
Degradation Profiling of Nardosinone at High Temperature and in Simulated Gastric and Intestinal Fluids

 and
and
Abstract
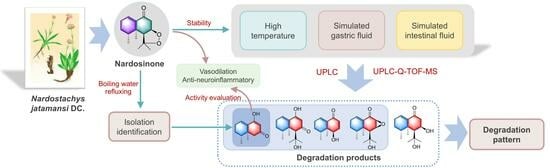
1. Introduction
2. Results and Discussion
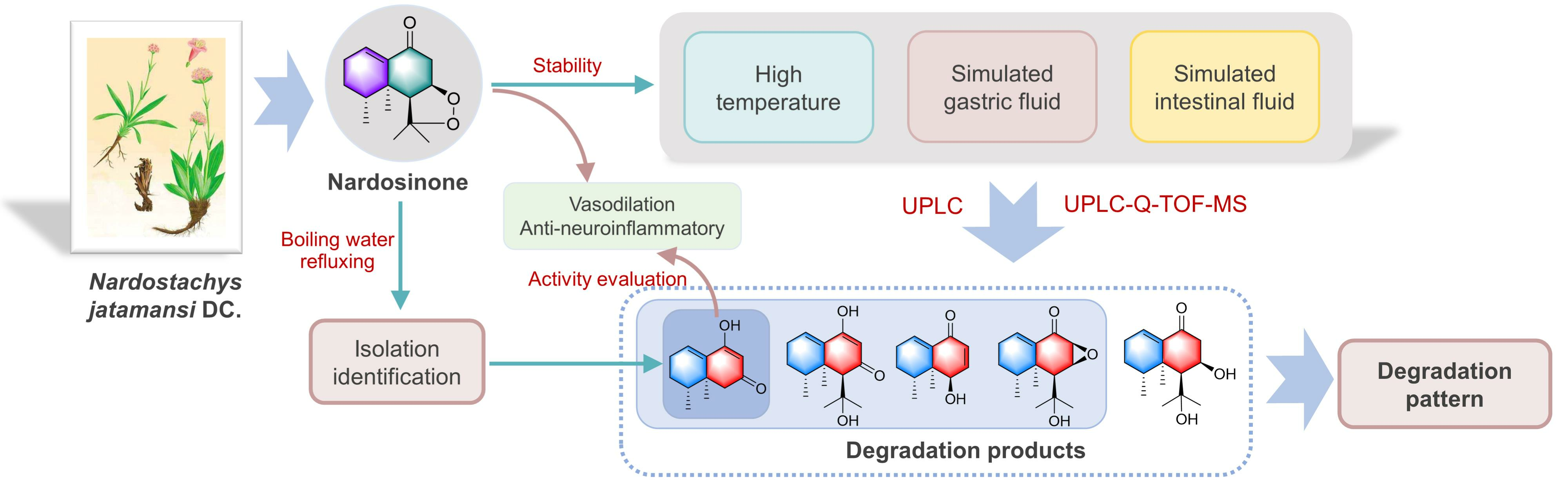
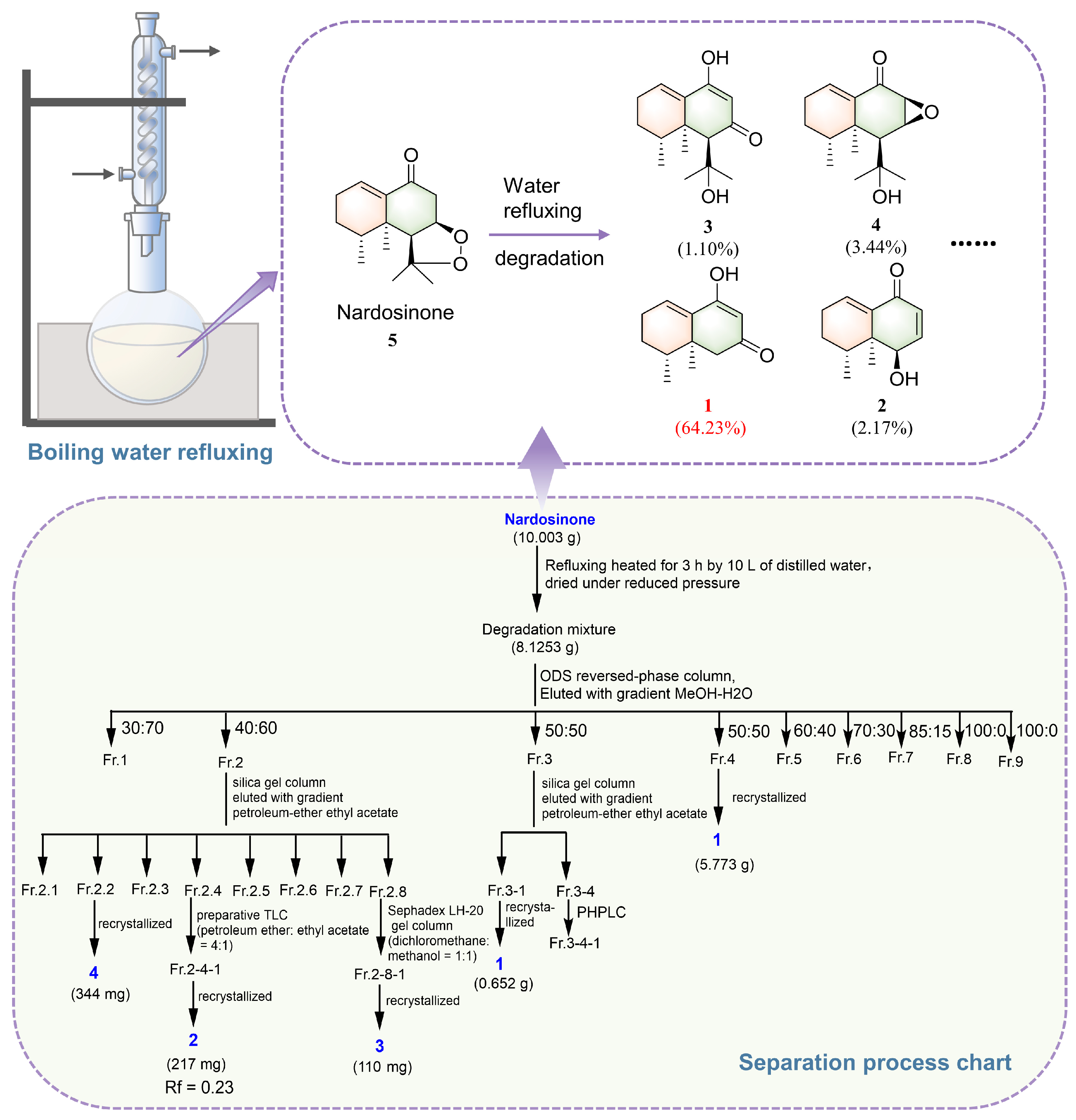
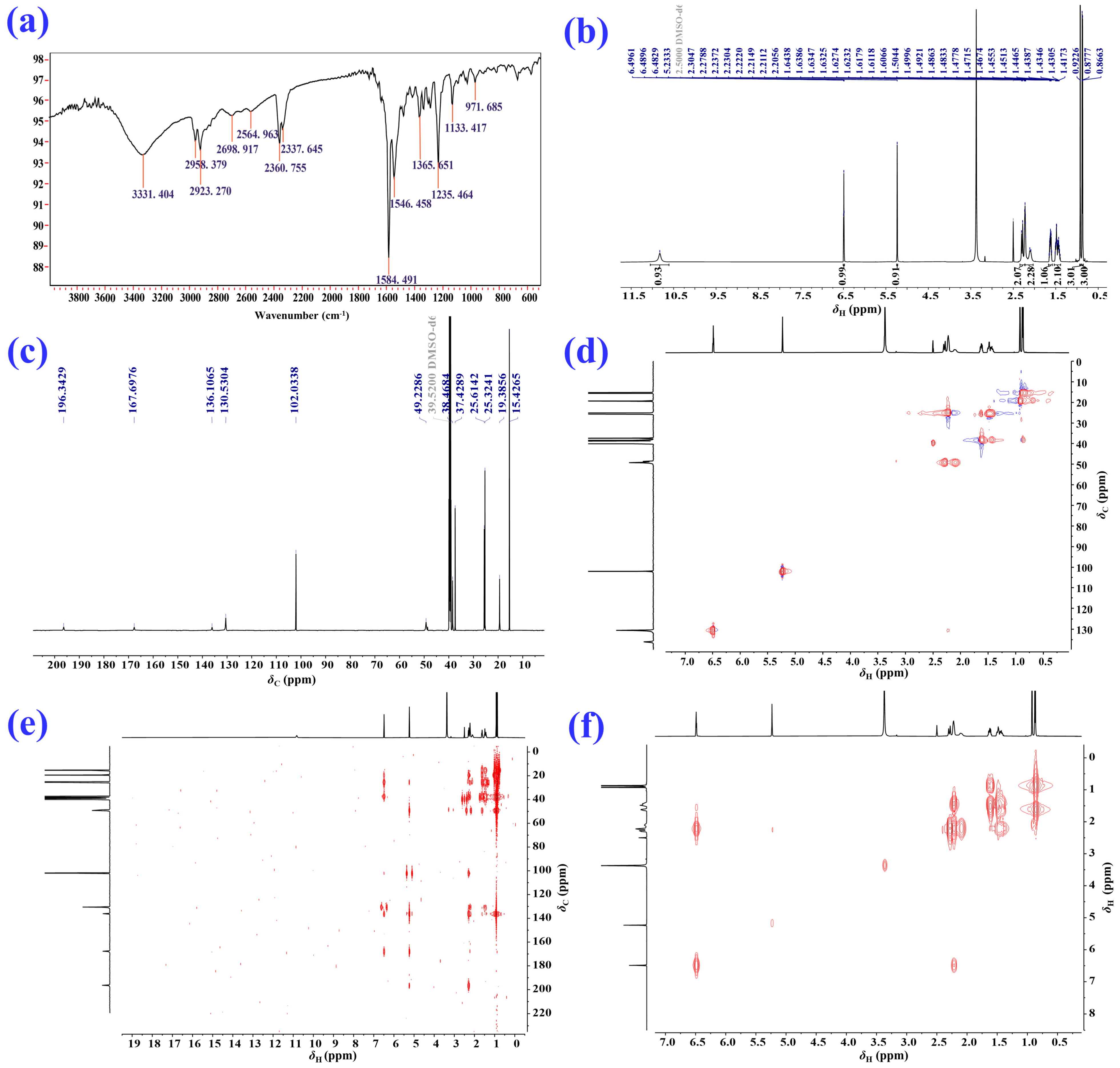
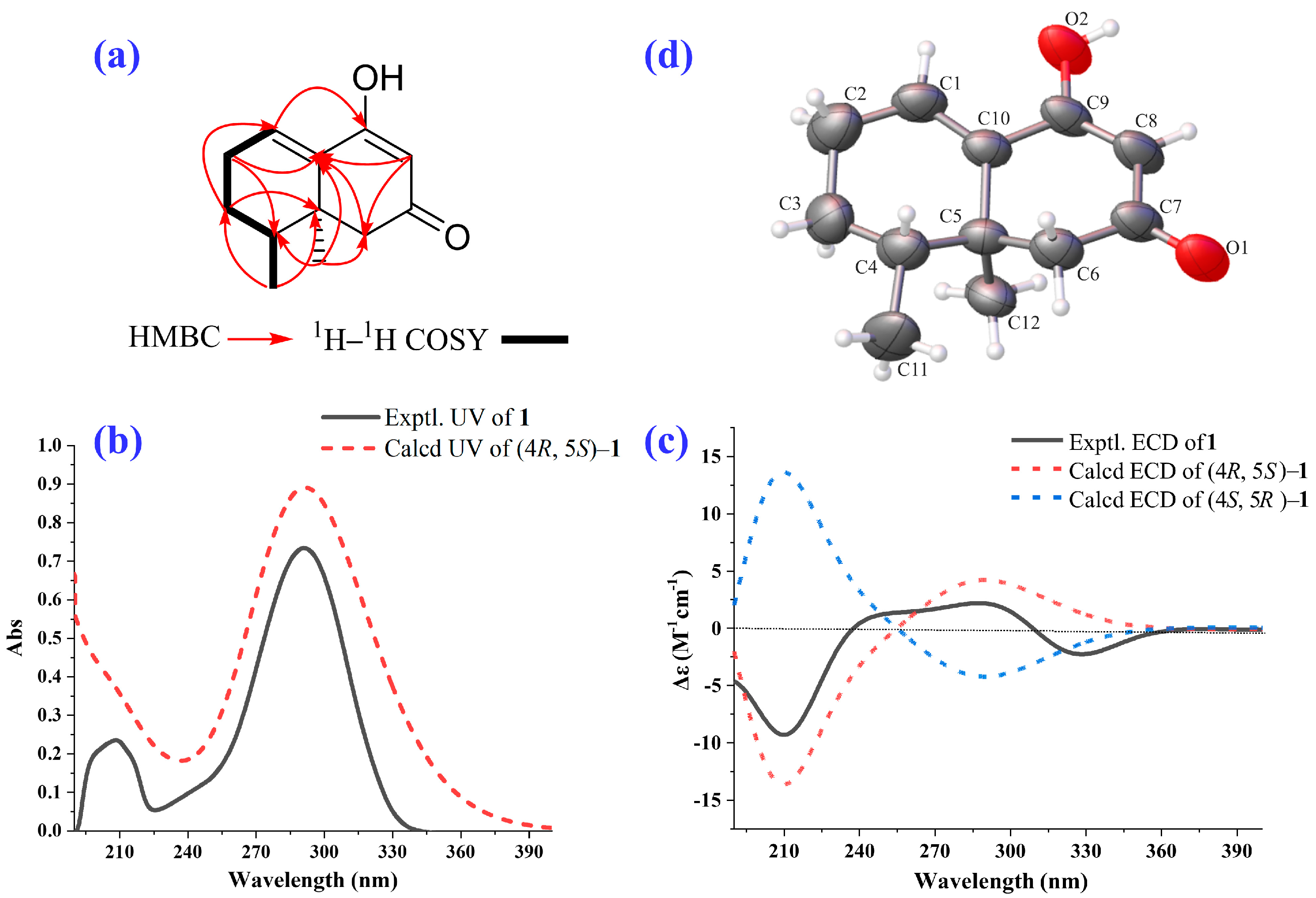
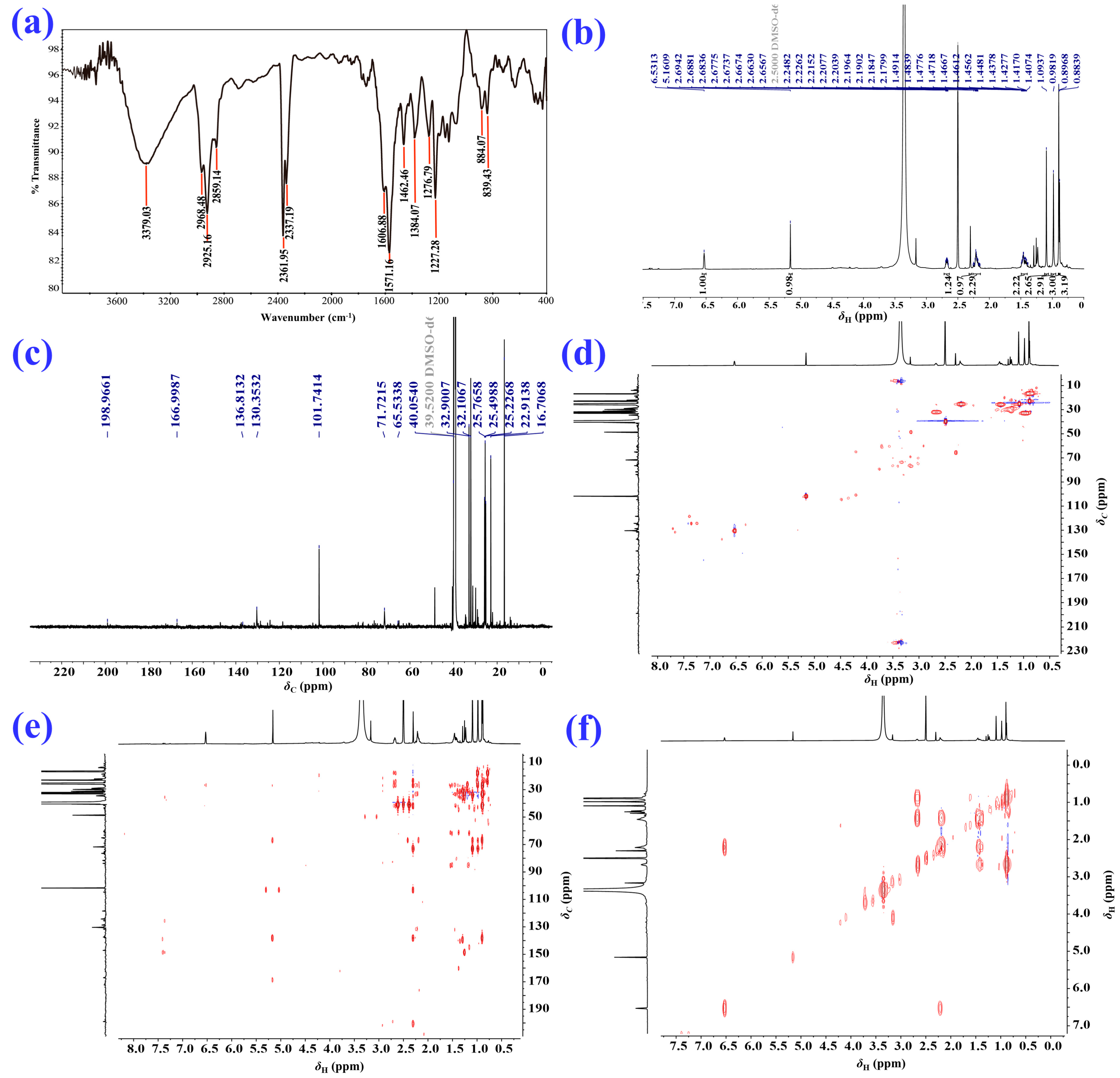
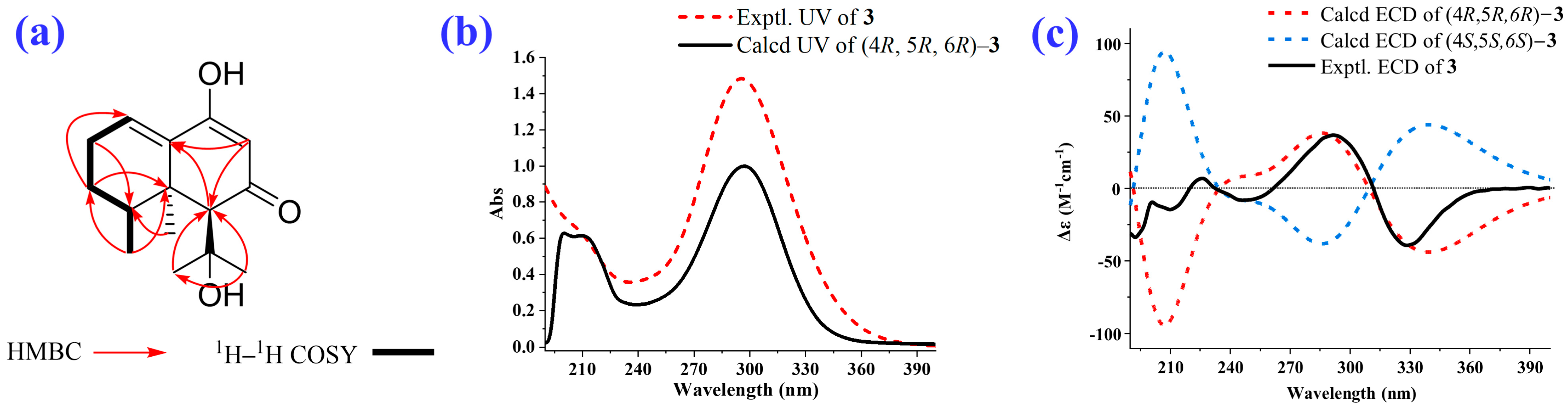
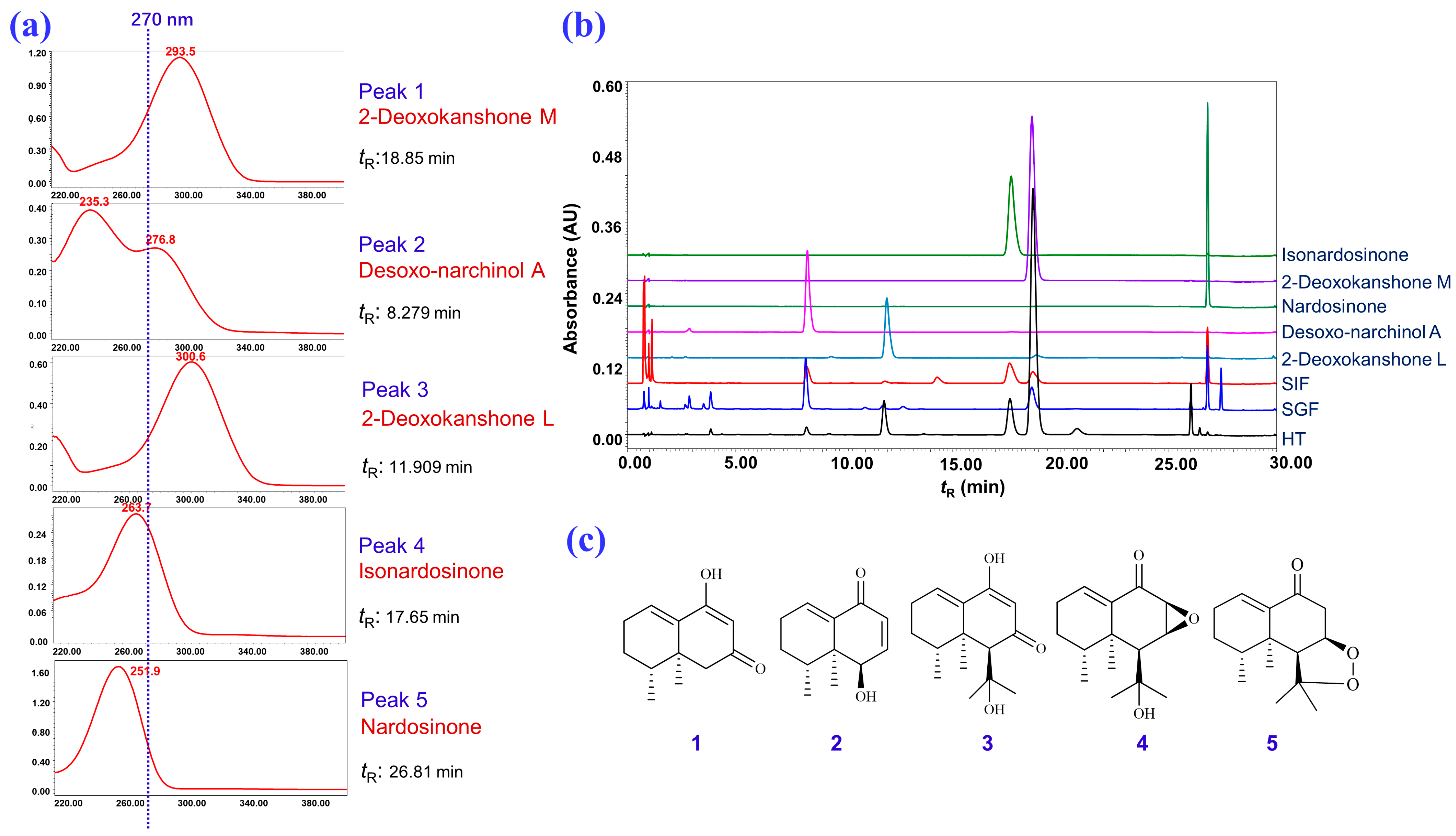
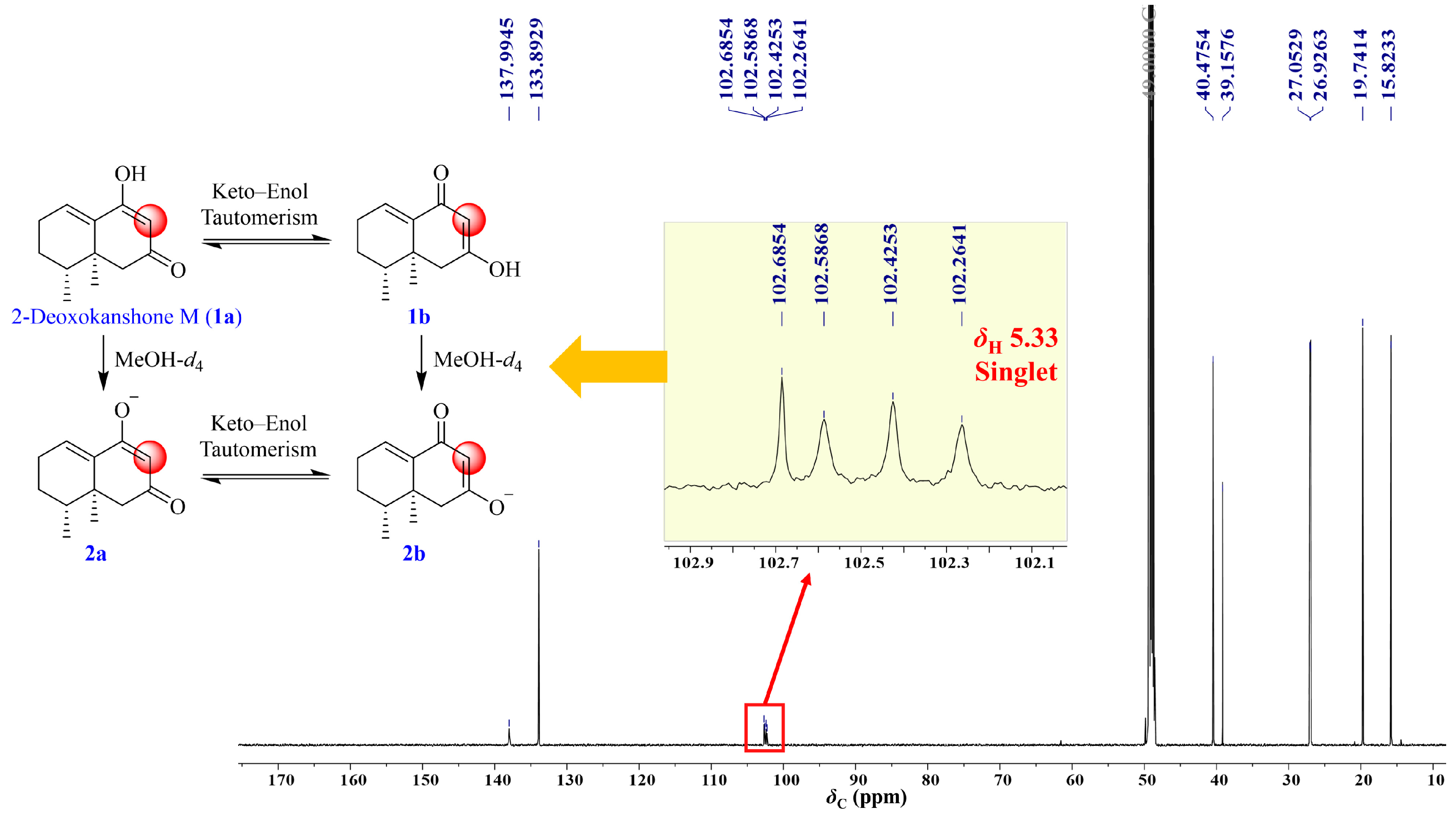
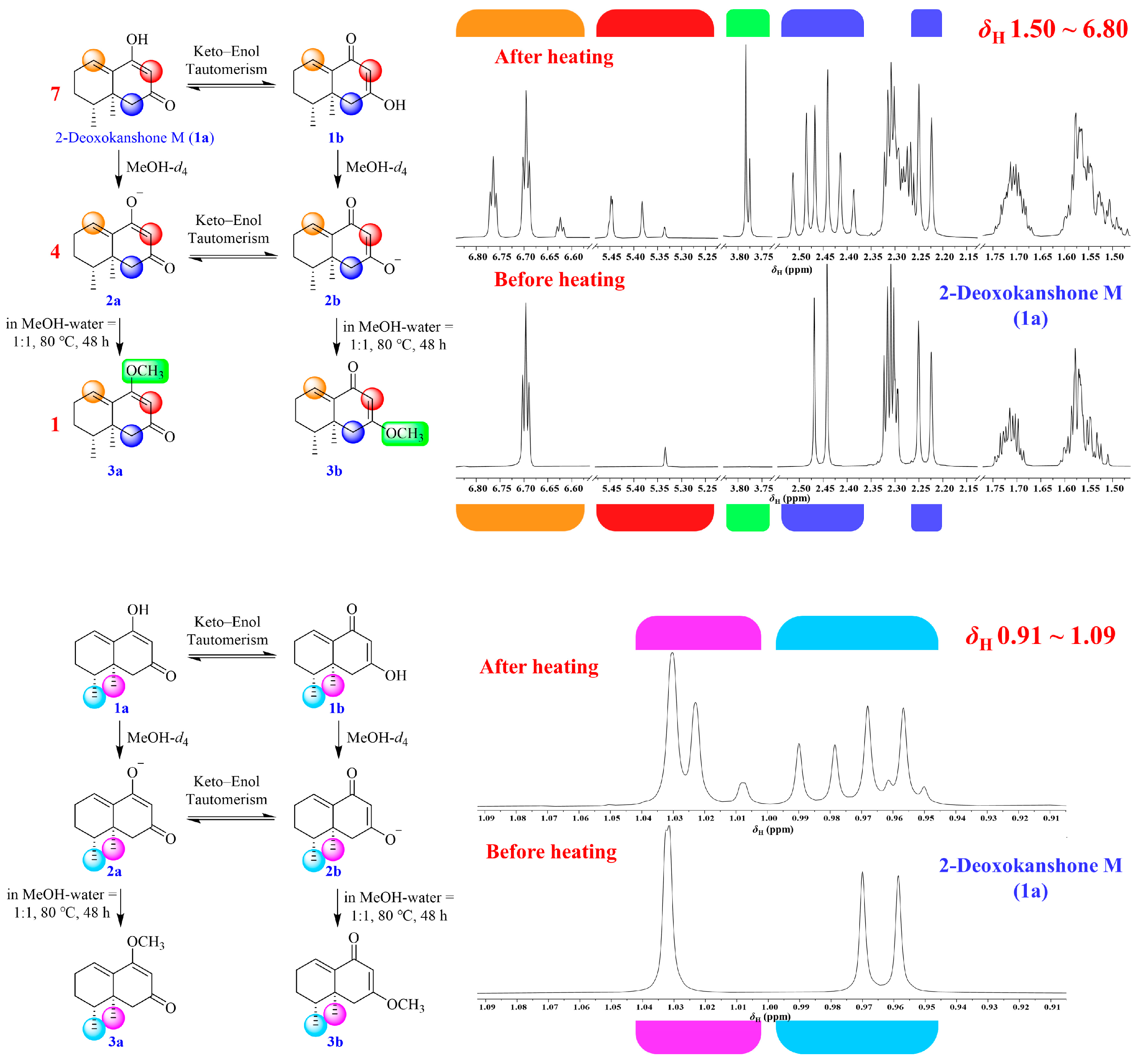
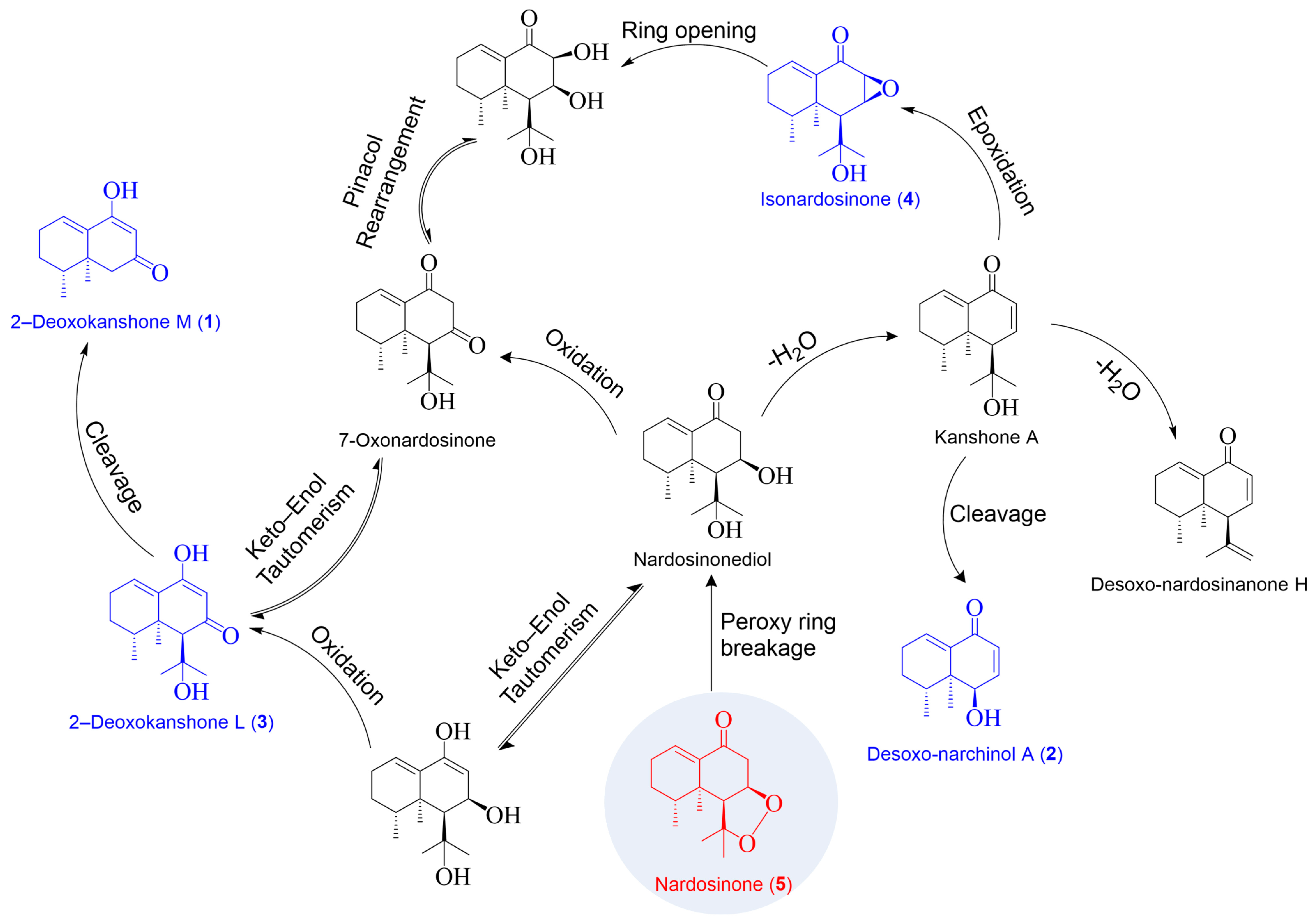
2.1. Isolation and Structural Elucidation of the Main Degradation Products of Nardosinone
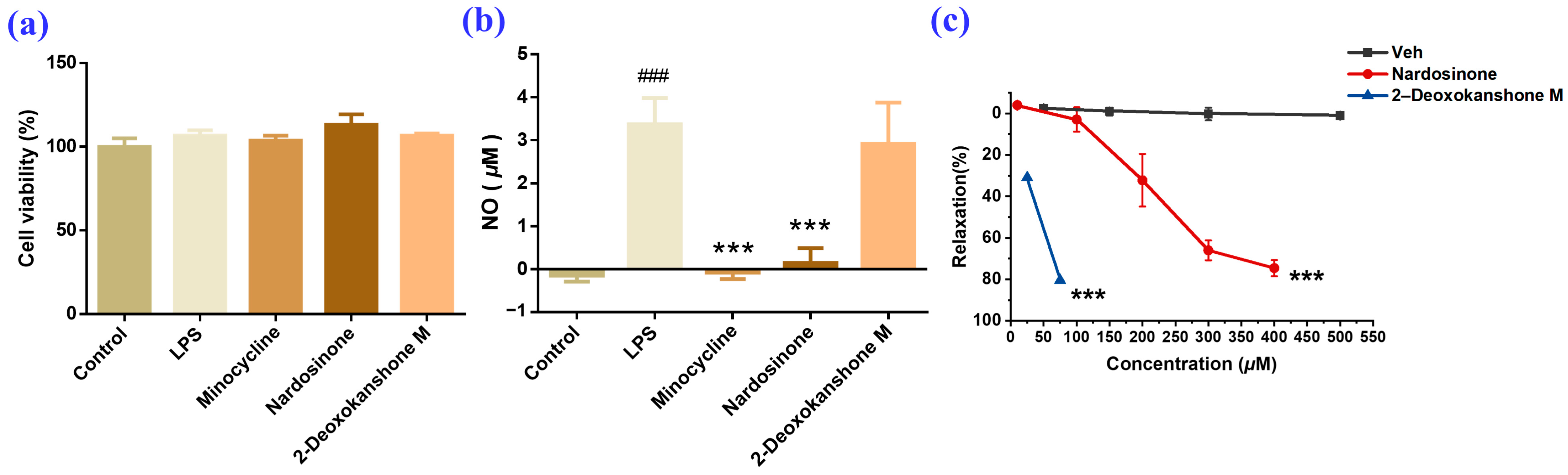
2.2. Evaluation of the Vasodilatory and Anti-Neuroinflammatory Activities of Nardosinone and the Main Degradation Products
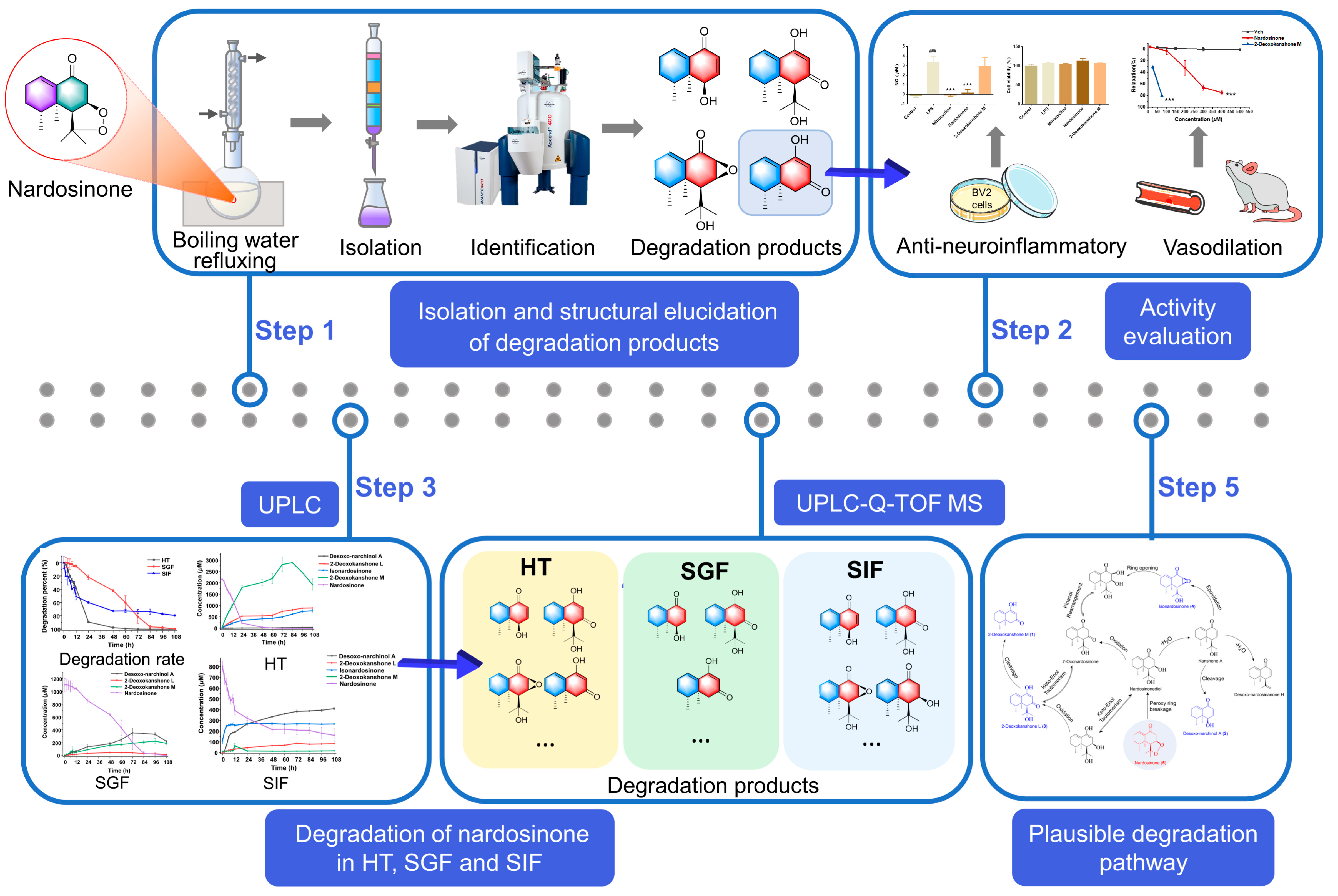
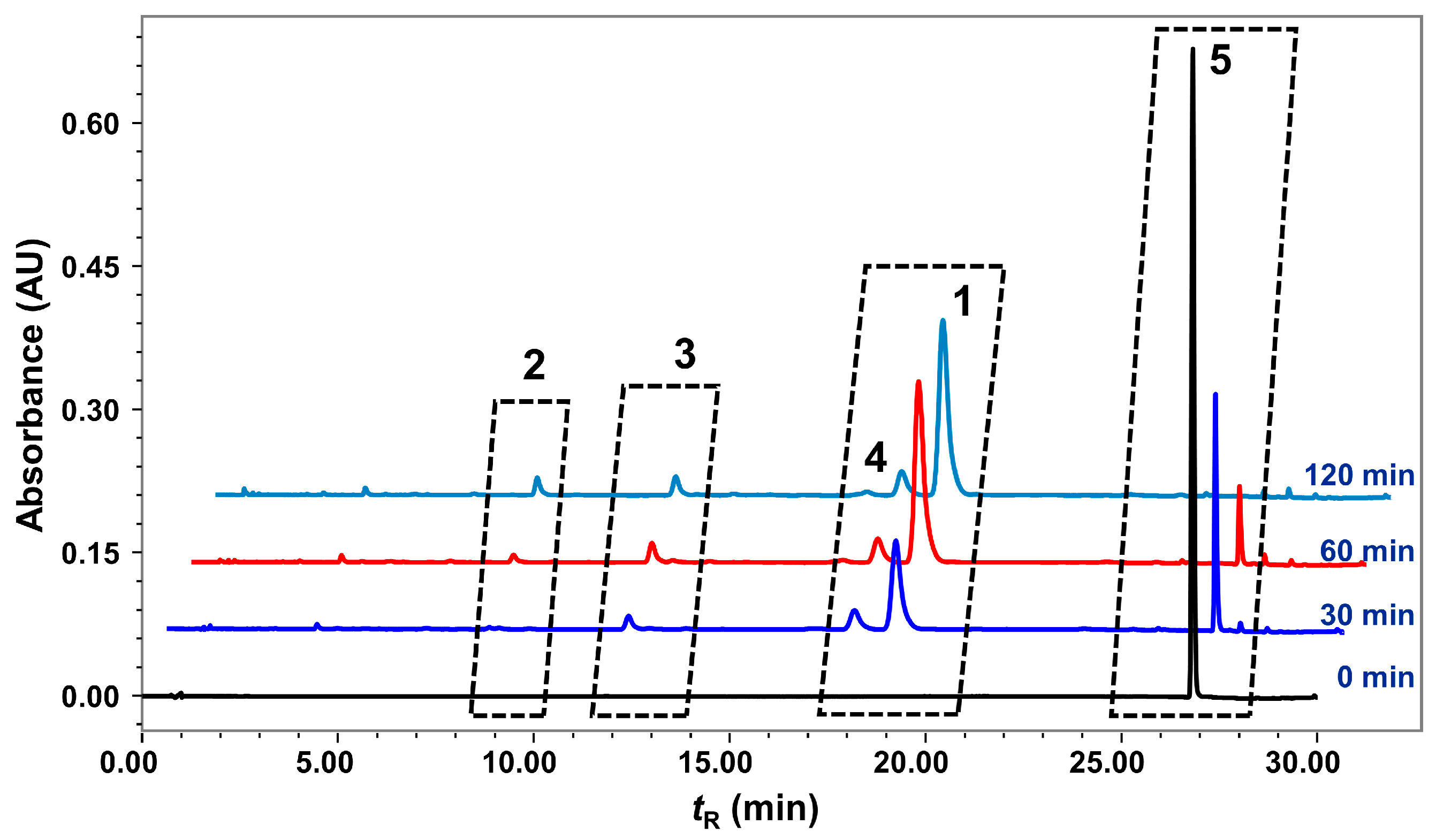
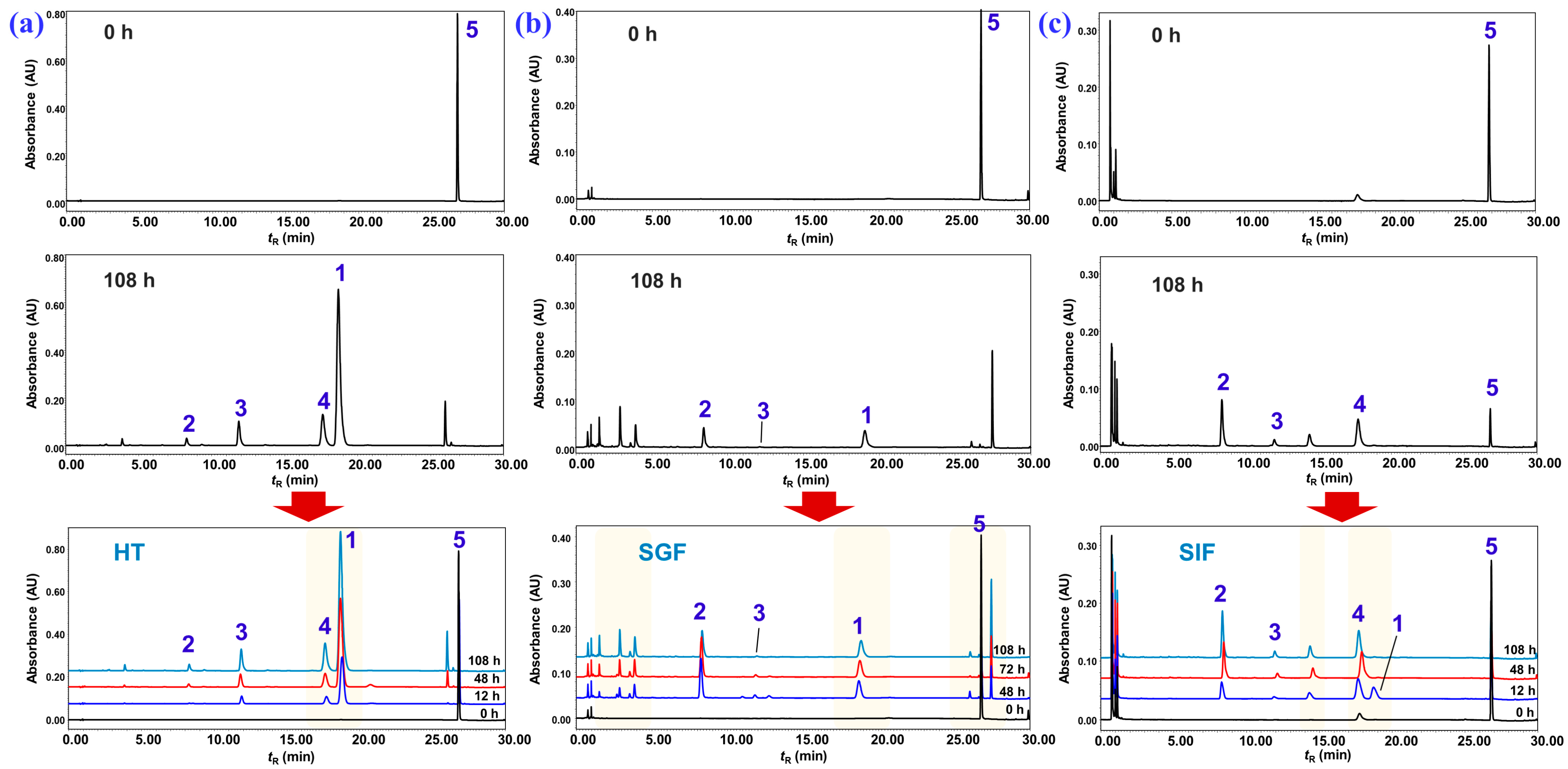
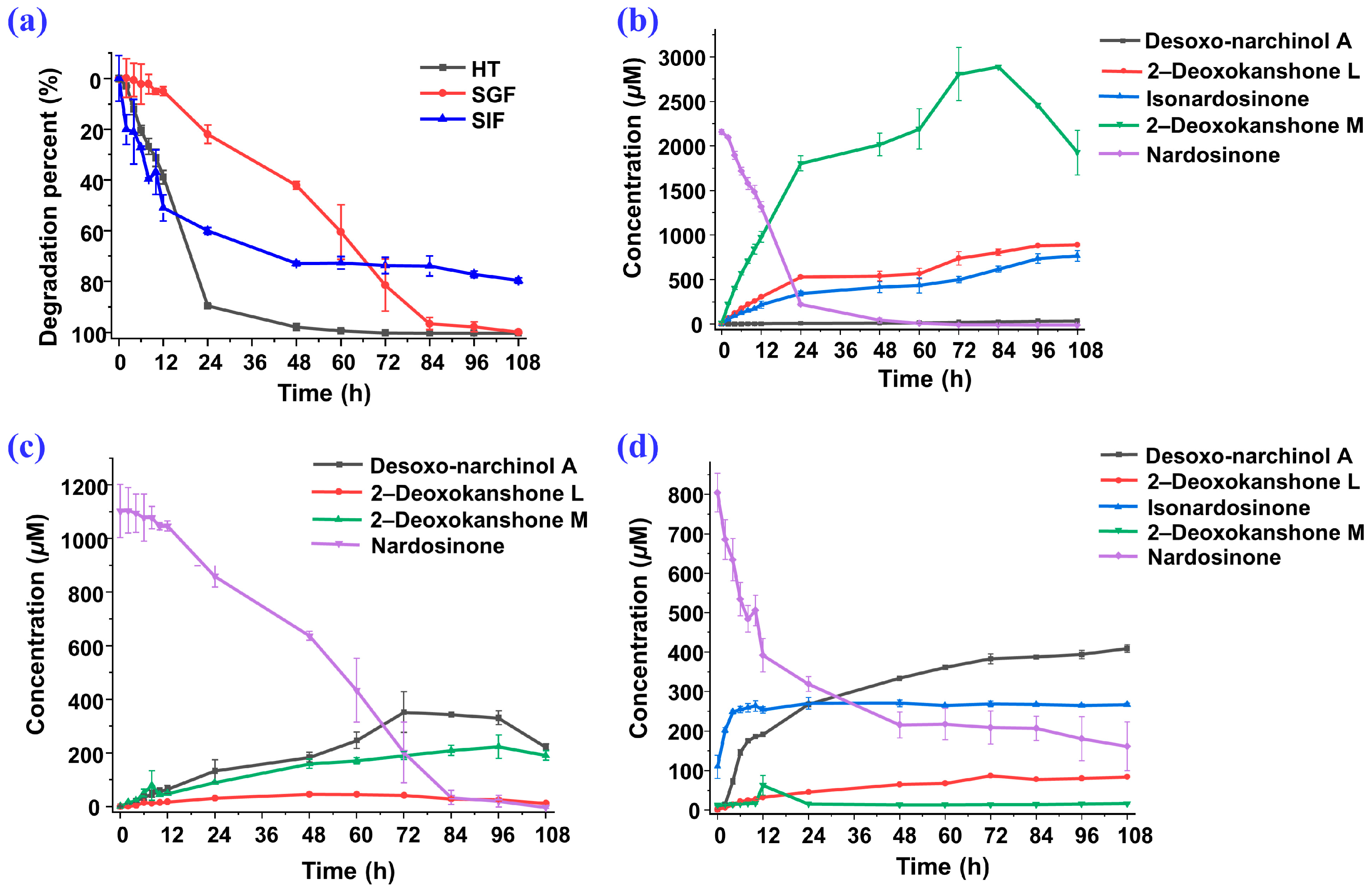
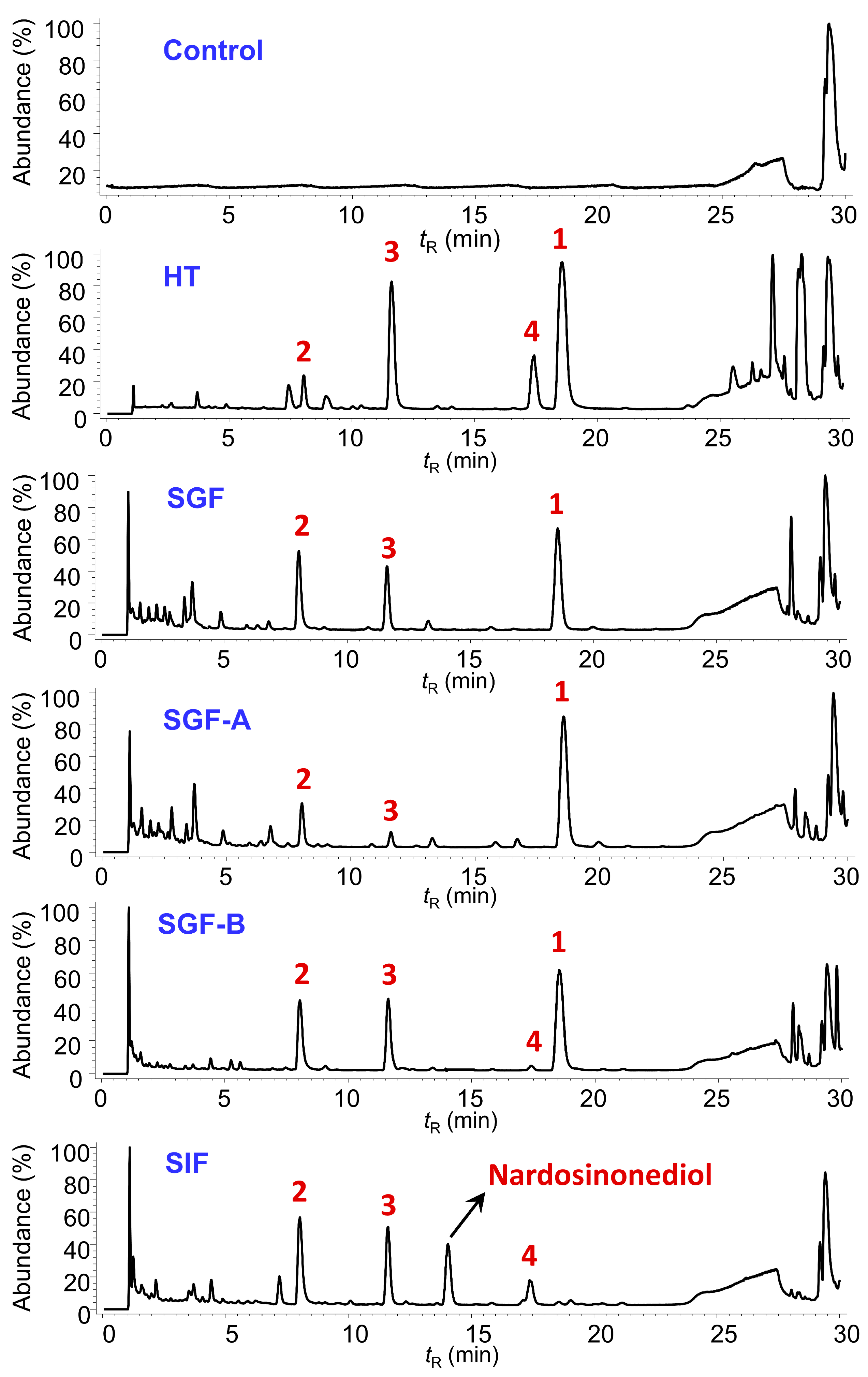
2.3. Degradation of Nardosinone in Different Conditions
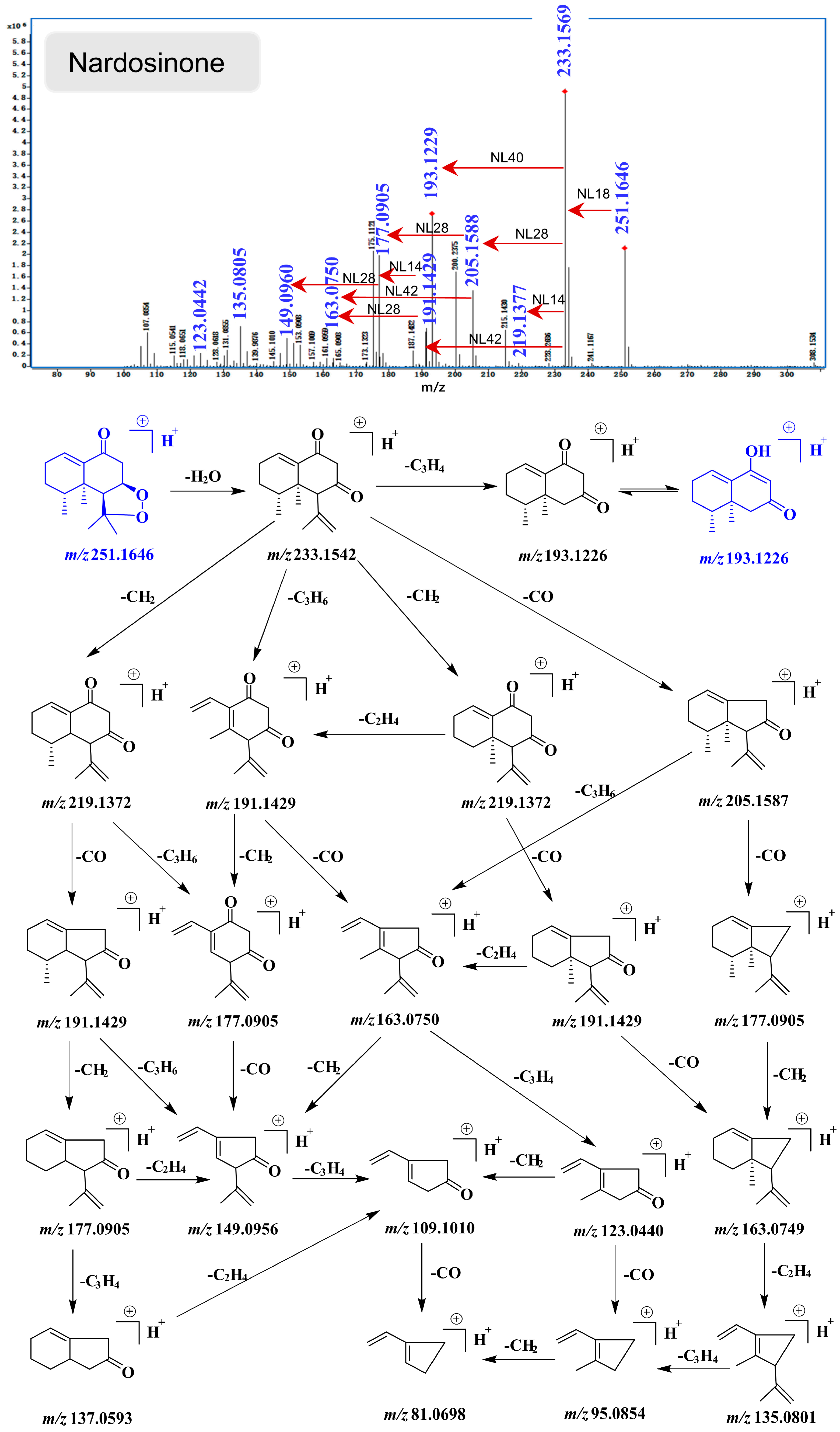
2.4. UHPLC-DAD/Q-TOF MS Analysis and Proposed Degradation Pathway of Nardosinone
3. Materials and Methods
3.1. General Experimental Procedures, Reagents, and Materials
3.2. NMR Spectroscopy
3.3. Isolation and Identification of the Main Degradation Products of Nardosinone (5)
3.4. Evaluation of the Vasodilatory Activity
3.5. Evaluation of the Anti-Neuroinflammatory Activity
3.6. Quantitative UPLC–PDA Analysis
3.7. UHPLC–DAD/Q–TOF MS Analysis
3.8. Preparation of Simulated Gastrointestinal Fluids
3.9. Incubations of Nardosinone at High-Temperature Condition and in Simulated Gastrointestinal Fluids
