

Effect of the Cu2+/1+ Redox Potential of Non-Macrocyclic Cu Complexes on Electrochemical CO2 Reduction


Abstract

1. Introduction
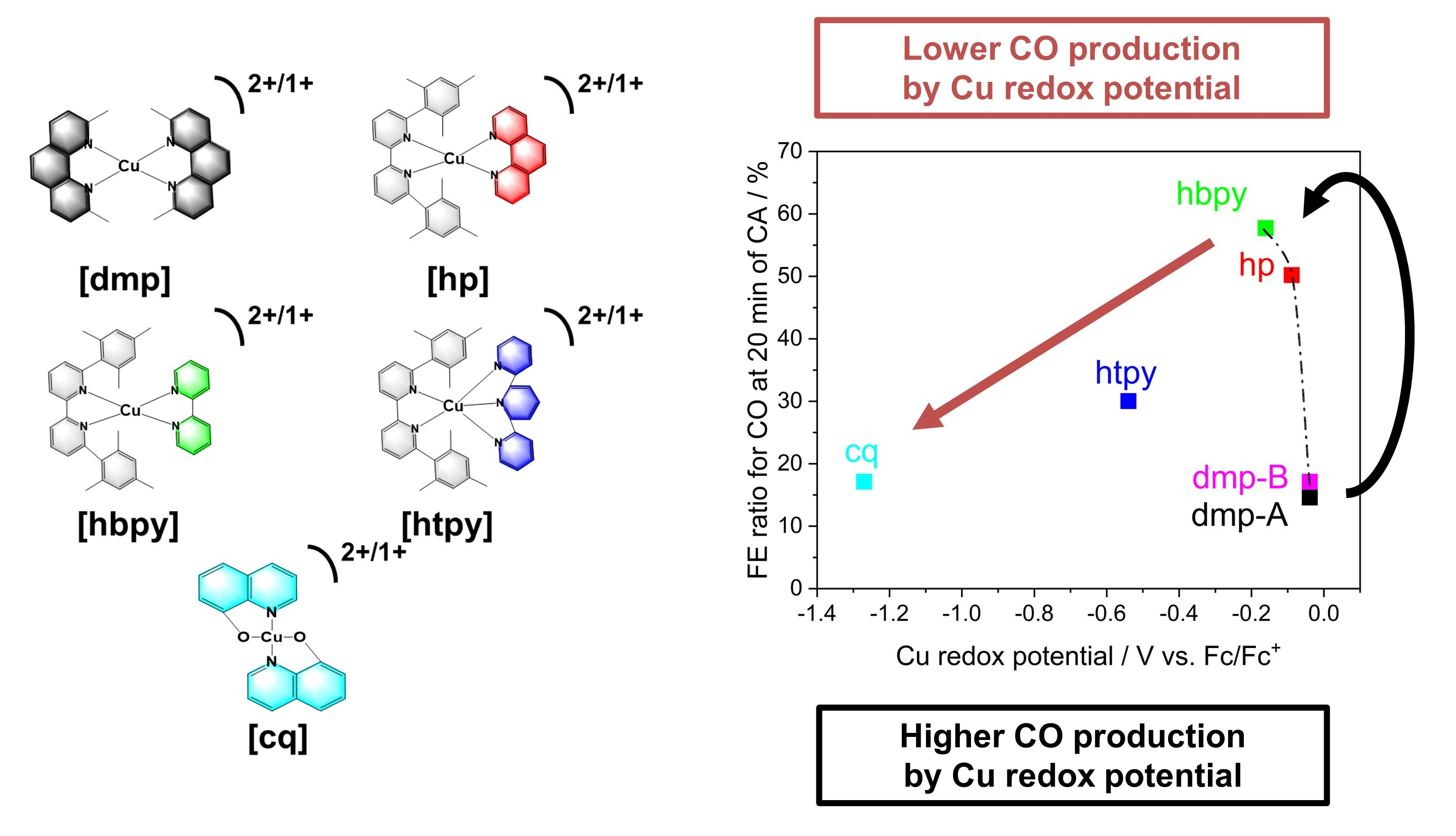
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Preparation of Cu Complexes
3.2.1. (6,6′-Bismesitil-2,2′-bipyridine) (1,10-phenanthroline) Acetonitrile Copper (II) TFSI (hp)
3.2.2. (6,6′-Bismesitil-2,2′-bipyridine) (2,2′-bispyridine) Acetonitrile Copper (II) BF4 (hbpy)
3.2.3. (6,6′-Bismesitil-2,2′-bipyridine) (2,2′:6′,2″-terpyridine) Copper (II) BF4 (htpy)
3.3. Electrochemical Measurements
3.4. Gas Product Analysis
3.5. Material Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kim, K.; Wagner, P.; Wagner, K.; Mozer, A.J. Electrochemical CO2 Reduction Catalyzed by Copper Molecular Complexes: The Influence of Ligand Structure. Energy Fuels 2022, 36, 4653–4676. [Google Scholar] [CrossRef]
- Eckert, N.A.; Vaddadi, S.; Stoian, S.; Lachicotte, R.J.; Cundari, T.R.; Holland, P.L. Coordination-Number Dependence of Reactivity in an Imidoiron (III) Complex. Angew. Chem. 2006, 118, 7022–7025. [Google Scholar] [CrossRef]
- Sarkar, A.; Dey, S.; Rajaraman, G. Role of Coordination Number and Geometry in Controlling the Magnetic Anisotropy in FeII, CoII, and NiII Single-Ion Magnets. Chem.-Eur. J. 2020, 26, 14036–14058. [Google Scholar] [CrossRef] [PubMed]
- Arana, C.; Yan, S.; Keshavarz-K, M.; Potts, K.; Abruna, H. Electrocatalytic reduction of carbon dioxide with iron, cobalt, and nickel complexes of terdentate ligands. Inorg. Chem. 1992, 31, 3680–3682. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, A. Comparative DNA Binding Abilities and Phosphatase-Like Activities of Mono-, Di-, and Trinuclear Ni (II) Complexes: The Influence of Ligand Denticity, Metal–Metal Distance, and Coordinating Solvent/Anion on Kinetics Studies. Inorg. Chem. 2014, 53, 10731–10742. [Google Scholar] [CrossRef]
- Caspar, J.V.; Westmoreland, T.D.; Allen, G.H.; Bradley, P.G.; Meyer, T.J.; Woodruff, W.H. Molecular and electronic structure in the metal-to-ligand charge-transfer excited states of d6 transition-metal complexes in solution. J. Am. Chem. Soc. 1984, 106, 3492–3500. [Google Scholar] [CrossRef]
