
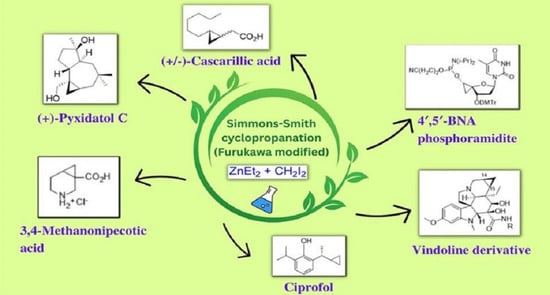
Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review

 , , ,
, , ,  ,
,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Review of Literature
2.1. Synthesis of Alkaloids Based Natural Products
2.1.1. Bisindole Alkaloids
2.1.2. Kopsia Alkaloids
2.1.3. Limonoid Alkaloids
2.1.4. Daphniphyllum Alkaloids
2.2. Synthesis of Terpenoid-Based Natural Products
2.2.1. Indole Terpenoids
2.2.2. Sesquiterpenes
2.2.3. Diterpenoids
2.2.4. Triterpenoids
2.3. Synthesis of Amino-Acid-Based Natural Products
2.4. Synthesis of Nucleosides
2.5. Synthesis of γ-Pyrone-Based Natural Product
2.6. Synthesis of Polyketide-Based Natural Product
2.7. Synthesis of Fatty-Acid-Based Natural Products
2.8. Synthesis of Drugs
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakamura, M.; Hirai, A.; Nakamura, E. Reaction pathways of the Simmons−Smith reaction. J. Am. Chem. Soc. 2003, 125, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Edwards, J.P.; Wilson, S.R. Solution and solid-state structure of the “Wittig-Furukawa” cyclopropanation reagent. J. Am. Chem. Soc. 1991, 113, 723–725. [Google Scholar] [CrossRef]
- Charette, A.B.; Juteau, H. Design of amphoteric bifunctional ligands: Application to the enantioselective Simmons-Smith cyclopropanation of allylic alcohols. J. Am. Chem. Soc. 1994, 116, 2651–2652. [Google Scholar] [CrossRef]
- Ukaji, Y.; Sada, K.; Inomata, K. Synthesis of silicon substituted cyclopropylmethyl alcohols in optically active form via asymmetric Simmons-Smith reaction of γ-silicon substituted allylic alcohols. Chem. Lett. 1993, 22, 1227–1230. [Google Scholar] [CrossRef]
- Gonzalez, J.; Gonzalez, J.; Perez-Calleja, C.; Lopez, L.A.; Vicente, R. Zinc-Catalyzed Synthesis of Functionalized Furans and Triarylmethanes from Enynones and Alcohols or Azoles: Dual X–H Bond Activation by Zinc. Angew. Chem. Int. Ed. 2013, 52, 5853–5857. [Google Scholar] [CrossRef]
- Simmons, H.E.; Smith, R.D. A new synthesis of cyclopropanes from olefins. J. Am. Chem. Soc. 1958, 80, 5323–5324. [Google Scholar] [CrossRef]
- Simmons, H.E.; Smith, R.D. A new synthesis of cyclopropanes1. J. Am. Chem. Soc. 1959, 81, 4256–4264. [Google Scholar] [CrossRef]
- Furukawa, J.; Kawabata, N.; Nishimura, J. Synthesis of cyclopropanes by the reaction of olefins with dialkylzinc and methylene iodide. Tetrahedron 1968, 24, 53–58. [Google Scholar] [CrossRef]
- Long, J.; Du, H.; Li, K.; Shi, Y. Catalytic asymmetric Simmons–Smith cyclopropanation of unfunctionalized olefins. Tetrahedron Lett. 2005, 46, 2737–2740. [Google Scholar] [CrossRef]
- Sawada, S.; Oda, J.; Inouye, Y. Partial asymmetric synthesis in the Simmons-Smith reaction. II. J. Org. Chem. 1968, 33, 2141–2143. [Google Scholar] [CrossRef]
- Fujii, K.; Misaki, T.; Sugimura, T. Another Role of Copper in the Simmons–Smith Reaction: Copper-catalyzed Nucleophilic Michael-type Cyclopropanation of α, β-Unsaturated Ketones. Chem. Lett. 2014, 43, 634–636. [Google Scholar] [CrossRef]
- Simmons, H.E.; Blanchard, E.P.; Smith, R.D. Cyclopropane synthesis from methylene iodide, zinc-copper couple, and olefins. III. The methylene-transfer reaction. J. Am. Chem. Soc. 1964, 86, 1347–1356. [Google Scholar] [CrossRef]
- Imamoto, T.; Takeyama, T.; Koto, H. The reaction of carbonyl compounds with diiodomethane in the presence of samarium: Novel syntheses of iodohydrins and cyclopropanols. Tetrahedron Lett. 1985, 27, 3243–3246. [Google Scholar] [CrossRef]
- Friedrich, E.C.; Biresaw, G. Zinc dust-cuprous chloride promoted cyclopropanations of allylic alcohols using ethylidene iodide. J. Org. Chem. 1982, 47, 1615–1618. [Google Scholar] [CrossRef]
- Fujii, K.; Shiine, K.; Misaki, T.; Sugimura, T. Efficient Simmons–Smith cyclopropanation with Zn/Cu and CH2I2. Appl. Organomet. Chem. 2013, 27, 69–72. [Google Scholar] [CrossRef]
- Denmark, S.E.; Edwards, J.P.; Wilson, S.R. Solution-and solid-state structural studies of (halomethyl) zinc reagents. J. Am. Chem. Soc. 1992, 114, 2592–2602. [Google Scholar] [CrossRef]
- Donaldson, W. Synthesis of cyclopropane containing natural products. Tetrahedron 2011, 27, 8589–8627. [Google Scholar] [CrossRef]
- Lu, T.; Hayashi, R.; Hsung, R.P.; DeKorver, K.A.; Lohse, A.G.; Song, Z.; Tang, Y. Synthesis of amido-spiro [2.2] pentanes via Simmons–Smith cyclopropanation of allenamides. Org. Biomol. Chem. 2009, 7, 3331–3337. [Google Scholar] [CrossRef]