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| 1H–1H COSY | 1H–1H correlation spectroscopy |
| CCK8 | Cell Counting Kit–8 |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | Dimethyl sulfoxide |
| ECD | Electronic circular dichroism |
| FBS | Fetal bovine serum |
| HMBC | Heteronuclear multiple bonding correlation |
| HRESI–MS | High-resolution electrospray ionization mass spectrometry |
| HSQC | Heteronuclear single-quantum correlation |
| HT | High temperature |
| IC50 | Half-maximal inhibitory concentration |
| IR | Infrared spectroscopy |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| LPS | Lipopolysaccharide |
| NMR | Nuclear magnetic resonance |
| NO | Nitric oxide |
| SEM | Standard errors of the mean |
| SGF | Simulated gastric fluid |
| SGF–A | Simulated gastric fluid without HCl |
| SGF–B | Simulated gastric fluid without pepsin |
| SIF | Simulated intestinal fluid |
| S/N | Signal–noise ratio |
| TDDFT | Time-dependent density functional theory |
| TLC | Thin-layer chromatography |
| UHPLC–DAD–Q–TOF MS | Ultra high-performance liquid chromatography-diode array detector-quadrupole time-of-flight mass spectrometry |
| UPLC–PDA | Ultra performance liquid chromatography-photo-diode array |
| UV | Ultraviolet absorption spectrum |
| U46619 | 9,11-Methanoepoxy PGH2 |
References
- Rehman, T.; Ahmad, S. Nardostachys chinensis Batalin: A review of traditional uses, phytochemistry, and pharmacology. Phytother. Res. 2019, 33, 2622–2648. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, T.T.; Rao, Y.; Wang, Z.M.; Dong, X.Q.; Zhang, L.H.; Han, L.F.; Zhang, Y.; Wang, T.; Zhu, Y.; et al. A review on traditional uses, phytochemistry, pharmacology, toxicology and the analytical methods of the genus Nardostachys. J. Ethnopharmacol. 