
Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate


Abstract
:1. Introduction
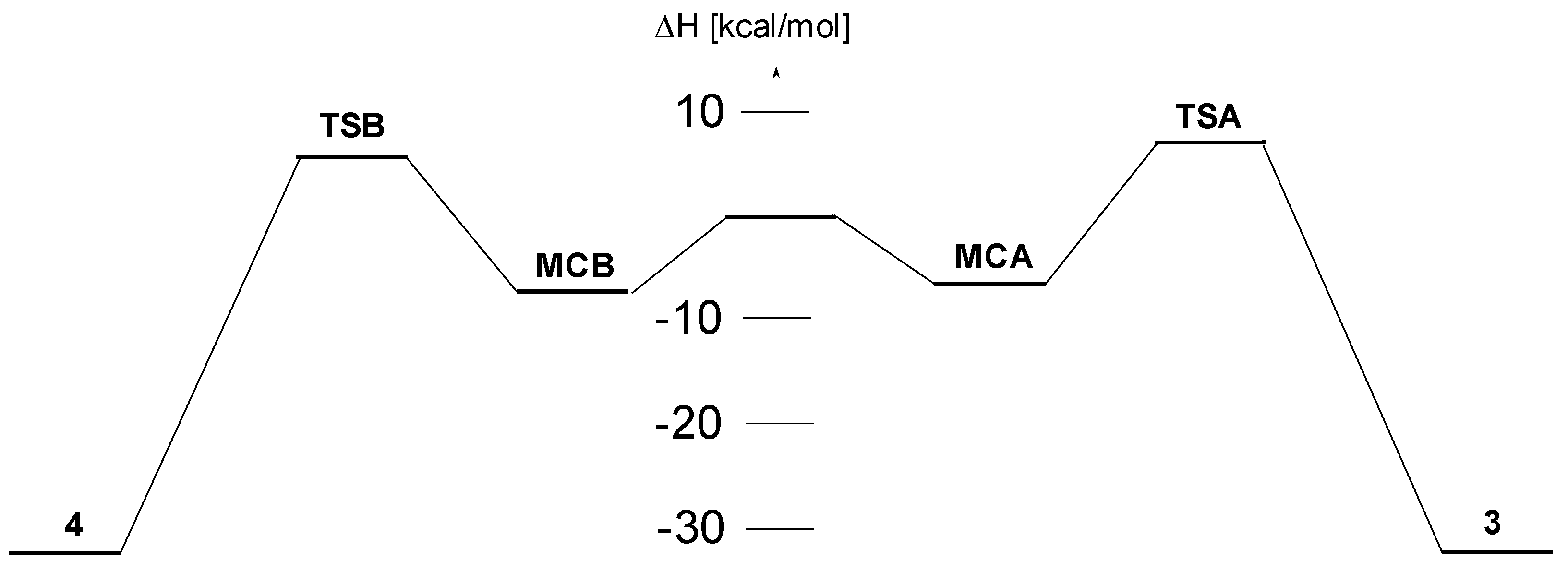
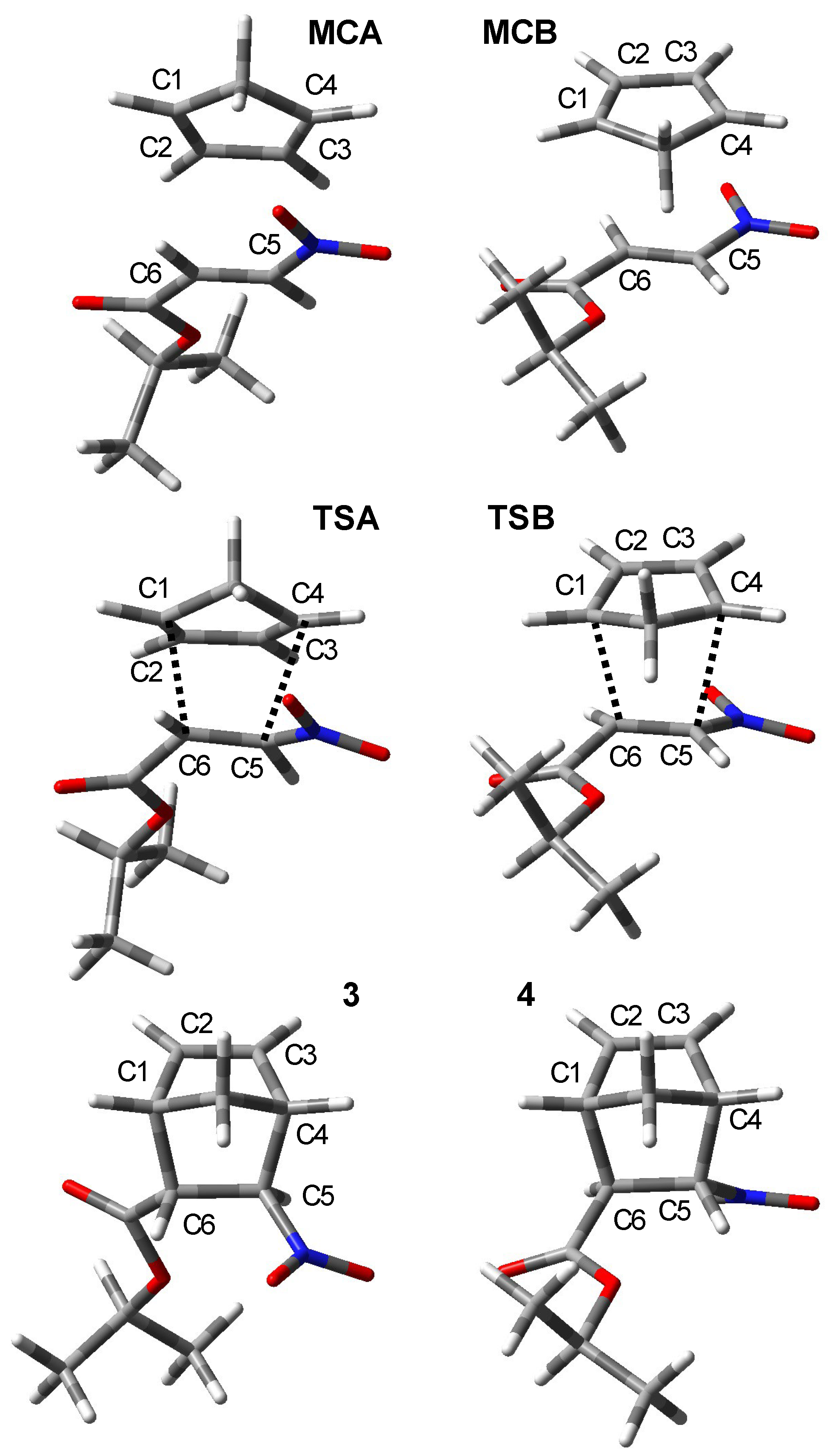
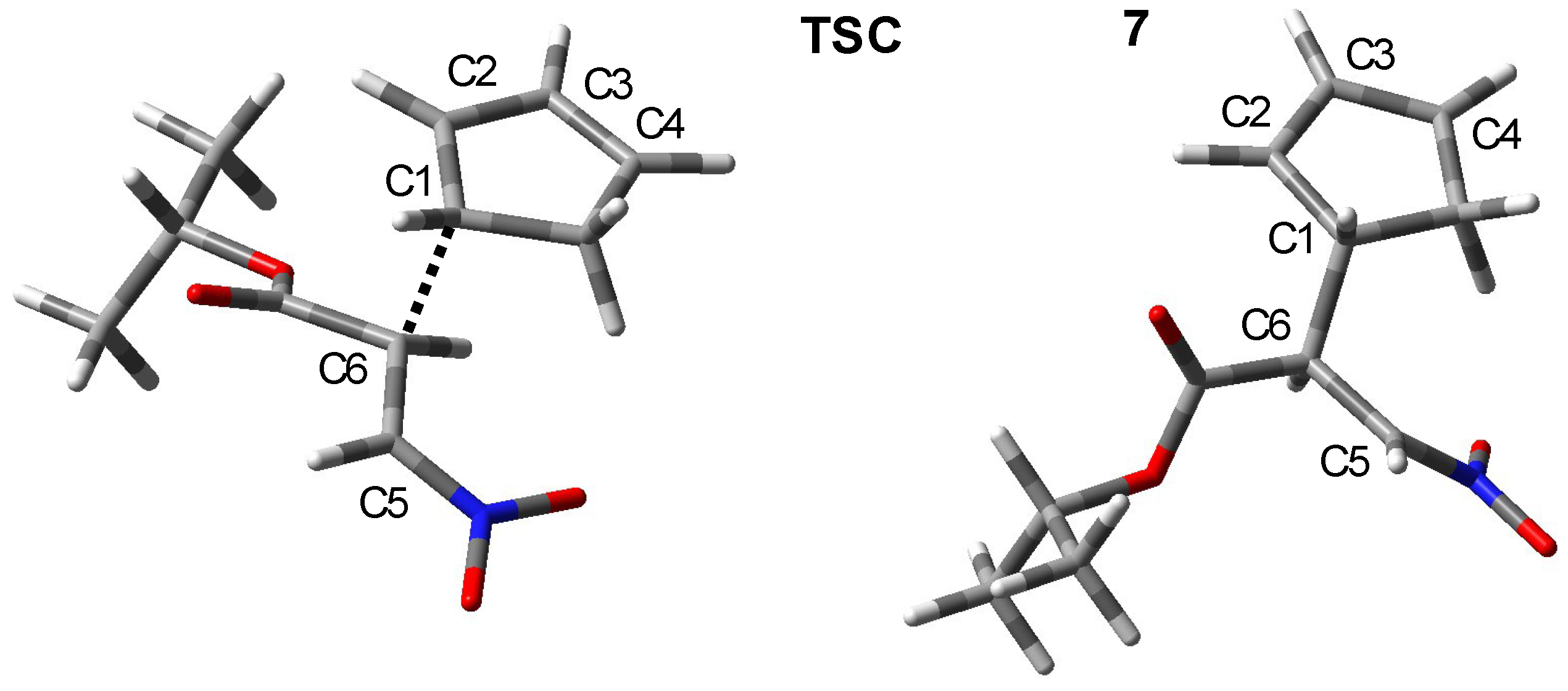
2. Results and Discussion
3. Computational Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels—Alder Reaction in Total Synthesis. Angew. Chem. 2002, 41, 1668–1698. [Google Scholar]
- Łapczuk-Krygier, A.; Kącka-Zych, A.; Kula, K. Recent progress in the field of cycloaddition reactions involving conjugated nitroalkenes. Curr. Chem. Lett. 2019, 8, 13–38. [Google Scholar]
- Grozav, A.N.; Kemskiy, S.V.; Fedoriv, M.Z.; Chornous, V.A.; Palamar, A.A.; Dorokhov, V.I.; Rusanov, E.B.; Vovk, M.V. Synthesis, hydrolysis, and reductive cyclization of ethyl 5-chloro-4-(4-nitropyrrolidin-3-yl)pyrrole-3-carboxylates. Chem. Heterocycl. Comp. 2022, 58, 24–31. [Google Scholar]
- Kącka-Zych, A.; Jasiński, R. Understanding the molecular mechanism of the stereoselective conversion of N-trialkylsilyloxy nitronates into bicyclic isoxazoline derivatives. New J. Chem. 2021, 45, 9491–9500. [Google Scholar]
- Inamoto, Y.; Nishimoto, Y.; Yasuda, M.; Baba, A. Direct Use of Esters in the Mukaiyama Aldol Reaction: A Powerful and Convenient Alternative to Aldehydes. Org. Lett. 2012, 14, 1168–1171. [Google Scholar]
- Lee, S.H.; Park, Y.J.; Yoon, C.M. Catalytic transfer hydrogenation of conjugated nitroalkenes using decaborane: Synthesis of oximes. Org. Biomol. Chem. 2003, 1, 1099–1100. [Google Scholar]
- Kącka-Zych, A.; Jasiński, R. Mechanistic aspects of the synthesis of seven-membered internal nitronates via stepwise [4 + 3] cycloaddition involving conjugated nitroalkenes: Molecular Electron Density Theory computational study. J. Comput. Chem. 2022, 43, 1221–1228. [Google Scholar]