- Mali, M.; Sharma, G.V.; Ghosh, S.; Roisnel, T.; Carboni, B.; Berree, F. Simmons–Smith Cyclopropanation of Alkenyl 1, 2-Bis (boronates): Stereoselective Access to Functionalized Cyclopropyl Derivatives. J. Org. Chem. 2022, 87, 7649–7657. [Google Scholar] [CrossRef] [PubMed]
- Pietruszka, J. Synthesis and properties of oligocyclopropyl-containing natural products and model compounds. Chem. Rev. 2003, 103, 1051–1070. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Brandt, W.; Thiemann, T. Biosynthesis and metabolism of cyclopropane rings in natural compounds. Chem. Rev. 2003, 103, 1625–1648. [Google Scholar] [CrossRef]
- Salaun, J. Cyclopropane derivatives and their diverse biological activities. In Small Ring Compounds in Organic Synthesis; Springer: Berlin/Heidelberg, Germany, 2000; Volume 207, ISBN 978-3-540-48255-0. [Google Scholar] [CrossRef]
- Kim, H.Y.; Walsh, P.J. Tandem catalytic enantio-and diastereoselective synthesis of cyclopropyl alcohols using aryl aldehydes. J. Phys. Org. Chem. 2012, 25, 933–938. [Google Scholar] [CrossRef]
- Long, J.; Yuan, Y.; Shi, Y. Asymmetric Simmons−Smith Cyclopropanation of Unfunctionalized Olefins. J. Am. Chem. Soc. 2003, 125, 13632–13633. [Google Scholar] [CrossRef] [PubMed]
- Arai, I.; Mori, A.; Yamamoto, H. An asymmetric Simmons-Smith reaction. J. Am. Chem. Soc. 1985, 107, 8254–8256. [Google Scholar] [CrossRef]
- Verpoorte, R.; van der Heijden, R.; Moreno, P.R. Biosynthesis of terpenoid indole alkaloids in Catharanthus roseus cells. In The Alkaloids: Chemistry and Pharmacology; Academic Press: Cambridge, MA, USA, 1997; Volume 49, pp. 221–299. [Google Scholar] [CrossRef]
- Sarfraz, I.; Rasul, A.; Hussain, G.; Shah, M.A.; Zahoor, A.F.; Asrar, M.; Selamoglu, Z.; Ji, X.Y.; Adem, S.; Sarker, S.D. 6-Phosphogluconate dehydrogenase fuels multiple aspects of cancer cells: From cancer initiation to metastasis and chemoresistance. BioFactors 2020, 46, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Zahoor, A.F.; Parveen, B.; Rasul, A.; Raza, Z.; Ahmad, S.; Irfan, A.; El-Hiti, G.A. Acefylline derivatives as a new class of anticancer agents: Synthesis, molecular docking, and anticancer, hemolytic, and thrombolytic activities of acefylline-triazole hybrids. J. Chem. 2022, 2022, 3502872. [Google Scholar] [CrossRef]
- Sears, J.E.; Boger, D.L. Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure–function properties. Acc. Chem. Res. 2015, 48, 653–662. [Google Scholar] [CrossRef]
- Keglevich, P.; Hazai, L.; Kalaus, G.; Szántay, C. Cyclopropanation of some alkaloids. Period. Polytech. Chem. Eng. 2015, 59, 3–15. [Google Scholar] [CrossRef]
- Keglevich, A.; Mayer, S.; Pspai, R.; Szigetvsri, A.; Santa, Z.; Dekany, M.; Szantay, C.; Keglevich, P.; Hazai, L. Attempted synthesis of vinca alkaloids condensed with three-membered rings. Molecules 2018, 23, 2574. [Google Scholar] [CrossRef]
- Kam, T.S.; Yoganathan, K.; Chuah, C.H. Lundurines A, B and C, new indole alkaloids with a novel carbon skeleton containing a cyclopropyl moiety. Tetrahedron Lett. 1995, 36, 759–762. [Google Scholar] [CrossRef]
- Kam, T.S.; Lim, K.H.; Yoganathan, K.; Hayashi, M.; Komiyama, K. Lundurines A–D, cytotoxic indole alkaloids incorporating a cyclopropyl moiety from Kopsia tenuis and revision of the structures of tenuisines A–C. Tetrahedron 2004, 60, 10739–10745. [Google Scholar] [CrossRef]
- Jin, S.; Gong, J.; Qin, Y. Total Synthesis of (−)-Lundurine A and Determination of its Absolute Configuration. Angew. Chem. Int. Ed. 2015, 127, 2256–2259. [Google Scholar] [CrossRef]
- Zhou, Z.F.; Liu, H.L.; Zhang, W.; Kurtan, T.; Mandi, A.; Benyei, A.; Li, J.; Taglialatela-Scafati, O.; Guo, Y.W. Bioactive rearranged limonoids from the Chinese mangrove Xylocarpus granatum Koenig. Tetrahedron 2014, 70, 6444–6449. [Google Scholar] [CrossRef]
- Pan, J.Y.; Chen, S.L.; Li, M.Y.; Li, J.; Yang, M.H.; Wu, J. Limonoids from the Seeds of a Hainan Mangrove, Xylocarpus granatum. J. Nat. Prod. 2010, 73, 1672–1679. [Google Scholar] [CrossRef]
- Schuppe, A.W.; Huang, D.; Chen, Y.; Newhouse, T.R. Total synthesis of (−)-Xylogranatopyridine B via a Palladium-catalyzed oxidative stannylation of enones. J. Am. Chem. Soc. 2018, 140, 2062–2066. [Google Scholar] [CrossRef]