2021, 280, 114446. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Liu, H.T.; Huang, Q.; Zhu, L.; Lei, P. Determination of the content of nardosinone in Nardostachys chinensis Batal. Chin. J. Tradit. Chin. Med. Pharm. 2015, 30, 249–251. [Google Scholar]
- Zhang, W.Z.; Nan, G.; Wu, H.H.; Jiang, M.M.; Li, T.X.; Wang, M.; Gao, X.M.; Zhu, Y.; Song, Y.S.; Wang, J.M.; et al. A simple and rapid UPLC-PDA method for quality control of Nardostachys jatamansi. Planta Med. 2018, 84, 536–543. [Google Scholar] [CrossRef]
- Li, Y.M.; Liu, G.L.; Qiao, J.; Liu, S.; Zhang, Y.; Qin, Z.X.; Liu, Y. Simultaneous determination of chlorogenic acid and nardosinone in Nardostachys chinensis DC. from different producing areas by HPLC. J. Inf. Tradit. Chin. Med. 2015, 32, 27–30. [Google Scholar]
- China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China. Part I; China Medical Science and Technology Press: Beijing, China, 2020; pp. 87–88.
- Hwang, J.S.; Lee, S.A.; Hong, S.S.; Han, X.H.; Lee, C.; Lee, D.; Lee, C.; Hong, J.T.; Kim, Y.; Lee, M.K.; et al. Inhibitory constituents of Nardostachys chinensis on nitric oxide production in RAW 264.7 macrophages. Bioorg. Med. Chem. Lett. 2012, 22, 706–708. [Google Scholar] [CrossRef]
- Chen, Y.P.; Ying, S.S.; Zheng, H.H.; Liu, Y.T.; Wang, Z.P.; Zhang, H.; Deng, X.; Wu, Y.J.; Gao, X.M.; Li, T.X.; et al. Novel serotonin transporter regulators: Natural aristolane- and nardosinane- types of sesquiterpenoids from Nardostachys chinensis Batal. Sci. Rep. 2017, 7, 15114. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhao, S.H.; Mei, X.Y.; Chen, Z.; Li, X.G.; Sun, F.Y. The effect of nardosinone on hypoxic injury of H9C2 cardiomyocytes and its mechanism. J. S. China Norm. Univ. 2021, 53, 51–58. [Google Scholar]