- Crutchley, R.J. Phenylcyanamide ligands and their metal complexes. Coord. Chem. Rev. 2001, 219, 125–155. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Saväant, J.-M. Dissection of electronic substituent effects in multielectron–multistep molecular catalysis. Electrochemical CO2-to-CO conversion catalyzed by iron porphyrins. J. Phys. Chem. C 2016, 120, 28951–28960. [Google Scholar] [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Fontecave, M. Turning it off! Disfavouring hydrogen evolution to enhance selectivity for CO production during homogeneous CO2 reduction by cobalt–terpyridine complexes. Chem. Sci. 2015, 6, 2522–2531. [Google Scholar] [CrossRef]
- Saygili, Y.; Söderberg, M.; Pellet, N.; Giordano, F.; Cao, Y.; Muñoz-García, A.B.; Zakeeruddin, S.M.; Vlachopoulos, N.; Pavone, M.; Boschloo, G. Copper bipyridyl redox mediators for dye-sensitized solar cells with high photovoltage. J. Am. Chem. Soc. 2016, 138, 15087–15096. [Google Scholar] [CrossRef]
- Singh, S.; Phukan, B.; Mukherjee, C.; Verma, A. Salen ligand complexes as electrocatalysts for direct electrochemical reduction of gaseous carbon dioxide to value added products. RSC Adv. 2015, 5, 3581–3589. [Google Scholar] [CrossRef]
- Shi, N.-N.; Xie, W.-J.; Zhang, D.-M.; Fan, Y.-H.; Cui, L.-S.; Wang, M. A mononuclear copper complex as bifunctional electrocatalyst for CO2 reduction and water oxidation. J. Electroanal. Chem. 2021, 886, 115106. [Google Scholar] [CrossRef]
- Haines, R.J.; Wittrig, R.E.; Kubiak, C.P. Electrocatalytic Reduction of Carbon Dioxide by the Binuclear Copper Complex [Cu2(6-(diphenylphosphino-2,2′-bipyridyl)2(MeCN)2][PF6]2. Inorg. Chem. 1994, 33, 4723–4728. [Google Scholar] [CrossRef]
- Smieja, J.M.; Kubiak, C.P. Re (bipy-tBu)(CO)3Cl−improved catalytic activity for reduction of carbon dioxide: IR-spectroelectrochemical and mechanistic studies. Inorg. Chem. 2010, 49, 9283–9289. [Google Scholar] [CrossRef]
- Nie, W.; Tarnopol, D.E.; McCrory, C.C. Enhancing a molecular electrocatalyst’s activity for CO2 reduction by simultaneously modulating three substituent effects. J. Am. Chem. Soc. 2021, 143, 3764–3778. [Google Scholar] [CrossRef]
- Nie, W.; McCrory, C.C. Strategies for breaking molecular scaling relationships for the electrochemical CO2 reduction reaction. Dalton Trans. 2022, 51, 6993–7010. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Wu, Y.; Wang, M.; Jiang, J.; Yang, K.; Huo, S.; Wang, X.-F.; Ma, Q.; Brudvig, G.W.; Batista, V.S. Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 2018, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, M.; Jeong, H.Y.; Choutipalli, V.S.K.; Hong, J.S.; Seo, H.; Saravanan, N.; Jang, J.H.; Lee, K.G.; Lee, Y.H.; Im, S.W. Electrocatalytic Reduction of CO2 to Ethylene by Molecular Cu-Complex Immobilized on Graphitized Mesoporous Carbon. Small 2020, 16, 2000955. [Google Scholar] [CrossRef]