- Munir, I.; Zahoor, A.F.; Rasool, N.; Naqvi, S.A.R.; Zia, K.M.; Ahmad, R. Synthetic applications and methodology development of Chan–Lam coupling: A review. Mol. Divers. 2019, 23, 215–259. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Chen, K.; Qiu, Y.; He, H.; Gao, S. A unified strategy to construct the tetracyclic ring of calyciphylline A alkaloids: Total synthesis of himalensine A. Org. Lett. 2019, 21, 3741–3745. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, J.; Guo, L.D.; Tian, P.; Xu, T.; Xu, J. Synthesis of the core structure of Daphnimacropodines. Org. Lett. 2019, 21, 4309–4312. [Google Scholar] [CrossRef]
- Hanessian, S.; Dorich, S.; Menz, H. Concise and stereocontrolled synthesis of the tetracyclic core of daphniglaucin C. Org. Lett. 2013, 15, 4134–4137. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, L.D.; Xu, J. Synthesis of the tricyclic skeleton of Daphniphyllum alkaloids daphnimacropodines. Tetrahedron Lett. 2021, 71, 153030. [Google Scholar] [CrossRef]
- Nakazawa, J.; Yajima, J.; Usui, T.; Ueki, M.; Takatsuki, A.; Imoto, M.; Toyoshima, Y.Y.; Osada, H. A novel action of terpendole E on the motor activity of mitotic Kinesin Eg5. Chem. Biol. 2003, 10, 131–137. [Google Scholar] [CrossRef]
- Tarui, Y.; Chinen, T.; Nagumo, Y.; Motoyama, T.; Hayashi, T.; Hirota, H.; Muroi, M.; Ishii, Y.; Kondo, H.; Osada, H.; et al. Terpendole E and its Derivative Inhibit STLC-and GSK-1-Resistant Eg5. ChemBioChem 2014, 15, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Teranishi, T.; Murokawa, T.; Enomoto, M.; Kuwahara, S. Synthesis of (±)-terpendole E. Biosci. Biotechnol. Biochem. 2015, 79, 11–15. [Google Scholar] [CrossRef]
- Ogata, M.; Ueda, J.Y.; Hoshi, M.; Hashimoto, J.; Nakashima, T.; Anzai, K.; Takagi, M.; Shin-ya, K. A novel indole-diterpenoid, JBIR-03 with anti-MRSA activity from Dichotomomyces cejpii var. cejpii NBRC 103559. J. Antibiot. 2007, 60, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.F.; Ji, N.Y.; Liu, X.H.; Li, K.; Zhu, Q.M.; Xue, Q.Z. Indoloditerpenes from an algicolous isolate of Aspergillus oryzae. Bioorg. Med. Chem. Lett. 2010, 20, 5677–5680. [Google Scholar] [CrossRef] [PubMed]
- Tetsuro, M.; Masaru, E.; Takaaki, T.; Yusuke, O.; Shigefumi, K. Total synthesis of JBIR-03 and asporyzin C. Tetrahedron Lett. 2018, 59, 4107–4109. [Google Scholar] [CrossRef]
- Liu, G.; Romo, D. Cover Picture: Total Synthesis of (+)-Omphadiol. Angew. Chem. Int. Ed. 2011, 50, 7449. [Google Scholar] [CrossRef]
- Zhou, L.; Yao, Y.; Xu, W.; Liang, G. Total syntheses of (±)-omphadiol and (±)-pyxidatol C through a cis-fused 5, 7-carbocyclic common intermediate. J. Org. Chem. 2014, 79, 5345–5350. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Eggert, U.; Kalesse, M. Synthesis of (+)-Omphadiol and (+)-Pyxidatol C. Org. Lett. 2016, 18, 2320–2322. [Google Scholar] [CrossRef]
- McMorris, T.C.; Lira, R.; Gantzel, P.K.; Kelner, M.J.; Dawe, R. Sesquiterpenes from the Basidiomycete Omphalotus i lludens. J. Nat. Prod. 2000, 63, 1557–1559. [Google Scholar] [CrossRef]
- Zheng, Y.B.; Lu, C.H.; Zheng, Z.H.; Lin, X.J.; Su, W.J.; Shen, Y.M. New sesquiterpenes from edible fungus Clavicorona pyxidata. Helv. Chim. Acta 2008, 91, 2174–2180. [Google Scholar] [CrossRef]
- Osler, J.D.; Unsworth, W.P.; Taylor, R.J. Synthetic Studies towards the Africanane Sesquiterpenes via the Cope Rearrangement of gem-Dimethyl-Substituted Divinyl Cyclopropanes. Synlett 2016, 27, 70–74. [Google Scholar] [CrossRef]
- Singh, V.; Thomas, B. Recent developments in general methodologies for the synthesis of linear triquinanest. Tetrahedron 1998, 54, 3647–3692. [Google Scholar] [CrossRef]
- Mehta, G.; Srikrishna, A. Synthesis of polyquinane natural products: An update. Chem. Rev. 1997, 97, 671–720. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Yuan, C.; Yu, Z.X. Tandem Rh (I)-catalyzed [(5+ 2)+ 1] cycloaddition/aldol reaction for the construction of linear triquinane skeleton: Total syntheses of (±)-hirsutene and (±)-1-desoxyhypnophilin. J. Am. Chem. Soc. 2008, 130, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Gao, Z.B.; Wang, F.D.; Liao, S.G.; Chen, H.D.; Zhang, C.R.; Hu, G.Y.; Yue, J.M. Chlorahololides A and B, two potent and selective blockers of the potassium channel isolated from Chloranthus holostegius. Org. Lett. 2007, 9, 903–906. [Google Scholar] [CrossRef]