- Ko, W.; Park, J.S.; Kim, K.W.; Kim, J.; Kim, Y.C.; Oh, H. Nardosinone-type sesquiterpenes from the hexane fraction of Nardostachys jatamansi attenuate NF-κB and MAPK signaling pathways in lipopolysaccharide-stimulated BV2 microglial cells. Inflammation 2018, 41, 1215–1228. [Google Scholar] [CrossRef]
- Jian, P.; Li, Q.H.; Fan, L.H. Experimental study on the inhibitory effect of nardosinone on myocardial cell in rats with tachyarrhythmia. Chin. J. Clin. Pharmacol. 2015, 31, 2240–2242. [Google Scholar]
- Li, W.; Shi, J.L.; Li, Q.; Duan, H.H.; Tang, M.K. Nardosinone reduces neuronal injury induced by oxygen-glucose deprivation in primary cortical cultures. Acta Pharm. Sin. 2013, 48, 1422–1429. [Google Scholar]
- Li, Z.H.; Li, W.; Shi, J.L.; Tang, M.K. Nardosinone improves the proliferation, migration and selective differentiation of mouse embryonic neural stem cells. PLoS ONE 2014, 9, e91260. [Google Scholar] [CrossRef]
- Niu, C.G.; Xiao, F.; Yuan, K.Y.; Hu, X.C.; Lin, W.Z.; Ma, R.; Zhang, X.L.; Huang, Z.W. Nardosinone suppresses RANKL-induced osteoclastogenesis and attenuates lipopolysaccharide-induced alveolar bone resorption. Front. Pharmacol. 2017, 8, 626. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.W.; Liu, L.Q.; Li, J.J.; He, Y. A review of nardosinone for pharmacological activities. Eur. J. Pharmacol. 2021, 90, 174343. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.H.; Sun, Y.; Liu, S.; Li, C.Y.; Chen, S.Q.; Qiu, R.J.; Zhang, X.Y.; Li, Y.P.; Li, M.; Shang, H.C. Therapeutic effects of Wenxin Keli in cardiovascular diseases: An experimental and mechanism overview. Front. Pharmacol. 2018, 9, 1005. [Google Scholar] [CrossRef]
