
Generating Potential Protein-Protein Interaction Inhibitor Molecules Based on Physicochemical Properties




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- Molecular weight ≤ 500
- Indicator of lipophilicity, LogP ≤ 5
- Number of hydrogen bond donors ≤ 10
- Number of hydrogen bond acceptors ≤ 5.
- Molecular weight > 400
- Indicator of lipophilicity, LogP > 4
- Number of cyclic structures > 4
- Number of hydrogen bond acceptors > 4
2. Results
2.1. Inducing Exploration through Reinforcement Learning
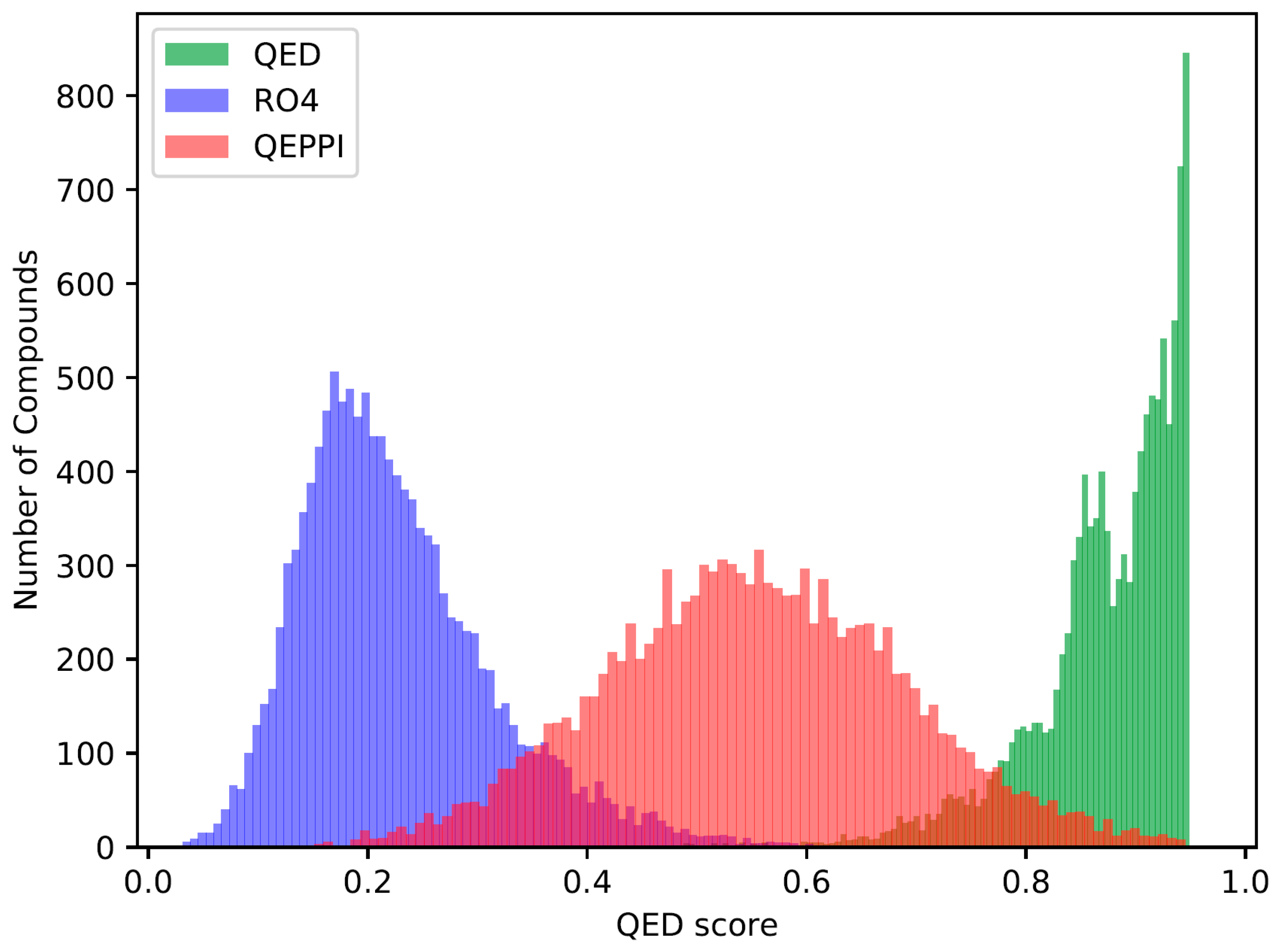
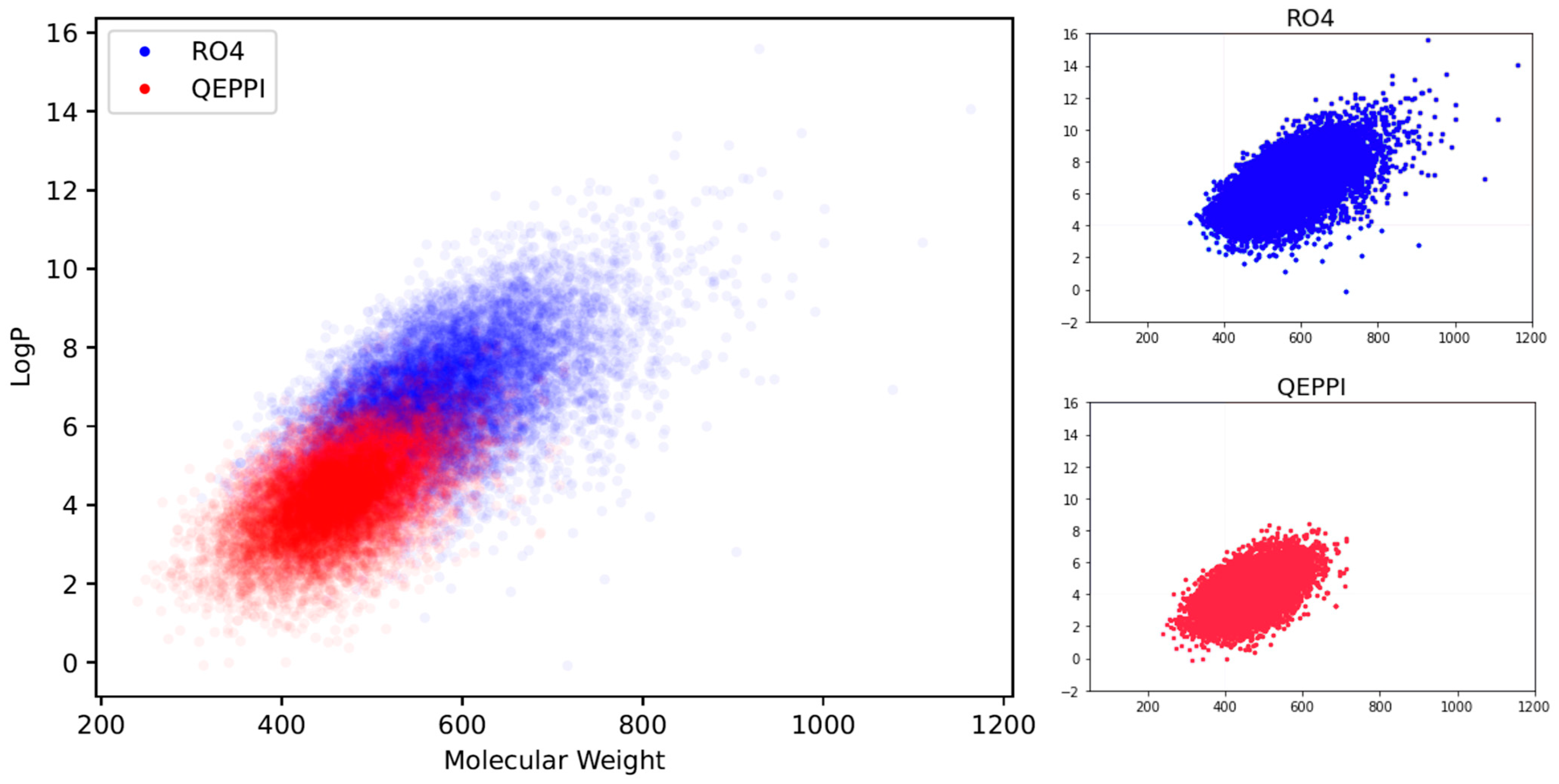
2.2. Distribution of Compounds Generated by REINVENT
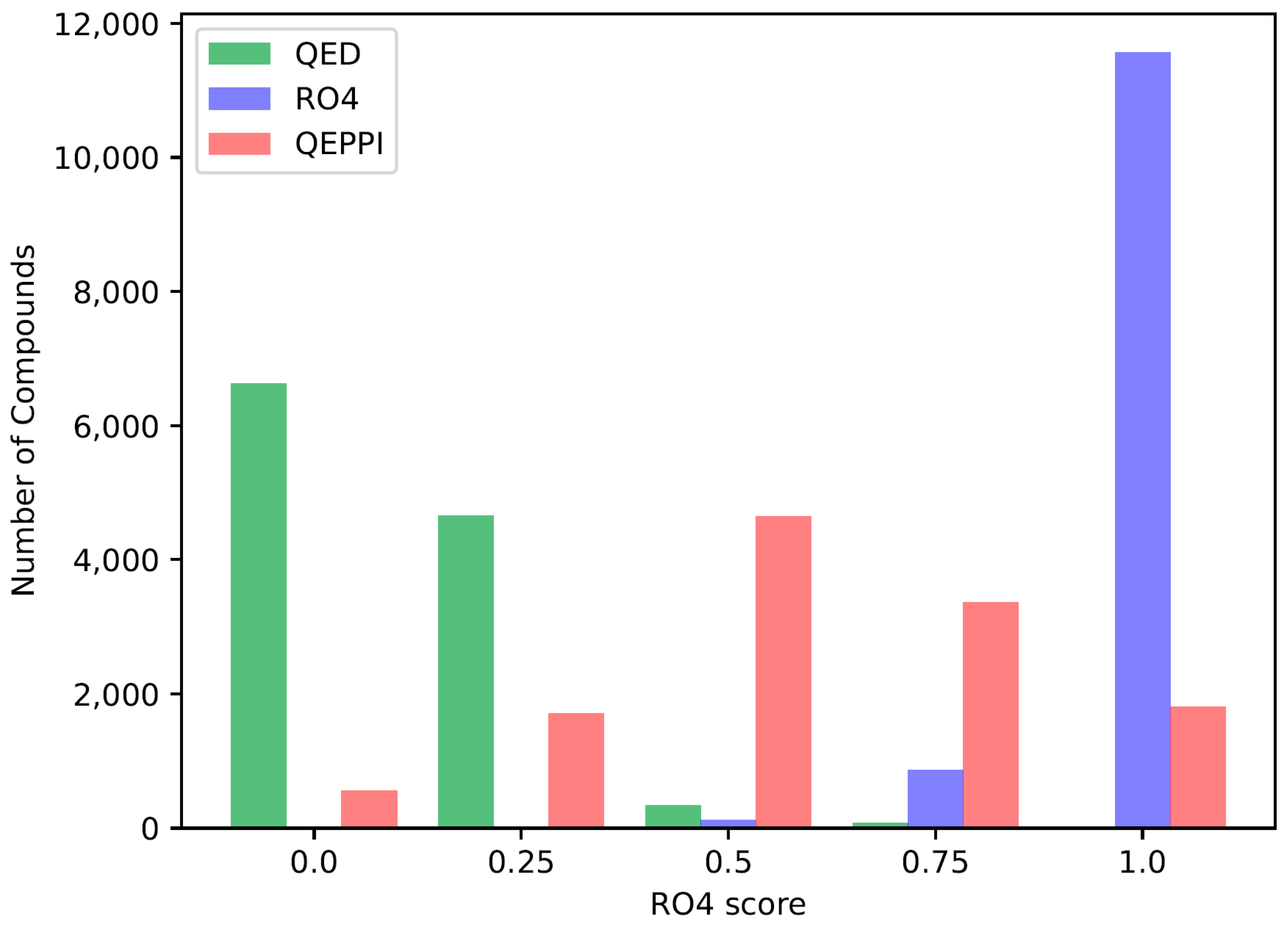
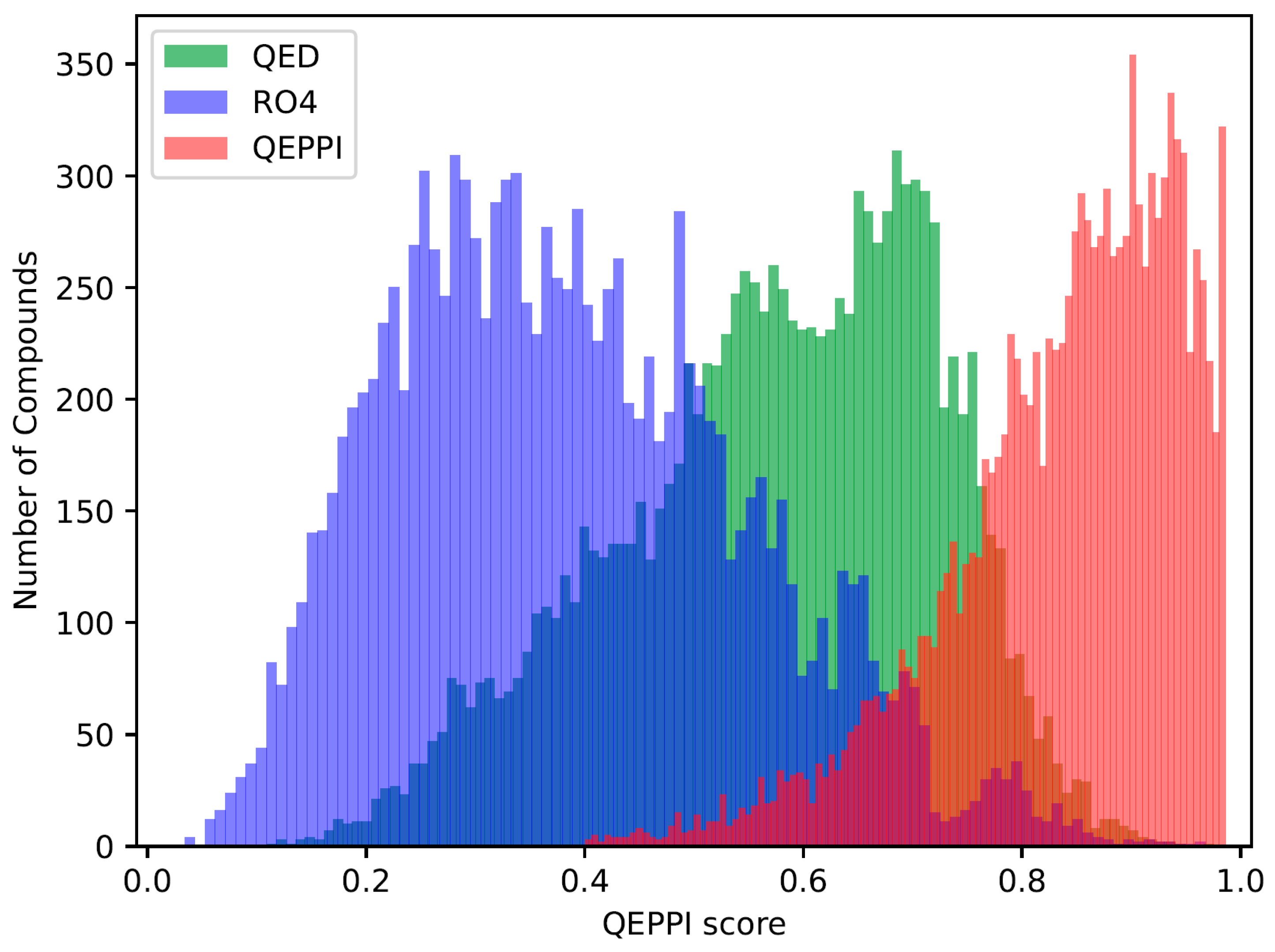
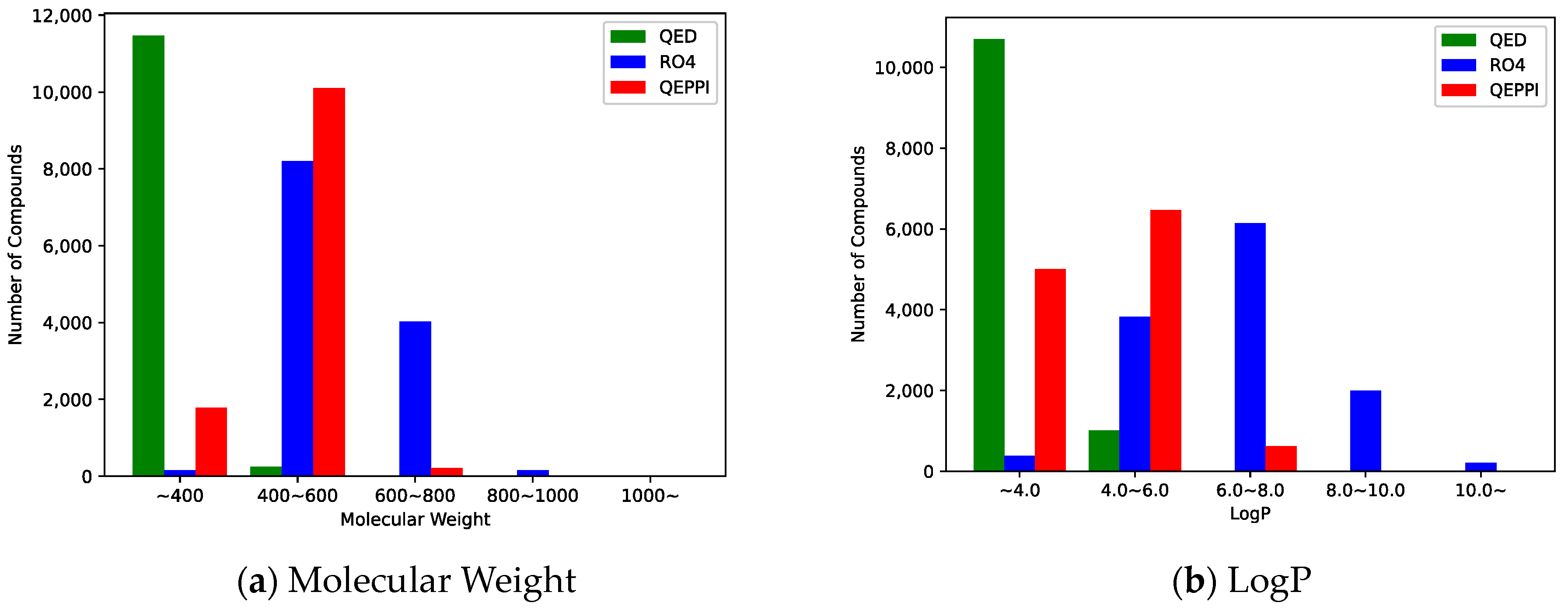
2.3. Indicators for Oral Bioavailability
- Number of rotatable bonds, Rbond ≤ 10