- Liu, Y.; Nan, F.J. Synthetic studies towards Chlorahololides A: Practical synthesis of a lindenane-type sesquiterpenoid core framework with a 5, 6-double bond. Tetrahedron Lett. 2010, 51, 1374–1376. [Google Scholar] [CrossRef]
- Qian, S.; Zhao, G. Synthetic studies toward the total synthesis of Chlorahololide A. Synlett 2011, 722–724. [Google Scholar] [CrossRef]
- Zhan, Z.J.; Ying, Y.M.; Ma, L.F.; Shan, W.G. Natural disesquiterpenoids. Nat. Prod. Rep. 2011, 28, 594–629. [Google Scholar] [CrossRef]
- Takeda, Y.; Yamashita, H.; Matsumoto, T.; Terao, H. Chloranthalactone F, a sesquiterpenoid from the leaves of Chloranthus glaber. Phytochemistry 1993, 33, 713–715. [Google Scholar] [CrossRef]
- Qian, S.; Zhao, G. Total synthesis of (+)-chloranthalactone F. Chem. Commun. 2012, 48, 3530–3532. [Google Scholar] [CrossRef] [PubMed]
- Kashiwabara, M.; Kamo, T.; Makabe, H.; Shibata, H.; Hirota, M. Repraesentins D, E and F, new plant growth promoters from Lactarius repraesentaneus. Biosci. Biotechnol. Biochem. 2006, 70, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Hirota, M.; Shimizu, Y.; Kamo, T.; Makabe, H.; Shibata, H. New plant growth promoters, repraesentins A, B and C, from Lactarius repraesentaneus. Biosci. Biotechnol. Biochem. 2003, 67, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, S.; Echavarren, A.M. Total synthesis of repraesentin F and configuration reassignment by a gold (I)-catalyzed cyclization cascade. Org. Lett. 2018, 20, 5784–5788. [Google Scholar] [CrossRef]
- Khatri Chhetri, B.; Lavoie, S.; Sweeney-Jones, A.M.; Mojib, N.; Raghavan, V.; Gagaring, K.; Dale, B.; McNamara, C.W.; Soapi, K.; Quave, C.L.; et al. Peyssonnosides A–B, unusual diterpene glycosides with a sterically encumbered cyclopropane motif: Structure elucidation using an integrated spectroscopic and computational workflow. J. Org. Chem. 2019, 84, 8531–8541. [Google Scholar] [CrossRef]
- Ebner, C.; Carreira, E.M. Cyclopropanation strategies in recent total syntheses. Chem. Rev. 2017, 117, 11651–11679. [Google Scholar] [CrossRef] [PubMed]
- Chesnokov, G.A.; Gademann, K. Concise Total Synthesis of Peyssonnoside A. J. Am. Chem. Soc. 2021, 143, 14083–14088. [Google Scholar] [CrossRef]
- Justice, M.C.; Hsu, M.J.; Tse, B.; Ku, T.; Balkovec, J.; Schmatz, D.; Nielsen, J. Elongation factor 2 as a novel target for selective inhibition of fungal protein synthesis. J. Biol. Chem. 1998, 273, 3148–3151. [Google Scholar] [CrossRef]
- Quesnelle, C.A.; Gill, P.; Dodier, M.; Laurent, D.S.; Serrano-Wu, M.; Marinier, A.; Martel, A.; Mazzucco, C.E.; Stickle, T.M.; Barrett, J.F.; et al. Sordaricin antifungal agents. Bioorg. Med. Chem. Lett. 2003, 13, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Kitamura, M.; Narasaka, K. Synthesis of (−)-sordarin. J. Am. Chem. Soc. 2006, 128, 6931–6937. [Google Scholar] [CrossRef]
- Block, S.; Gerkens, P.; Peulen, O.; Jolois, O.; Mingeot-Leclercq, M.P.; De Pauw-gillet, M.C.; Quetin-Leclercq, J. Induction of apoptosis in human promyelocytic leukemia cells by a natural trachylobane diterpene. Anticancer Res. 2005, 25, 363–368. [Google Scholar] [PubMed]
- Block, S.; Baccelli, C.; Tinant, B.; Van Meervelt, L.; Rozenberg, R.; Jiwan, J.L.H.; Llabres, G.; Pauw-Gillet, M.C.D.; Quetin-Leclercq, J. Diterpenes from the leaves of Croton zambesicus. Phytochemistry 2004, 65, 1165–1171. [Google Scholar] [CrossRef]
- Abad, A.; Agullo, C.; Cunat, A.C.; de Alfonso Marzal, I.; Navarro, I.; Gris, A. A unified synthetic approach to trachylobane-, beyerane-, atisane-and kaurane-type diterpenes. Tetrahedron 2006, 62, 3266–3283. [Google Scholar] [CrossRef]
- Eddy, N.A.; Fenteany, G. Model studies directed to the synthesis of cucurbitacin IC/D rings. Tetrahedron Lett. 2015, 56, 5079–5081. [Google Scholar] [CrossRef]
- Chen, J.C.; Chiu, M.H.; Nie, R.L.; Cordell, G.A.; Qiu, S.X. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat. Prod. Rep. 2005, 22, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Ramalhete, C.; Molnar, J.; Mulhovo, S.; Rosario, V.E.; Ferreira, M.J.U. New potent P-glycoprotein modulators with the cucurbitane scaffold and their synergistic interaction with doxorubicin on resistant cancer cells. Bioorg. Med. Chem. 2009, 17, 6942–6951. [Google Scholar] [CrossRef] [PubMed]