- Cao, X.F.; Zhou, M.X.; Liu, H.X.; Chen, X.F.; Li, X.; Jia, S.H. Clinical efficacy and safety of Shensong Yangxin Capsule-amiodarone combination on heart failure complicated by ventricular arrhythmia: A meta-analysis of randomized controlled trials. Front. Pharmacol. 2021, 12, 613922. [Google Scholar] [CrossRef]
- Lu, X.L.; Ma, Q.D.; Wang, X.Y.; Tian, S.G. Songbuli Oral Solution quality standards. Glob. Tradit. Chin. Med. 2013, 6, 577–580. [Google Scholar]
- Riicker, G.; Olbrich, A. Darstellung von nardosinon aus 1(10)-aristolenon-(9) (gansongon). Tetrahedron Lett. 1988, 29, 4703–4704. [Google Scholar] [CrossRef]
- Rwker, G.; Dyck, E. Bildung von “desoxo-narchinol A” aus nardosinon. Phytochemistry 1974, 13, 1907–1909. [Google Scholar] [CrossRef]
- Liu, G.L.; Liu, Y.; Shi, J.L.; Fang, N.; Li, J.; Lv, Y.W.; Wu, M.X. Study on stability of nardosinone. Chin. J. Pharm. Anal. 2015, 35, 360–363. [Google Scholar]
- Lu, Z.H.; Zhou, P.; Zhan, Y.Z.; Su, J.R.; Yi, D.L. Quantification of nardosinone in rat plasma using liquid chromatography-tandem mass spectrometry and its pharmacokinetics application. J. Chromatogr. Sci. 2015, 53, 1725–1729. [Google Scholar] [CrossRef]
- Zhang, J.; Lv, Y.; Zhang, J.; Bai, Y.S.; Li, M.Y.; Wang, S.Q.; Wang, L.L.; Liu, G.X.; Xu, F.; Sha, M.Y.; et al. Analysis of in vivo existence forms of nardosinone in mice by UHPLC-Q-TOF-MS technique. Molecules 2022, 27, 7267. [Google Scholar] [CrossRef]
- Deng, X.; Wang, Y.; Wu, H.H.; Zhang, W.Z.; Dong, X.Q.; Wang, Z.M.; Zhu, Y.; Gao, X.M.; Li, L.; Wang, Y.N.; et al. Six kanshone C-derived sesquiterpenoid hybrids nardochalaristolones A–D, nardoflavaristolone A and dinardokanshone F from Nardostachys jatamansi DC. Bioorg. Chem. 2018, 81, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, H.; Masuyama, K.; Morita, H.; Takeya, K. Cytotoxic sesquiterpenes from Nardostachys chinensis. Chem. Pharm. Bull. 1993, 41, 1183–1184. [Google Scholar] [CrossRef]
