 15, 738–752. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: Fast Generation of Probabilistic Residue Interaction Networks from Structural Ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef]
- Taricska, N.; Horváth, D.; Menyhárd, D.K.; Ákontz-Kiss, H.; Noji, M.; So, M.; Goto, Y.; Fujiwara, T.; Perczel, A. The Route from the Folded to the Amyloid State: Exploring the Potential Energy Surface of a Drug-Like Miniprotein. Chemistry 2020, 26, 1968–1978. [Google Scholar] [CrossRef]
- Baytshtok, V.; Fei, X.; Grant, R.A.; Baker, T.A.; Sauer, R.T. A Structurally Dynamic Region of the HslU Intermediate Domain Controls Protein Degradation and ATP Hydrolysis. Structure 2016, 24, 1766–1777. [Google Scholar] [CrossRef]
- Apgar, J.R.; Mader, M.; Agostinelli, R.; Benard, S.; Bialek, P.; Johnson, M.; Gao, Y.; Krebs, M.; Owens, J.; Parris, K.; et al. Beyond CDR-Grafting: Structure-Guided Humanization of Framework and CDR Regions of an Anti-Myostatin Antibody. MAbs 2016, 8, 1302–1318. [Google Scholar] [CrossRef]
- Cash, J.N.; Rejon, C.A.; McPherron, A.C.; Bernard, D.J.; Thompson, T.B. The Structure of Myostatin:Follistatin 288: Insights into Receptor Utilization and Heparin Binding. EMBO J. 2009, 28, 2662–2676. [Google Scholar] [CrossRef]
- Dagbay, K.B.; Treece, E.; Streich, F.C.; Jackson, J.W.; Faucette, R.R.; Nikiforov, A.; Lin, S.C.; Boston, C.J.; Nicholls, S.B.; Capili, A.D.; et al. Structural Basis of Specific Inhibition of Extracellular Activation of Pro- or Latent Myostatin by the Monoclonal Antibody SRK-015. J. Biol. Chem. 2020, 295, 5404–5418. [Google Scholar] [CrossRef]
- Fazekas, Z.; Menyhárd, D.K.; Perczel, A. Omicron Binding Mode: Contact Analysis and Dynamics of the Omicron Receptor-Binding Domain in Complex with ACE2. J. Chem. Inf. Model. 2022, 62, 3844–3853. [Google Scholar] [CrossRef]
- Abraham, L.; van der Spoel, H. GROMACS Documentation Release 2021.4; GROMACS Development Team: Uppsala, Sweden, 2021. [Google Scholar]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentialst. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional Timescale Predictions by Molecular Dynamics Simulations: Case Study Using Proline and Hydroxyproline Sidechain Dynamics. Proteins Struct. Funct. Bioinform. 2014, 82, 195–215. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L. PISCES: A Protein Sequence Culling Server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]
- PyGraphViz. Available online: https://github.com/pygraphviz/pygraphviz (accessed on 20 July 2023).












{kind=link}
{kind=link}
{kind=link}
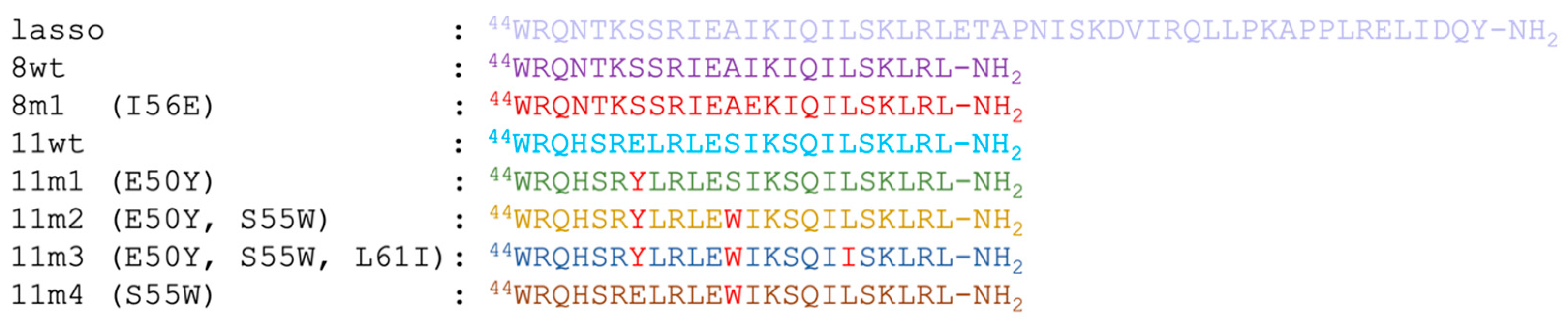{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Number of the Different Interaction Types | |||||
|---|---|---|---|---|---|
| Van der Waals | H-Bond | Ionic | Cation-π | π-π Stack | |
| 8wt | 25.3 | 14.5 | 1.0 | 0.4 | 0.0 |
| 8m1 | 23.2 | 10.4 | 0.9 | 0.5 | 0.1 |
| 11wt | 31.0 | 13.5 | 1.1 | 0.2 | 0.0 |
| 11m1 | 28.4 | 9.4 | 0.7 | 0.7 | 0.5 |
| 11m2 | 29.1 | 12.7 | 0.6 | 0.5 | 0.3 |
| 11m3 | 29.7 | 8.5 | 1.1 | 0.7 | 0.6 |
| 11m4 | 29.8 | 9.2 | 1.0 | 0.4 | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy-Fazekas, D.; Fazekas, Z.; Taricska, N.; Stráner, P.; Karancsiné Menyhárd, D.; Perczel, A. Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes. Molecules 2023, 28, 5655. https://doi.org/10.3390/molecules28155655
Nagy-Fazekas D, Fazekas Z, Taricska N, Stráner P, Karancsiné Menyhárd D, Perczel A. Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes. Molecules. 2023; 28(15):5655. https://doi.org/10.3390/molecules28155655
Chicago/Turabian StyleNagy-Fazekas, Dóra, Zsolt Fazekas, Nóra Taricska, Pál Stráner, Dóra Karancsiné Menyhárd, and András Perczel. 2023. "Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes" Molecules 28, no. 15: 5655. https://doi.org/10.3390/molecules28155655
APA StyleNagy-Fazekas, D., Fazekas, Z., Taricska, N., Stráner, P., Karancsiné Menyhárd, D., & Perczel, A. (2023). Inhibitor Design Strategy for Myostatin: Dynamics and Interaction Networks Define the Affinity and Release Mechanisms of the Inhibited Complexes. Molecules, 28(15), 5655. https://doi.org/10.3390/molecules28155655