- Kącka-Zych, A. Understanding the uniqueness of the stepwise [4 + 1] cycloaddition reaction between conjugated nitroalkenes and electrophilic carbene systems with a molecular electron density theory perspective. Int. J. Quantum Chem. 2021, 121, e26440. [Google Scholar]
- Jasiński, R.; Kula, K.; Kącka, A.; Mirosław, B. Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: Why is there no 1,3-dipolar cycloaddition? Monatsh. Chem. 2017, 148, 909–915. [Google Scholar]
- Bykova, L.S.; Kochnev, I.A.; Barkov, A.Y.; Zimnitskiy, N.S.; Korotaev, V.Y.; Sosnovskikh, V.Y. An AgOAc-catalyzed reaction of 3-nitro-2H-chromenes with ethyl diazoacetate: An efficient one-pot synthesis of ethyl 3,4-dihydrochromeno [3,4-c]pyrazole-1-carboxylates. Chem. Heterocycl. Comp. 2022, 58, 646–650. [Google Scholar]
- Zawadzińska, K.; Gostyński, B. Nitrosubstituted analogs of isoxazolines and isoxazolidines: A surprising estimation of their biological activity via molecular docking. Sci. Rad. 2023, 2, 25–46. [Google Scholar]
- Fryźlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O.M.; Dresler, E.; Jasiński, R. Regio—And stereoselective synthesis of nitrofunctionalized 1,2-oxazolidine analogs of nicotine. Chem. Heterocycl. Comp. 2020, 56, 120–122. [Google Scholar]
- Obernilhina, N.N.; Kachaeva, M.V.; Kachkovsky, O.D.; Brovarets, V.S. In silico study of conjugated nitrogen heterocycles affinity in their biological complexes. Chem. Heterocycl. Comp. 2022, 58, 412–420. [Google Scholar]
- Bonner, T.G.; Hancock, R.A.; Rolle, F.R.; Yousif, G. Nitration of aromatic hydrocarbons by anhydrous nitric acid in carbon tetrachloride. J. Chem. Soc. B. 1970, 314–318. [Google Scholar]