- Yoon, C.S.; Kim, D.C.; Park, J.S.; Kim, K.W.; Kim, Y.C.; Oh, H. Isolation of novel sesquiterpeniods and anti-neuroinflammatory metabolites from Nardostachys jatamansi. Molecules 2018, 23, 2367. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.M.; Li, R.; Zhang, Y.; Kwabena Oduro, P.; Li, S.; Leng, L.; Wang, Z.M.; Rao, Y.; Niu, L.; Wu, H.H.; et al. Aristolone in Nardostachys jatamansi DC. induces mesenteric vasodilation and ameliorates hypertension via activation of the KATP channel and PDK1-Akt-eNOS pathway. Phytomedicine 2022, 104, 154257. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.X.; Wang, Z.; Wang, Y.H.; Xu, J.W.; He, X.J. Anti-neuroinflammatory triterpenoids from the seeds of Quercus serrata Thunb. Fitoterapia 2020, 142, 104523. [Google Scholar] [CrossRef]
- Yoon, C.S.; Kim, K.W.; Lee, S.C.; Kim, Y.C.; Oh, H. Anti-neuroinflammatory effects of sesquiterpenoids isolated from Nardostachys jatamansi. Bioorg. Med. Chem. Lett. 2018, 28, 140–144. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Li, S.W.; Xue, B.X.; Yang, T.T.; Li, R.; Zhang, M.J.; Wang, M.; Zhang, L.H.; Zhang, P.; Zhang, Y.; Wang, T.; et al. Sesquiterpenoids and monoterpenoids from the water decoction of Valeriana officinalis L. Phytochemistry 2023, 205, 113474. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.J.; Oppong, M.B.; Di, J.R.; Yuan, Q.; Chang, Y.X.; Jiang, M.M.; Cao, S.J.; Dong, P.Z.; Li, L.; Xie, Y.; et al. Steroidal saponins with anti-inflammatory activity from Tribulus terrestris L. AHM 2022, 2, 41–48. [Google Scholar] [CrossRef]