- Topological polar surface area TPSA ≤ 140
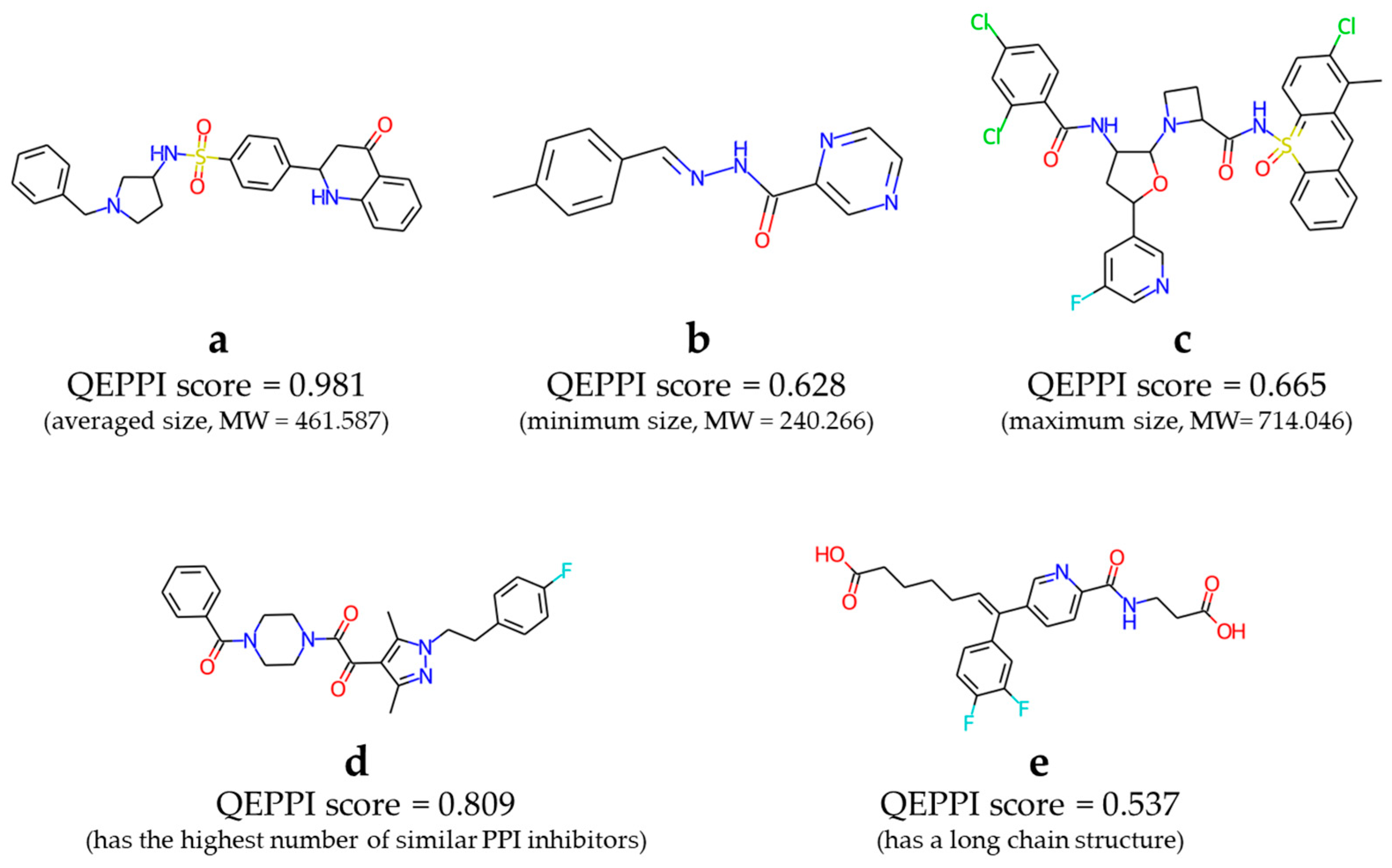
2.4. Constructing Virtual Libraries of PPI-Target Compounds
3. Discussion
3.1. Chemical Space of Generated Compounds
3.2. Comparison with Known PPI Modulators
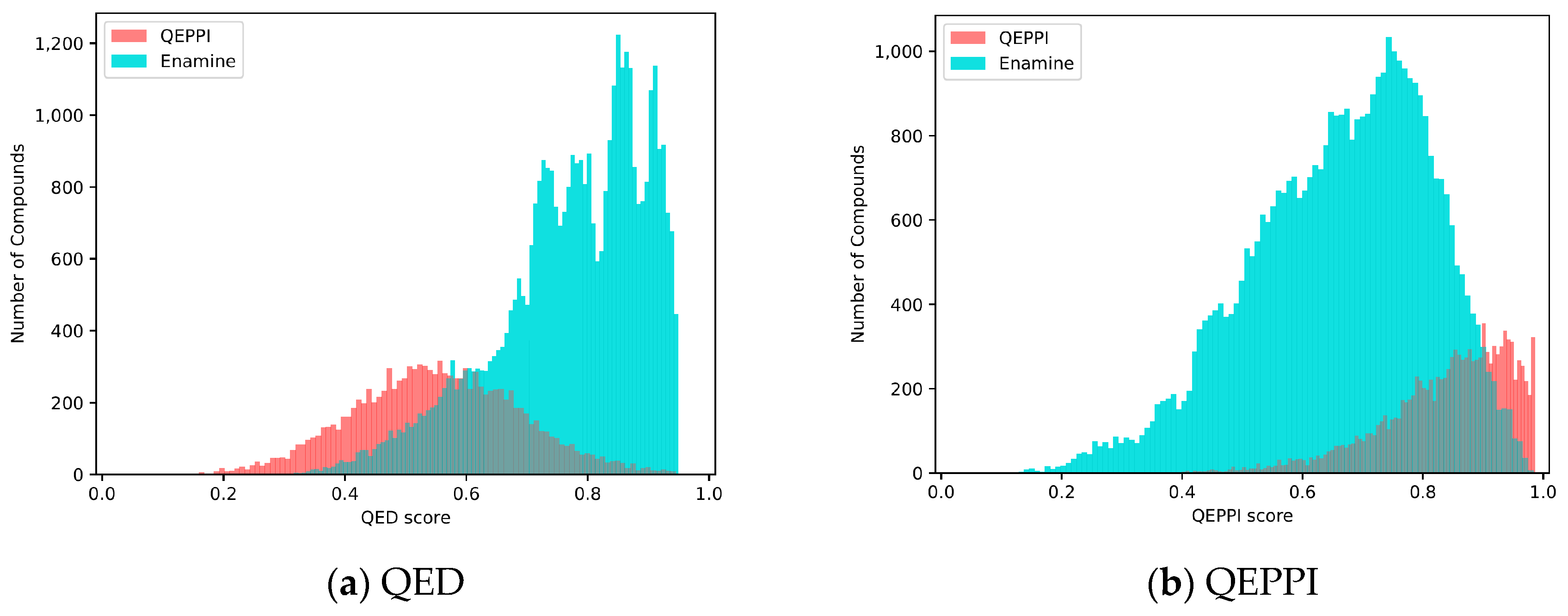
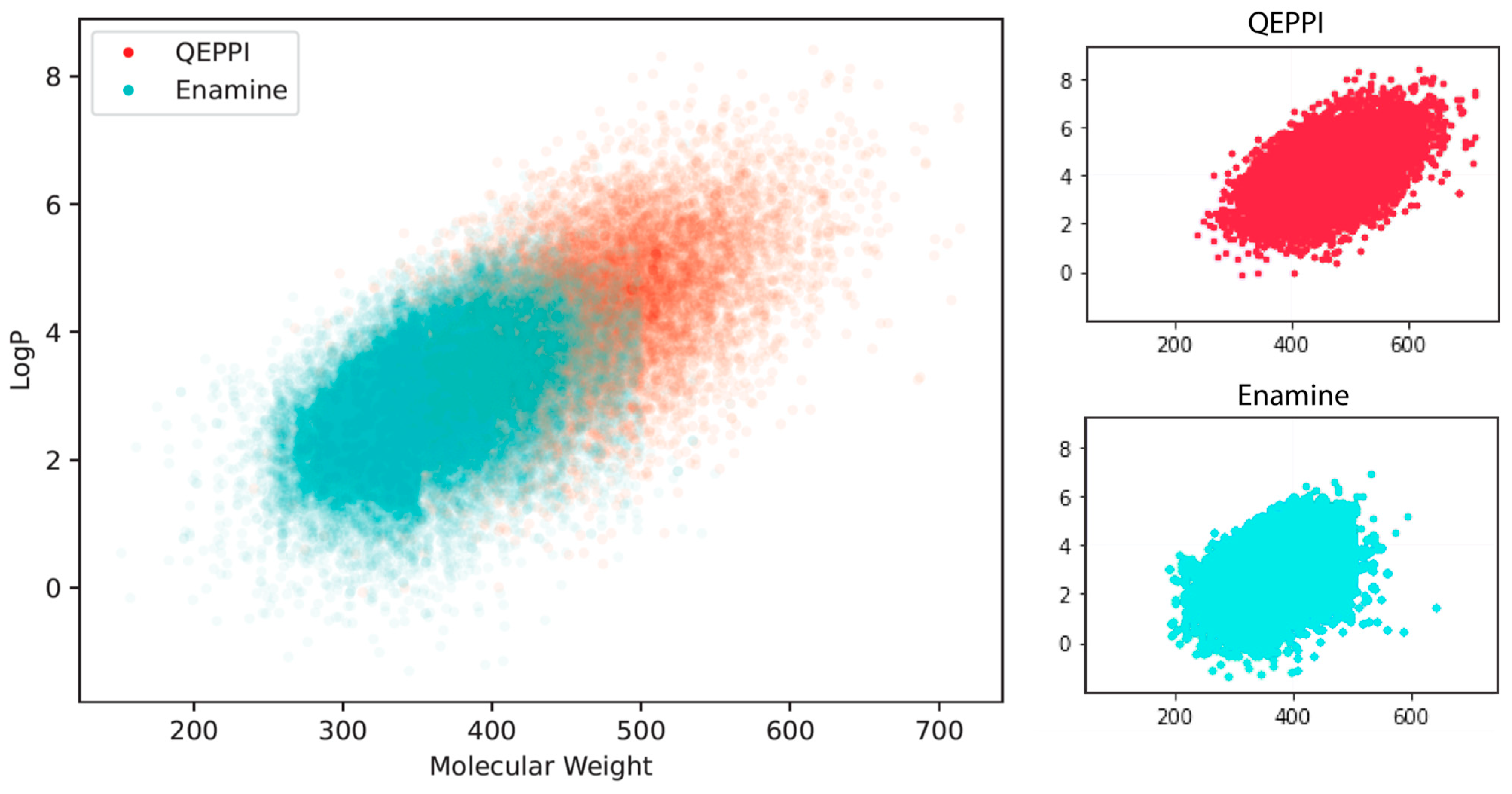
3.3. Comparison with Existing PPI Libraries
3.4. Limitations and Future Directions
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S.; Huys, I. R&D Costs of New Medicines: A Landscape Analysis. Front. Med. 2021, 8, 760762. [Google Scholar] [CrossRef]
- Toogood, P.L. Inhibition of protein-protein association by small molecules: Approaches and progress. J. Med. Chem. 2002, 45, 1543–1558. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Dev, K.K. Making protein interactions druggable: Targeting PDZ domains. Nat. Rev. Drug Discov. 