- Kącka, A.; Dresler, E.; Jasiński, R. New implementations of Meyer process, Nowe realizacje procesu Meyera. Przem. Chem. 2016, 11, 204–207. [Google Scholar]
- Jasiński, R. β-Trifluoromethylated nitroethenes in Diels-Alder reaction with cyclopentadiene: A DFT computational study. J. Fluor. Chem. 2018, 206, 1–7. [Google Scholar]
- Jasiński, R. One-step versus two-step mechanism of Diels-Alder reaction of 1-chloro-1-nitroethene with cyclopentadiene and furan. J. Mol. Graph. Model. 2017, 75, 55–61. [Google Scholar]
- Jasiński, R. First example of stepwise, zwitterionic mechanism for bicyclo [2.2.1]hept-5-ene (norbornene) formation process catalyzed by the 1-butyl-3-methylimidazolium cations. Monatsh. Chem. 2016, 147, 1207–1213. [Google Scholar]
- Łapczuk-Krygier, A.; Ponikiewski, Ł.; Jasiński, R. The crystal structure of (1RS,4RS,5RS,6SR)-5-cyano-5-nitro-6-phenyl-bicyclo [2.2.1]hept-2-ene. Crystallogr. Rep. 2014, 59, 961–963. [Google Scholar]
- Mukherjee, S.; Corey, E.J. [4 + 2] Cycloaddition reactions catalyzed by a chiral oxazaborolidinium cation. Reaction rates and diastereo-, regio-, and enantioselectivity depend on whether both bonds are formed simultaneously. Org. Lett. 2010, 12, 1024–1027. [Google Scholar]
- Sadowski, M.; Utnicka, J.; Wójtowicz, A.; Kula, K. The global and local Reactivity of C,N-diarylnitryle imines in [3 + 2] cycloaddition processes with trans-β-nitrostyrene according to Molecular Electron Density Theory: A computational study. Curr. Chem. Lett. 2023, 12, 421–430. [Google Scholar]