- Bucknam, A.R.; Micalizio, G.C. Asymmetric de novo synthesis of a cucurbitane triterpenoid: Total synthesis of octanorcucurbitacin B. J. Am. Chem. Soc. 2022, 144, 8493–8497. [Google Scholar] [CrossRef] [PubMed]
- Ghany, M.G.; Strader, D.B.; Thomas, D.L.; Seeff, L.B. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2009, 49, 1335–1374. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, F.R.; Levinskas, G.J. Subchronic toxicity of tetramethylsuccinonitrile. Fundam. Appl. Toxicol. 1986, 7, 41–48. [Google Scholar] [CrossRef]
- Young, I.S.; Qiu, Y.; Smith, M.J.; Hay, M.B.; Doubleday, W.W. Preparation of a tricyclopropylamino acid derivative via Simmons–Smith cyclopropanation with downstream intramolecular aminoacetoxylation for impurity control. Org. Process Res. Dev. 2016, 20, 2108–2115. [Google Scholar] [CrossRef]
- Tandon, M.; Wu, M.; Begley, T.P.; Myllyharju, J.; Pirskanen, A.; Kivirikko, K. Substrate specificity of human prolyl-4-hydroxylase. Bioorg. Med. Chem. Lett. 1998, 8, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Grygorenko, O.O.; Radchenko, D.S.; Volochnyuk, D.M.; Tolmachev, A.A.; Komarov, I.V. Bicyclic conformationally restricted diamines. Chem. Rev. 2011, 111, 5506–5568. [Google Scholar] [CrossRef] [PubMed]
- Tymtsunik, A.V.; Bilenko, V.A.; Ivon, Y.M.; Grygorenko, O.O.; Komarov, I.V. Synthesis of a novel Boc-protected cyclopropane-modified proline analogue. Tetrahedron Lett. 2012, 53, 3847–3849. [Google Scholar] [CrossRef]
- Savage, S.A.; Jones, G.S.; Kolotuchin, S.; Ramrattan, S.A.; Vu, T.; Waltermire, R.E. Preparation of saxagliptin, a novel DPP-IV inhibitor. Org. Process Res. Dev. 2009, 13, 1169–1176. [Google Scholar] [CrossRef]
- Oliveira, D.F.; Miranda, P.C.; Correia, C.R. Efficient and expeditious protocols for the synthesis of racemic and enantiomerically pure endocyclic enecarbamates from N-acyl lactams and N-acyl pyrrolidines. J. Org. Chem. 1999, 64, 6646–6652. [Google Scholar] [CrossRef]
- Tymtsunik, A.V.; Ivon, Y.M.; Komarov, I.V.; Grygorenko, O.O. Synthesis of Boc-protected 4, 5-methano-β-proline. Tetrahedron Lett. 2014, 55, 3312–3315. [Google Scholar] [CrossRef]
- Rizzo, S.; Waldmann, H. Development of a Natural-Product-Derived Chemical Toolbox for Modulation of Protein Function. Chem. Rev. 2014, 114, 4621–4639. [Google Scholar] [CrossRef]
- Spiteller, P.; Von Nussbaum, F. β-Amino Acids in Natural Products. In Enantioselective Synthesis of β-Amino Acids; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 19–91. [Google Scholar] [CrossRef]
- Tymtsunik, A.V.; Ivon, Y.M.; Komarov, I.V.; Grygorenko, O.O. Synthesis of racemic and enantiopure 3, 4-methanonipecotic acid. Tetrahedron Asymmetry 2015, 26, 1268–1272. [Google Scholar] [CrossRef]
- Frantz, M.C.; Wipf, P. Mitochondria as a target in treatment. Environ. Mol. Mutagen. 2010, 51, 462–475. [Google Scholar] [CrossRef]
- Epperly, M.W.; Goff, J.P.; Li, S.; Gao, X.; Wipf, P.; Dixon, T.; Wang, H.; Franicola, D.; Shen, H.; Rwigema, J.C.M.; et al. Intraesophageal administration of GS-nitroxide (JP4-039) protects against ionizing irradiation-induced esophagitis. In Vivo 2010, 24, 811–819. [Google Scholar]
- Frantz, M.C.; Pierce, J.G.; Pierce, J.M.; Kangying, L.; Qingwei, W.; Johnson, M.; Wipf, P. Large-scale asymmetric synthesis of the bioprotective agent JP4-039 and analogs. Org. Lett. 2011, 13, 2318–2321. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, A.N.; Radchenko, D.S.; Mykhailiuk, P.K.; Grygorenko, O.O.; Komarov, I.V. 4-Fluoro-2, 4-methanoproline. Org. Lett. 2009, 11, 5674–5676. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R.; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; James, C.A.; Meanwell, N.A.; Hamann, L.G.; Belema, M. A scalable synthesis of (1R, 3S, 5R)-2-(tert-butoxycarbonyl)-2-azabicyclo [3.1. 0] hexane-3-carboxylic acid. Tetrahedron Lett. 2013, 54, 6722–6724. [Google Scholar] [CrossRef]
- Saenger, W. Defining terms for the nucleic acids. In Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1984; pp. 9–28. [Google Scholar] [CrossRef]
- Gagneron, J.; Gosselin, G.; Mathe, C. Synthesis of nucleoside analogues bearing the five naturally occurring nucleic acid bases built on a 2-oxabicylo [3.1. 0] hexane scaffold. J. Org. Chem. 2005, 70, 6891–6897. [Google Scholar] [CrossRef]
- Toti, K.S.; Osborne, D.; Ciancetta, A.; Boison, D.; Jacobson, K.A. South (S)-and North (N)-methanocarba-7-deazaadenosine analogues as inhibitors of human adenosine kinase. J. Med. Chem. 2016, 59, 6860–6877. [Google Scholar] [CrossRef]