2004, 3, 1047–1056. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein–protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Fisher, D.W.; Yang, S.; Keszycki, R.M.; Dong, H. Protein-protein interactions underlying the behavioral and psychological symptoms of dementia (BPSD) and Alzheimer’s disease. PLoS ONE 2020, 15, e0226021. [Google Scholar] [CrossRef]
- Shin, W.H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current challenges and opportunities in designing protein-protein interaction targeted drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar] [CrossRef]
- Shin, W.H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Basse, M.J.; Betzi, S.; Morelli, X.; Roche, P. 2P2Idb v2: Update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database 2016, 2016, baw007. [Google Scholar] [CrossRef]
- Jnoff, E.; Albrecht, C.; Barker, J.J.; Barker, O.; Beaumont, E.; Bromidge, S.; Brookfield, F.; Brooks, M.; Bubert, C.; Ceska, T.; et al. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem 2014, 9, 699–705. [Google Scholar] [CrossRef]
- Bosc, N.; Muller, C.; Hoffer, L.; Lagorce, D.; Bourg, S.; Derviaux, C.; Gourdel, M.E.; Rain, J.C.; Miller, T.W.; Villoutreix, B.O.; et al. Fr-PPIChem: An academic compound library dedicated to protein-protein interactions. ACS Chem. Biol. 2020, 15, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yonezawa, T.; Sakamoto, J.; Furuya, T.; Osawa, M.; Ikeda, K. Identification of novel inhibitors of Keap1/Nrf2 by a promising method combining protein–protein interaction-oriented library and machine learning. Sci. Rep. 2021, 11, 7420. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Quantitative estimate index for early-stage screening of compounds targeting protein-protein interactions. Int. J. Mol. Sci. 2021, 22, 10925. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Torchet, R.; Druart, K.; Ruano, L.C.; Moine-Franel, A.; Borges, H.; Doppelt-Azeroual, O.; Brancotte, B.; Mareuil, F.; Nilges, M.; Ménager, H.; et al. The iPPI-DB initiative: A community-centered database of protein-protein interaction modulators. Bioinformatics 2021, 37, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Enamine. PPI Library. Available online: https://enamine.net/compound-libraries/targeted-libraries/ppi-library (accessed on 20 June 2023).
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Thakkar, A.; Chadimová, V.; Reymond, J.L.; Engkvist, O.; Bjerrum, E. AiZynthFinder: A fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 2020, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Kengkanna, A.; Ohue, M. Enhancing Model Learning and Interpretation Using Multiple Molecular Graph Representations for Compound Property and Activity Prediction. arXiv 2023, arXiv:2304.06253. [Google Scholar] [CrossRef]
- Mochizuki, M.; Suzuki, S.D.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. QEX: Target-specific druglikeness filter enhances ligand-based virtual screening. Mol. Divers. 2019, 23, 11–18. [Google Scholar] [CrossRef]
- Jeon, W.; Kim, D. Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci. Rep. 2020, 10, 22104. [Google Scholar] [CrossRef]
- Danel, T.; Łęski, J.; Podlewska, S.; Podolak, I.T. Docking-based generative approaches in the search for new drug candidates. Drug Discov. Today 2023, 28, 103439. [Google Scholar] [CrossRef]