- Kula, K.; Zawadzińska, K. Local nucleophile-electrophile interactions in [3 + 2] cycloaddition reactions between benzonitrile N-oxide and selected conjugated nitroalkenes in the light of MEDT computational study. Curr. Chem. Lett. 2021, 10, 9–16. [Google Scholar]
- Jasiński, R.; Żmigrodzka, M.; Dresler, E.; Kula, K. A full regio- and stereoselective synthesis of 4-nitroisoxazolidines via stepwise [3 + 2] cycloaddition reactions between (Z)-C-(9-anthryl)-N-arylnitrones and (E)-3,3,3-trichloro-1-nitroprop-1-ene: Comprehensive experimental and theoretical study. J. Heterocyclic. Chem. 2017, 54, 3314–3320. [Google Scholar]
- Jasiński, R. On the Question of Stepwise [4 + 2] Cycloaddition Reactions and Their Stereochemical Aspects. Symmetry 2021, 13, 1911. [Google Scholar]
- Jasiński, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3 + 2] Cycloaddition Reactions: A Critical Review. Organics 2020, 1, 49–69. [Google Scholar]
- Raji, H.; Aitouna, A.O.; Barhoumi, A.; Hammal, R.; Chekroun, A.; Zeroual, A.; Benharref, A.; Mazoir, N. [2 + 1] Cycloaddition reaction of α-atlantone with m-CPBA in the light of experimental and MEDT quantum-chemical study. Chem. Heterocycl. Comp. 2023, 59, 112–117. [Google Scholar]
- Jasiński, R. Stepwise, zwitterionic course of hetero-Diels–Alder reaction between 1,2,4-triazine molecular systems and 2-cyclopropylidene-1,3-dimethylimidazoline. Chem. Heterocycl. Comp. 2022, 58, 260–262. [Google Scholar]
- Mondal, A.; Acharjee, N. Unveiling the exclusive stereo and site selectivity in [3 + 2] cycloaddition reactions of a tricyclic strained alkene with nitrile oxides from the molecular electron density theory perspective. Chem. Heterocycl. Comp. 2023, 59, 145–154. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Rad. 2023, 2, 1–24. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [PubMed]