- An, S.; Kim, G.; Kim, H.J.; Ahn, S.; Kim, H.Y.; Ko, H.; Hyun, Y.E.; Nguyen, M.; Joeng, J.; Liu, Z.; et al. Discovery and structure–activity relationships of novel template, truncated 1′-homologated adenosine derivatives as pure dual PPARγ/δ modulators. J. Med. Chem. 2020, 63, 16012–16027. [Google Scholar] [CrossRef]
- Rasool, I.; Ahmad, M.; Khan, Z.A.; Mansha, A.; Maqbool, T.; Zahoor, A.F.; Aslam, S. Recent advancements in oxadiazole-based anticancer agents. Trop. J. Pharm. Res. 2017, 16, 723–733. [Google Scholar] [CrossRef]
- Hyun, Y.E.; Kim, H.R.; Jeong, L.S. Stereoselective Synthesis of (S)-and (N)-Cyclopropyl-Fused Carbocyclic Nucleosides using stereoselective cyclopropanation. J. Org. Chem. 2021, 86, 9828–9837. [Google Scholar] [CrossRef]
- Munawar, S.; Zahoor, A.F.; Ali, S.; Javed, S.; Irfan, M.; Irfan, A.; Mojzych, K.K.; Mojzych, M. Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review. Molecules 2022, 27, 6953. [Google Scholar] [CrossRef]
- Bessieres, M.; Chevrier, F.; Roy, V.; Agrofoglio, L.A. Recent progress for the synthesis of selected carbocyclic nucleosides. Future Med. Chem. 2015, 7, 1809–1828. [Google Scholar] [CrossRef]
- Singh, U.S.; Mulamoottil, V.A.; Chu, C.K. 2′-Fluoro-6′-methylene carbocyclic adenosine and its phosphoramidate prodrug: A novel anti-HBV agent, active against drug-resistant HBV mutants. Med. Res. Rev. 2018, 38, 977–1002. [Google Scholar] [CrossRef]
- Singh, U.S.; Chu, C.K. Synthesis of 2′-deoxy-2′-fluoro-2′-C-methyl spiro cyclopentyl carbocyclic uridine analog as potential inhibitors of HCV NS5B polymerase. Nucleosides Nucleotides Nucleic Acids 2020, 39, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Tong, C.Y.W.; Wong, T.; Wilkinson, M. New therapies for chronic hepatitis C infection: A systematic review of evidence from clinical trials. Int. J. Clin. Pract. 2012, 66, 342–355. [Google Scholar] [CrossRef]
- Suhara, Y.; Nihei, K.I.; Kurihara, M.; Kittaka, A.; Yamaguchi, K.; Fujishima, T.; Kommo, K.; Miyata, N.; Takayama, H. Efficient and versatile synthesis of novel 2α-substituted 1α, 25-dihydroxyvitamin D3 analogues and their docking to vitamin D receptors. J. Org. Chem. 2001, 66, 8760–8771. [Google Scholar] [CrossRef] [PubMed]
- Komsta, Z.; Mayes, B.A.; Moussa, A.; Shelbourne, M.; Stewart, A.; Tyrrell, A.J.; Wallis, L.L.; Weymouth-Wilson, A.C.; Yurek-George, A. Stereoselective Cyclopropanation in the Synthesis of 3′-Deoxy-3′-C-hydroxymethyl-2′, 3′-methylene-uridine. Org. Lett. 2014, 16, 4878–4880. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef]
- Panayides, J.L.; Mathieu, V.; Banuls, L.M.Y.; Apostolellis, H.; Dahan-Farkas, N.; Davids, H.; Harmse, L.; Rey, M.E.C.; Green, I.R.; Pelly, S.C.; et al. Synthesis and in vitro growth inhibitory activity of novel silyl-and trityl-modified nucleosides. Bioorg. Med. Chem. 2016, 24, 2716–2724. [Google Scholar] [CrossRef]
- Kollmann, C.; Wiechert, S.M.; Jones, P.G.; Pietschmann, T.; Werz, D.B. Synthesis of 4′/5′-Spirocyclopropanated Uridine and d-Xylouridine Derivatives and Their Activity against the Human Respiratory Syncytial Virus. Org. Lett. 2019, 21, 6966–6971. [Google Scholar] [CrossRef]
- Qiu, Y.L.; Ksebati, M.B.; Ptak, R.G.; Fan, B.Y.; Breitenbach, J.M.; Lin, J.S.; Cheng, Y.C.; Kern, E.R.; Drach, J.C.; Zemlicka, J. (Z)-and (E)-2-((hydroxymethyl) cyclopropylidene) methyladenine and-guanine. New nucleoside analogues with a broad-spectrum antiviral activity. J. Med. Chem. 1998, 41, 10–23. [Google Scholar] [CrossRef]
- Kim, A.; Hong, J.H. Synthesis and antiviral activity of C-fluoro-branched cyclopropyl nucleosides. Eur. J. Med. Chem. 2007, 42, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, R.D.; Salinas, J.C.; Ostergaard, M.E.; Swayze, E.E.; Seth, P.P.; Hanessian, S. Design, synthesis, and duplex-stabilizing properties of conformationally constrained tricyclic analogues of LNA. Org. Biomol. Chem. 2016, 14, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Hari, Y.; Osawa, T.; Obika, S. Synthesis and duplex-forming ability of oligonucleotides containing 4′-carboxythymidine analogs. Org. Biomol. Chem. 2012, 10, 9639–9649. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamamoto, C.; Horiba, M.; Aoyama, H.; Obika, S. Synthesis and duplex-forming ability of oligonucleotides modified with 4′-C, 5′-C-methylene-bridged nucleic acid (4′, 5′-BNA). Bioorg. Med. Chem. 2021, 46, 116359. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Guguloth, N.; Yadav, J.S. The first asymmetric total syntheses of 3 ((1R, 2R)-and 3 ((1S, 2R)-2-(12-methyltridecyl) cyclopropyl) propanoic acid. Tetrahedron Lett. 2012, 53, 6718–6720. [Google Scholar] [CrossRef]