- China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China. Part IV; China Medical Science and Technology Press: Beijing, China, 2020; p. 130.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH/ppm (J in Hz) a | δC, Type b | HMBC | 1H–1H COSY |
|---|---|---|---|---|
| 1 | 6.49 (1H, t, 4.0) | 130.6, CH | C–3, 5, 9, 10 | H–2 |
| 2 | 2.20 (2H, m) | 25.6, CH2 | C–1, 3, 4, 9, 10 | H–1, H–3 |
| 3 | 1.46 (2H, m) | 25.3, CH2 | C–1, 2, 5, 11 | H–2, H–4 |
| 4 | 1.62 (1H, m) | 38.5, CH | C–3, 5, 6, 11, 12 | H–3, H–11 |
| 5 | – | 37.4, C | – | – |
| 6 | 2.28 (2H, m) | 49.2, CH2 | C–5, 7, 8, 10, 12 | – |
| 7 | – | 196.3, C | – | – |
| 8 | 5.23 (1H, s) | 102.0, CH | C–6, 7, 9, 10 | – |
| 9 | – | 167.7, C | – | – |
| 10 | – | 136.1, C | – | – |
| 11 | 0.87 (3H, d, 6.8) | 15.4, CH3 | C–3, 5 | H–4 |
| 12 | 0.92 (3H, s) | 19.4, CH3 | C–4, 6, 10 | – |
| -OH | 10.83 (1H, s) | – | – | – |
| Position | δH (J in Hz) a | δC, Type b | HMBC | 1H–1H COSY |
|---|---|---|---|---|
| 1 | 6.53 (1H, br s) | 130.4, CH | C–3 | H–2 |
| 2 | 2.20 (2H, m) | 25.5, CH2 | C–3, 4 | H–1, H–3 |
| 3 | 1.45 (2H, m) | 25.8, CH2 | C–1, 2, 4, 5, 14 | H–2, H–4 |
| 4 | 2.68 (1H, m) | 32.1, CH | C–2, 5, 15 | H–3, H–14 |
| 5 | – | 40.1, C | – | – |
| 6 | 2.30 (1H, s) | 65.5, CH | C–5, 8, 10, 11, 13, 15 | – |
| 7 | – | 199.0, C | – | – |
| 8 | 5.16 (1H, s) | 101.7, CH | C–6, 9, 10 | – |
| 9 | – | 167.0, C | – | – |
| 10 | – | 136.8, C | – | – |
| 11 | – | 71.7, C | – | – |
| 12 | 0.98 (3H, s) | 32.9, CH3 | C–6, 11, 13 | – |
| 13 | 1.10 (3H, s) | 25.2, CH3 | C–6, 11, 12 | – |
| 14 | 0.89 (3H, d, 7.8) | 16.7, CH3 | C–3, 4, 5 | H–4 |
| 15 | 0.90 (3H, s) | 22.9, CH3 | C–4, 5, 6, 10 | – |
| Compound | Regression Equation | r2 | Linear Range (μg/mL) | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|---|