- Kacka-Zych, A. Push-pull nitronates in the [3 + 2] cycloaddition with nitroethylene: Molecular Electron Density Theory study. J. Mol. Graph. Model. 2020, 97, 1075492. [Google Scholar]
- Jasiński, R. Searching for zwitterionic intermediates in Hetero Diels–Alder reactions between methyl α,p-dinitrocinnamate and vinyl-alkyl ethers. Comput. Theor. Chem. 2014, 1046, 93–98. [Google Scholar]
- Jasiński, R.; Kubik, M.; Łapczuk-Krygier, A.; Kącka, A.; Dresler, E.; Boguszewska-Czubara, A. An experimental and theoretical study of the hetero Diels–Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: One-step or zwitterionic, two-step mechanism? Reac. Kinet. Mech. Cat. 2014, 113, 333–345. [Google Scholar]
- Chupakhin, E.; Babich, O.; Prosekov, A.; Asyakina, L.; Krasavin, M. Spirocyclic Motifs in Natural Products. Molecules 2019, 24, 4165. [Google Scholar]
- Kącka-Zych, A.; Ríos-Gutiérrez, M.; Domingo, L.R. A molecular electron density theory study of the Lewis acid–catalyzed decomposition reaction of nitroethyl benzoate using aluminum derivatives. J. Phys. Org. Chem. 2019, 32, e3938. [Google Scholar]
- Lapczuk-Krygier, A.; Jaskowska, J.; Jasinski, R. The influence of Lewis acid catalyst on the kinetic and molecular mechanism of nitrous acid extrusion from 3-phenyl-5-nitro-2-isoxazoline: DFT computational study. Chem. Heterocycl. Compd. 2018, 54, 1172. [Google Scholar]
- Kącka, A.; Domingo, L.R.; Jasiński, R. Does a fluorinated Lewis acid catalyst change the molecular mechanism of the decomposition process of nitroethyl carboxylates? Res Chem. Intermed. 2018, 44, 325. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. The Participation of 3,3,3-Trichloro-1-nitroprop-1-ene in the [3 + 2] Cycloaddition Reaction with Selected Nitrile N-Oxides in the Light of the Ex-perimental and MEDT Quantum Chemical Study. Molecules 2021, 26, 6774. [Google Scholar]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzińska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the Question of the Formation of Nitro-Functionalized 2,4-Pyrazole Analogs on the Basis of Nitrylimine Molecular Systems and 3,3,3-Trichloro-1-Nitroprop-1-Ene. Molecules 2022, 27, 8409. [Google Scholar]
- Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Jasiński, R. Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes. Pure Appl. Chem. 2021, 93, 427–446. [Google Scholar]
- Żmigrodzka, M.; Sadowski, M.; Kras, J.; Desler, E.; Demchuk, O.M.; Kula, K. Polar [3 + 2] cycloaddition between N-methyl azomethine ylide and trans-3,3,3-trichloro-1-nitroprop-1-ene. Sci. Rad. 2022, 1, 26–35. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar]
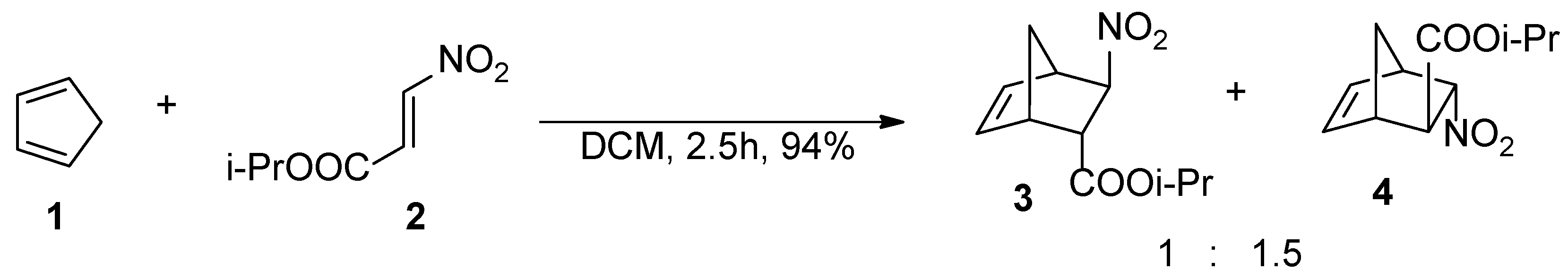



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Global Properties | Local Properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ω [eV] | N [eV] | P−C1 | P−C2 | NC1 [eV] | NC2 [eV] | P+C5 | P+C6 | ω C5 [eV] | ω C6 [eV] | |
| 1 | 0.83 | 3.36 | 0.47 | 0.08 | 1.59 | 0.27 | ||||
| 2 | 2.31 | 1.36 | 0.005 | 0.414 | 0.01 | 0.95 | ||||
| Solvent | Path | Transition | ΔH | ΔS | ΔG |
|---|---|---|---|---|---|
| DCM | A | 1 + 2 → MCA | −6.3 | −40.2 | 5.7 |
| 1 + 2 → TSA | 6.9 | −51.0 | 22.1 | ||
| 1 + 2 → 3 | −32.6 | −52.6 | −16.9 | ||
| B | 1 + 2 → MCB | −6.7 | −40.3 | 5.3 | |
| 1 + 2 → TSB | 5.9 | −51.1 | 21.1 | ||
| 1 + 2 → 4 | −32.2 | −50.4 | −17.2 | ||
| Nitromethane | A | 1 + 2 → MCA | −6.1 | −39.8 | 5.7 |