- Hegde, V.R.; Pu, H.; Patel, M.; Das, P.R.; Strizki, J.; Gullo, V.P.; Chou, C.C.; Buevich, A.V.; Chan, T.M. Three new compounds from the plant Lippia alva as inhibitors of chemokine receptor 5 (CCR5). Bioorg. Med. Chem. Lett. 2004, 14, 5339–5342. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Kanikarapu, S.; Naidu, P.R.; Yadav, J.S. Toward the synthesis of brevipolide H. Tetrahedron Lett. 2016, 56, 1041–1044. [Google Scholar] [CrossRef]
- Gesinski, M.R.; Rychnovsky, S.D. Total synthesis of the cyanolide A aglycon. J. Am. Chem. Soc. 2011, 133, 9727–9729. [Google Scholar] [CrossRef]
- Trost, B.M.; Quintard, A. Asymmetric Catalytic Alkynylation of Acetaldehyde: Application to the Synthesis of (+)-Tetrahydropyrenophorol. Angew. Chem. Int. Ed. 2012, 124, 6808–6812. [Google Scholar] [CrossRef]
- Ralston, K.J.; Ramstadius, H.C.; Brewster, R.C.; Niblock, H.S.; Hulme, A.N. Self-Assembly of Disorazole C1 through a One-Pot Alkyne Metathesis Homodimerization Strategy. Angew. Chem. Int. Ed. 2015, 127, 7192–7196. [Google Scholar] [CrossRef][Green Version]
- Haydl, A.M.; Breit, B. Atom-Economical Dimerization Strategy by the Rhodium-Catalyzed Addition of Carboxylic Acids to Allenes: Protecting-Group-Free Synthesis of Clavosolide A and Late-Stage Modification. Angew. Chem. Int Ed. 2015, 127, 15750–15754. [Google Scholar] [CrossRef]
- Salim, H.; Piva, O. Sequential cross-metathesis/cyclopropanation: Short syntheses of (+/−)-cascarillic acid and (+/−)-grenadamide. Tetrahedron Lett. 2007, 48, 2059–2062. [Google Scholar] [CrossRef]
- Seo, Y.; Cho, K.W.; Rho, J.R.; Shin, J.; Kwon, B.M.; Bok, S.H.; Song, J.I. Solandelactones AI, lactonized cyclopropyl oxylipins isolated from the hydroid Solanderia secunda. Tetrahedron 1996, 52, 10583–10596. [Google Scholar] [CrossRef]
- White, J.D.; Lincoln, C.M.; Yang, J.; Martin, W.H.; Chan, D.B. Total Synthesis of Solandelactones A, B, E, and F Exploiting a Tandem Petasis− Claisen Lactonization Strategy. J. Org. Chem. 2008, 73, 4139–4150. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Montano, N.; Vicente, J.; Rodriguez, A.D. Novel cyclopropane fatty acids from the phospholipids of the Caribbean sponge Pseudospongosorites suberitoides. Lipids 2007, 42, 519–524. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Montano, N.; Reguera, R.M.; Balana-Fouce, R. The first total synthesis of the (±)-17-methyl-trans-4, 5-methyleneoctadecanoic acid and related analogs with antileishmanial activity. Tetrahedron Lett. 2010, 51, 6153–6155. [Google Scholar] [CrossRef][Green Version]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins—Suppressors of apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef]
- Erdogan, M.; Daştan, A. Synthesis of N-substituted dibenzoazepine–pyridazine derivatives as potential neurologically active drugs. Synth. Commun. 2020, 50, 3845–3853. [Google Scholar] [CrossRef]
- Asano, M.; Hashimoto, K.; Saito, B.; Shiokawa, Z.; Sumi, H.; Yabuki, M.; Yoshimatsu, M.; Aoyama, K.; Hamada, T.; Morishita, N.; et al. Design, stereoselective synthesis, and biological evaluation of novel tri-cyclic compounds as inhibitor of apoptosis proteins (IAP) antagonists. Bioorg. Med. Chem. 2013, 21, 5725–5737. [Google Scholar] [CrossRef]
- Singh, H.; Gupta, N.; Kumar, P.; Dubey, S.K.; Sharma, P.K. A new industrial process for 10-methoxyiminostilbene: Key intermediate for the synthesis of oxcarbazepine. Org. Process Res. Dev. 2009, 13, 870–874. [Google Scholar] [CrossRef]
- Tian, M.; Abdelrahman, A.; Weinhausen, S.; Hinz, S.; Weyer, S.; Dosa, S.; El-Tayeb, A.; Muller, C.E. Carbamazepine derivatives with P2X4 receptor-blocking activity. Bioorg. Med. Chem. 2014, 22, 1077–1088. [Google Scholar] [CrossRef]
- Werth, J.; Uyeda, C. Cobalt-catalyzed Reductive Dimethylcyclopropanation of 1, 3-dienes. Angew. Chem. Int. Ed. 2018, 57, 13902–13906. [Google Scholar] [CrossRef] [PubMed]
- Bali, M.; Akabas, M.H. Defining the propofol binding site location on the GABAA receptor. Mol. Pharm. 2004, 65, 68–76. [Google Scholar] [CrossRef]