| Desoxo-narchinol A | y = 7310.7x + 2169.9 | 0.9999 | 0.9766–62.5 | 0.216 | 0.720 |
| 2–Deoxokanshone L | y = 5960.1x + 1819.5 | 1 | 0.9766–250 | 0.208 | 0.693 |
| Isonardosinone | y = 12,564x – 20,490 | 0.9996 | 1.9531–250 | 0.403 | 1.342 |
| 2–Deoxokanshone M | y = 20,365x – 47,345 | 0.9999 | 0.9766–1000 | 0.264 | 0.880 |
| Nardosinone | y = 5926.7x + 20,602 | 0.9999 | 0.9766–1000 | 0.244 | 0.813 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, B.-X.; Yang, T.-T.; He, R.-S.; Gao, W.-K.; Lai, J.-X.; Liu, S.-X.; Duan, C.-Y.; Wang, S.-X.; Yu, H.-J.; Yang, W.-Z.; et al. Degradation Profiling of Nardosinone at High Temperature and in Simulated Gastric and Intestinal Fluids. Molecules 2023, 28, 5382. https://doi.org/10.3390/molecules28145382
Xue B-X, Yang T-T, He R-S, Gao W-K, Lai J-X, Liu S-X, Duan C-Y, Wang S-X, Yu H-J, Yang W-Z, et al. Degradation Profiling of Nardosinone at High Temperature and in Simulated Gastric and Intestinal Fluids. Molecules. 2023; 28(14):5382. https://doi.org/10.3390/molecules28145382
Chicago/Turabian StyleXue, Bian-Xia, Tian-Tian Yang, Ru-Shang He, Wen-Ke Gao, Jia-Xin Lai, Si-Xia Liu, Cong-Yan Duan, Shao-Xia Wang, Hui-Juan Yu, Wen-Zhi Yang, and et al. 2023. "Degradation Profiling of Nardosinone at High Temperature and in Simulated Gastric and Intestinal Fluids" Molecules 28, no. 14: 5382. https://doi.org/10.3390/molecules28145382
APA StyleXue, B.-X., Yang, T.-T., He, R.-S., Gao, W.-K., Lai, J.-X., Liu, S.-X., Duan, C.-Y., Wang, S.-X., Yu, H.-J., Yang, W.-Z., Zhang, L.-H., Wang, Q.-L., & Wu, H.-H. (2023). Degradation Profiling of Nardosinone at High Temperature and in Simulated Gastric and Intestinal Fluids. Molecules, 28(14), 5382. https://doi.org/10.3390/molecules28145382