| 1 + 2 → TSA | 6.8 | −50.8 | 21.9 | ||
| 1 + 2 → 3 | −32.4 | −52.7 | −16.7 | ||
| B | 1 + 2 → MCB | −6.5 | −40.1 | 5.4 | |
| 1 + 2 → TSB | 5.7 | −50.5 | 20.7 | ||
| 1 + 2 → 4 | −32.1 | −50.4 | −17.1 | ||
| C | 1 + 2 → MCA | −6.1 | −39.8 | 5.7 | |
| 1 + 2 → TSC | 18.9 | −47.6 | 33.1 | ||
| 1 + 2 → 7 | 11.7 | −46.4 | 25.6 | ||
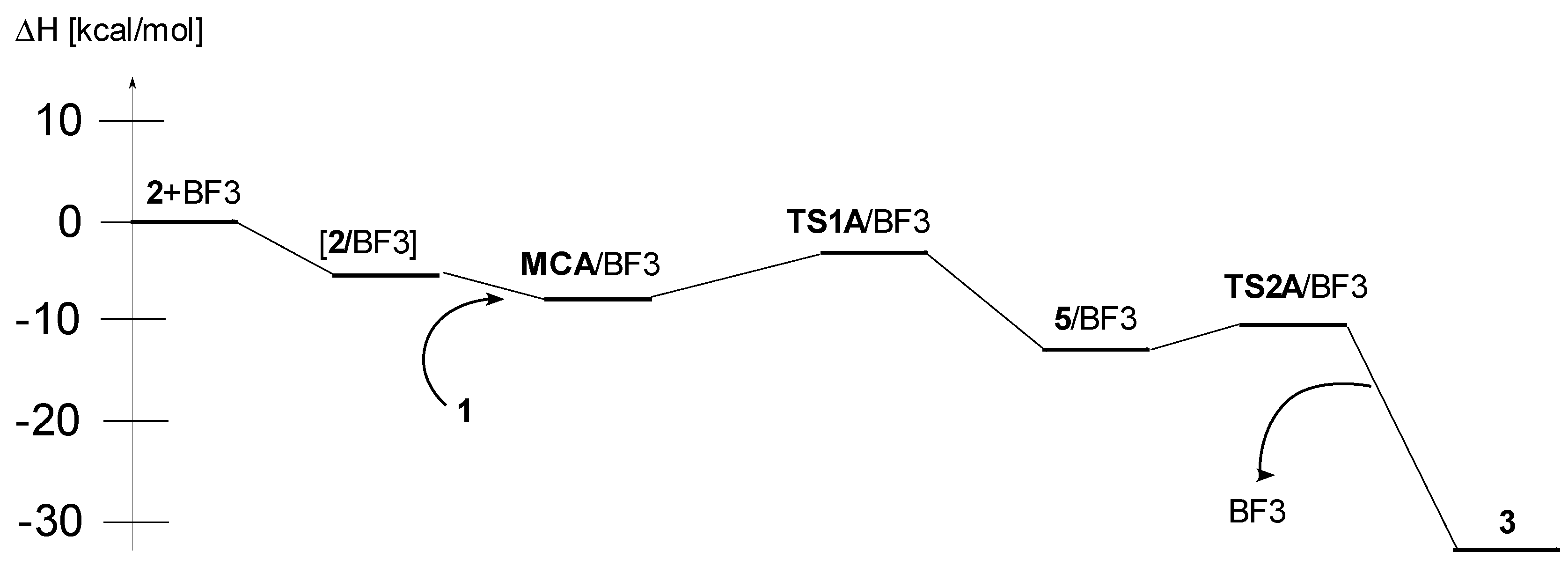
| DCM | A | 2 + BF3 → [2/BF3] | −6.1 | −36.5 | 4.7 |
| 1 + [2/BF3] → MCA/BF3 | −7.7 | −41.2 | 4.6 | ||
| 1 + [2/BF3] → TS1A/BF3 | −2.7 | −50.6 | 12.4 | ||
| 1 + [2/BF3] → 5/BF3 | −13.3 | −52.2 | 2.2 | ||
| 1 + [2/BF3] → TS2A/BF3 | −11.0 | −57.3 | 6.1 | ||
| 1 + 2+BF3 → 3 + BF3 | −32.6 | −52.6 | −16.9 | ||
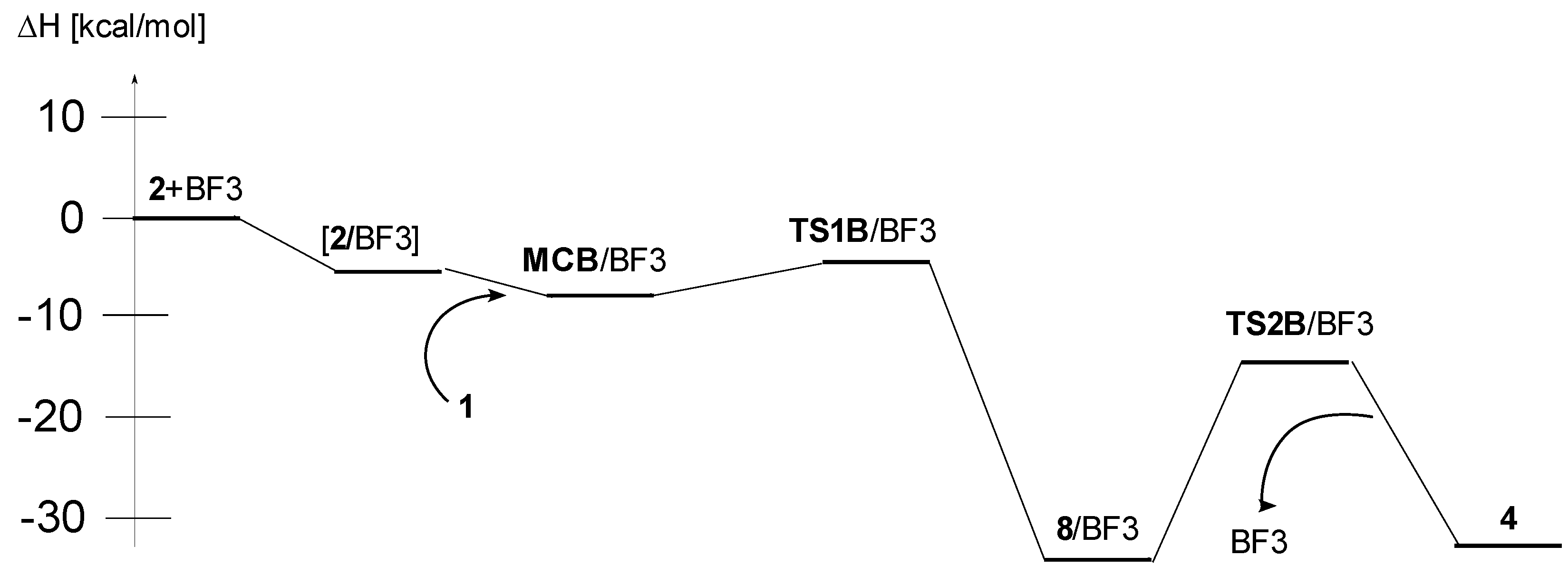
| B | 1 + [2/BF3] → MCB/BF3 | −8.5 | −44.2 | 4.6 | |
| 1 + [2/BF3] → TS1B/BF3 | −5.3 | −51.4 | 10.0 | ||
| 1 + [2/BF3] → 8/BF3 | −37.7 | −55.1 | −21.2 | ||
| 1 + [2/BF3] → TS2A/BF3 | −14.8 | −58.8 | 2.7 | ||
| 1 + 2 + BF3 → 4 + BF3 | −32.2 | −50.4 | −17.2 |
| Solvent | Structure | Interatomic Distances [Å] | GEDT | |||||
|---|---|---|---|---|---|---|---|---|
| C1–C2 | C2–C3 | C3–C4 | C4–C5 | C5–C6 | C6–C1 | [e] | ||
| DCM | 1 | 1.344 | 1.468 | 1.344 | ||||
| 2 | 1.323 | |||||||
| MCA | 1.346 | 1.463 | 1.346 | 3.210 | 1.325 | 3.231 | 0.00 | |
| TSA | 1.398 | 1.398 | 1.389 | 2.336 | 1.388 | 2.166 | 0.32 | |
| 3 | 1.520 | 1.333 | 1.520 | 1.565 | 1.543 | 1.557 | ||
| MCB | 1.346 | 1.464 | 1.345 | 3.235 | 1.324 | 3.227 | ||
| TSB | 1.397 | 1.401 | 1.385 | 2.379 | 1.387 | 2.147 | 0.32 | |
| 4 | 1.515 | 1.334 | 1.517 | 1.565 | 1.538 | 1.579 | 0.00 | |
| Nitromethane | 1 | 1.344 | 1.468 | 1.344 | ||||
| 2 | 1.323 | |||||||
| MCA | 1.346 | 1.463 | 1.346 | 3.211 | 1.325 | 3.231 | 0.00 | |