- White, P.F.; Warner, D.S. Propofol: Its role in changing the practice of anesthesia. Anesthesiologists 2008, 109, 1132–1136. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, S.; Liu, Z.; Long, Y.; Zhao, J.; Xu, W.; Zhang, H. Development of a Kilogram-Scale Route for Clinical Sample Production of the Intravenous Anesthetic Cipepofol. Org. Process Res. Dev. 2022, 26, 1054–1062. [Google Scholar] [CrossRef]
- Majounie, E.; Abramzon, Y.; Renton, A.E.; Perry, R.; Bassett, S.S.; Pletnikova, O.; Traynor, B.J. Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, A.; Prezzavento, O.; Pappalardo, M.S.; Bousquet, E.; Iadanza, M.; Pike, V.W.; Ronsisvalle, G. Synthesis of (+)-and (−)-cis-2-[(1-adamantylamino)-methyl]-1-phenylcyclopropane derivatives as high affinity probes for σ1 and σ2 binding sites. Il Farm. 2002, 57, 45–53. [Google Scholar] [CrossRef]
- Kawashima, Y.; Ezawa, T.; Yamamura, M.; Harada, T.; Noguchi, T.; Imai, N. Convenient synthesis of (+)-cis-4-(N-adamantyl-N-methylamino)-2, 3-methano-2-phenylbutan-1-ol as a candidate of anti-Alzheimer’s medicine via catalytic enantioselective Simmons–Smith reaction using l-phenylalanine-derived disulfonamide. Tetrahedron Lett. 2016, 57, 668–671. [Google Scholar] [CrossRef]
- Dilmaç, A.M.; Wezeman, T.; Bsr, R.M.; Brase, S. Occurrence, synthesis and applications of natural and designed [3.3. 3] propellanes. Nat. Prod. Rep. 2020, 37, 224–245. [Google Scholar] [CrossRef]
- Belzner, J.; Gareib, B.; Polborn, K.; Schmid, W.; Semmler, K.; Szeimies, G. Synthesen substituierter [1.1. 1] Propellane. Chem. Berichte 1989, 122, 1509–1529. [Google Scholar] [CrossRef]
- Nassar, Y.; Piva, O. A short route to access oxaspiro [n, 3, 3] propellanes. Org. Biomol. Chem. 2020, 18, 5811–5815. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Quinke, K.; Spahn, H.; Mutschler, E. (−)-(S)-Flunoxaprofen and (−)-(S)-naproxen isocyanate: Two new fluorescent chiral derivatizing agents for an enantiospecific determination of primary and secondary amines. Chirality 1989, 1, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Murakami, Y.; Imai, N. Syntheses of (R)-(+)-cibenzoline and analogues via catalytic enantioselective cyclopropanation using (S)-phenylalanine-derived disulfonamide. Tetrahedron Asymmetry 2006, 17, 3067–3069. [Google Scholar] [CrossRef]
- Burger, A.; Yost, W.L. Arylcycloalkylamines. I. 2-phenylcyclopropylamine. J. Am. Chem. Soc. 1948, 70, 2198–2201. [Google Scholar] [CrossRef]
- Alliot, J.; Gravel, E.; Pillon, F.; Buisson, D.A.; Nicolas, M.; Doris, E. Enantioselective synthesis of levomilnacipran. Chem. Commun. 2012, 48, 8111–8113. [Google Scholar] [CrossRef]
- Ishizuka, Y.; Fujimori, H.; Noguchi, T.; Kawasaki, M.; Kishida, M.; Nagai, T.; Imai, N.; Kirihara, M. Asymmetric Syntheses of Pharmaceuticals Containing a Cyclopropane Moiety Using Catalytic Asymmetric Simmons–Smith Reactions of Allylalcohols: Syntheses of Optically Active Tranylcypromine and Milnacipran. Chem. Lett. 2013, 42, 1311–1313. [Google Scholar] [CrossRef]

















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munir, R.; Zahoor, A.F.; Javed, S.; Parveen, B.; Mansha, A.; Irfan, A.; Khan, S.G.; Irfan, A.; Kotwica-Mojzych, K.; Mojzych, M. Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review. Molecules 2023, 28, 5651. https://doi.org/10.3390/molecules28155651
Munir R, Zahoor AF, Javed S, Parveen B, Mansha A, Irfan A, Khan SG, Irfan A, Kotwica-Mojzych K, Mojzych M. Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review. Molecules. 2023; 28(15):5651. https://doi.org/10.3390/molecules28155651
Chicago/Turabian StyleMunir, Ramsha, Ameer Fawad Zahoor, Sadia Javed, Bushra Parveen, Asim Mansha, Ahmad Irfan, Samreen Gul Khan, Ali Irfan, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2023. "Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review" Molecules 28, no. 15: 5651. https://doi.org/10.3390/molecules28155651
APA StyleMunir, R., Zahoor, A. F., Javed, S., Parveen, B., Mansha, A., Irfan, A., Khan, S. G., Irfan, A., Kotwica-Mojzych, K., & Mojzych, M. (2023). Simmons–Smith Cyclopropanation: A Multifaceted Synthetic Protocol toward the Synthesis of Natural Products and Drugs: A Review. Molecules, 28(15), 5651. https://doi.org/10.3390/molecules28155651