- Perišić, O. Recognition of Potential COVID-19 Drug Treatments through the Study of Existing Protein-Drug and Protein-Protein Structures: An Analysis of Kinetically Active Residues. Biomolecules 2020, 10, 1346. [Google Scholar] [CrossRef]
- Blaschke, T.; Aŕus-Pous, J.; Chen, H.; Margreitter, C.; Tyrchan, C.; Engkvist, O.; Papadopoulos, K.; Patronov, A. REINVENT 2.0: An AI tool for de novo drug design. J. Chem. Inf. Model. 2020, 60, 5918–5922. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrían-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- MolecularAI. ReinventCommunity. Available online: https://github.com/MolecularAI/ReinventCommunity (accessed on 20 June 2023).
- Wang, J.; Chu, Y.; Mao, J.; Jeon, H.N.; Jin, H.; Zeb, A.; Jang, Y.; Cho, K.H.; Song, T. De novo molecular design with deep molecular generative models for PPI inhibitors. Brief Bioinform. 2022, 23, bbac285. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohue, M.; Kojima, Y.; Kosugi, T. Generating Potential Protein-Protein Interaction Inhibitor Molecules Based on Physicochemical Properties. Molecules 2023, 28, 5652. https://doi.org/10.3390/molecules28155652
Ohue M, Kojima Y, Kosugi T. Generating Potential Protein-Protein Interaction Inhibitor Molecules Based on Physicochemical Properties. Molecules. 2023; 28(15):5652. https://doi.org/10.3390/molecules28155652
Chicago/Turabian StyleOhue, Masahito, Yuki Kojima, and Takatsugu Kosugi. 2023. "Generating Potential Protein-Protein Interaction Inhibitor Molecules Based on Physicochemical Properties" Molecules 28, no. 15: 5652. https://doi.org/10.3390/molecules28155652
APA StyleOhue, M., Kojima, Y., & Kosugi, T. (2023). Generating Potential Protein-Protein Interaction Inhibitor Molecules Based on Physicochemical Properties. Molecules, 28(15), 5652. https://doi.org/10.3390/molecules28155652