| TSA | 1.399 | 1.398 | 1.388 | 2.364 | 1.388 | 2.150 | 0.33 | |
| 3 | 1.520 | 1.333 | 1.519 | 1.565 | 1.544 | 1.557 | ||
| MCB | 1.346 | 1.464 | 1.346 | 3.236 | 1.324 | 3.228 | 0.00 | |
| TSB | 1.398 | 1.402 | 1.383 | 2.421 | 1.388 | 2.127 | 0.34 | |
| 4 | 1.515 | 1.334 | 1.517 | 1.565 | 1.538 | 1.579 | ||
| MCA | 0.00 | |||||||
| TSC | 1.415 | 1.411 | 1.367 | 4.371 | 1.416 | 1.855 | 0.61 | |
| 7 | 1.472 | 1.379 | 1.388 | 4.351 | 1.492 | 1.561 | 0.99 | |
| DCM | [2/BF3] | 1.325 | ||||||
| MCA/BF3 | 1.348 | 1.460 | 1.348 | 3.150 | 1.330 | 3.109 | 0.00 | |
| TS1A/BF3 | 1.388 | 1.418 | 1.368 | 2.870 | 1.382 | 2.181 | 0.39 | |
| 5/BF3 | 1.481 | 1.372 | 1.392 | 3.398 | 1.489 | 1.579 | 0.93 | |
| TS2A/BF3 | 1.483 | 1.361 | 1.417 | 2.347 | 1.488 | 1.606 | 0.61 | |
| MCB/BF3 | 1.350 | 1.460 | 1.348 | 3.162 | 1.331 | 3.029 | 0.00 | |
| TS1B/BF3 | 1.385 | 1.426 | 1.360 | 2.966 | 1.377 | 2.233 | 0.35 | |
| 8/BF3 | 1.546 | 1.491 | 1.328 | 3.688 | 1.488 | 1.548 | 0.34 | |
| TS2A/BF3 | 1.482 | 1.369 | 1.406 | 2.379 | 1.484 | 1.606 | 0.47 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules 2023, 28, 5289. https://doi.org/10.3390/molecules28145289
Dresler E, Wróblewska A, Jasiński R. Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules. 2023; 28(14):5289. https://doi.org/10.3390/molecules28145289
Chicago/Turabian StyleDresler, Ewa, Aneta Wróblewska, and Radomir Jasiński. 2023. "Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate" Molecules 28, no. 14: 5289. https://doi.org/10.3390/molecules28145289
APA StyleDresler, E., Wróblewska, A., & Jasiński, R. (2023). Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules, 28(14), 5289. https://doi.org/10.3390/molecules28145289