- Karapinar, D.; Zitolo, A.; Huan, T.N.; Zanna, S.; Taverna, D.; Galvão Tizei, L.H.; Giaume, D.; Marcus, P.; Mougel, V.; Fontecave, M. Carbon-nanotube-supported copper polyphthalocyanine for efficient and selective electrocatalytic CO2 reduction to CO. ChemSusChem 2020, 13, 173–179. [Google Scholar] [CrossRef]
- Xu, Y.; Li, F.; Xu, A.; Edwards, J.P.; Hung, S.-F.; Gabardo, C.M.; O’Brien, C.P.; Liu, S.; Wang, X.; Li, Y. Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly. Nat. Commun. 2021, 12, 2932. [Google Scholar] [CrossRef]
- Du, J.; Cheng, B.; Jiang, L.; Han, Z. Copper phenanthroline for selective electrochemical CO2 reduction on carbon paper. Chem. Commun. 2023, 59, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sahil, S.T.; Mazumder, M.M.R.; Stephens, A.M.; Cronin, B.; Duin, E.C.; Jurss, J.W.; Farnum, B.H. Synthesis, characterization, and electrocatalytic activity of bis (pyridylimino) isoindoline Cu (II) and Ni (II) complexes. Dalton Trans. 2021, 50, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Zhao, J.; Xue, S.; Barber, J.; Zhou, Y.; Meng, J.; Ke, X. An overview of Cu-based heterogeneous electrocatalysts for CO2 reduction. J. Mater. Chem. 2020, 8, 4700–4734. [Google Scholar] [CrossRef]
- Ma, Y.; Fang, Y.; Fu, X.; Wang, X. Photoelectrochemical conversion of CO2 into HCOOH using a polymeric carbon nitride photoanode and Cu cathode. Sustain. Energy Fuels 2020, 4, 5812–5817. [Google Scholar] [CrossRef]
- Kim, K.; Wagner, P.; Wagner, K.; Mozer, A.J. Electrochemical CO2 Reduction to Methane by Cu complex-derived Catalysts in Non-aqueous Media. ChemCatChem 2023, e202300396. [Google Scholar] [CrossRef]
- Leandri, V.; Daniel, Q.; Chen, H.; Sun, L.; Gardner, J.M.; Kloo, L. Electronic and structural effects of inner sphere coordination of chloride to a homoleptic copper (II) diimine complex. Inorg. Chem. 2018, 57, 4556–4562. [Google Scholar] [CrossRef]
- Kim, K.; Wagner, P.; Wagner, K.; Mozer, A.J. Catalytic Decomposition of Organic Electrolyte to Methane by a Cu-complex Derived In-situ CO2 Reduction Catalyst. ACS Omega, 2023; under review. [Google Scholar]
- Schmittel, M.; Ganz, A. Stable mixed phenanthroline copper (i) complexes. Key building blocks for supramolecular coordination chemistry. Chem. Commun. 1997, 999–1000. [Google Scholar] [CrossRef]
- Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Synthesis of sterically encumbered 2, 9-diaryl substituted phenanthrolines. Key building blocks for the preparation of mixed (bis-heteroleptic) phenanthroline copper (I) complexes. Heterocycl Comm 1997, 3, 493–498. [Google Scholar] [CrossRef]
- Fraser, M.G.; van der Salm, H.; Cameron, S.A.; Blackman, A.G.; Gordon, K.C. Heteroleptic Cu(I) bis-diimine complexes of 6, 6′-dimesityl-2, 2′-bipyridine: A structural, theoretical and spectroscopic study. Inorg. Chem. 2013, 52, 2980–2992. [Google Scholar] [CrossRef] [PubMed]
- Kavungathodi, M.F.M.; Wagner, P.; Mori, S.; Mozer, A.J. Four Orders of Magnitude Acceleration of Electron Recombination at the Dye-TiO2/Electrolyte Interface Severely Limiting Photocurrent with High-Oxidation-Potential Cu2+/1+ Complexes. J. Phys. Chem. C 2023, 127, 7618–7627. [Google Scholar] [CrossRef]
- De, S.; Mahata, K.; Schmittel, M. Metal-coordination-driven dynamic heteroleptic architectures. Chem. Soc. Rev. 2010, 39, 1555–1575. [Google Scholar] [CrossRef]
- Schmittel, M.; Kalsani, V.; Bats, J.W. Metal-driven and covalent synthesis of supramolecular grids from racks: A convergent approach to heterometallic and heteroleptic nanostructures. Inorg. Chem. 2005, 44, 4115–4117. [Google Scholar] [CrossRef] [PubMed]
- Pape, V.F.; May, N.V.; Gál, G.T.; Szatmári, I.; Szeri, F.; Fülöp, F.; Szakács, G.; Enyedy, É.A. Impact of copper and iron binding properties on the anticancer activity of 8-hydroxyquinoline derived Mannich bases. Dalton Trans. 2018, 47, 17032–17045. [Google Scholar] [CrossRef] [PubMed]
- Scaltrito, D.V.; Thompson, D.W.; O’Callaghan, J.A.; Meyer, G.J. MLCT excited states of cuprous bis-phenanthroline coordination compounds. Coord. Chem. Rev. 2000, 208, 243–266. [Google Scholar] [CrossRef]
- Roy, S.; Bhandari, S.; Chattopadhyay, A. Quantum Dot Surface Mediated Unprecedented Reaction of Zn2+ and Copper Quinolate Complex. J. Phys. Chem. C 2015, 119, 21191–21197. [Google Scholar] [CrossRef]
- Hashmi, S.G.; Sonai, G.G.; Iftikhar, H.; Lund, P.D.; Nogueira, A.F. Printed single-walled carbon-nanotubes-based counter electrodes for dye-sensitized solar cells with copper-based redox mediators. Semicond. Sci. Technol. 2019, 34, 105001. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A.; Schenk, W.A.; Hagel, M. Synthesis and coordination properties of 6, 6′-dimesityl-2, 2′-bipyridine. J. Z. Für Nat. B 1999, 54, 559–564. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cu Complex | Cu2+/1+ Redox Potential (V vs. Fc/Fc+) in Argon | Cu2+/1+ Redox Potential (V vs. NHE) in Argon | FE Ratio for CO (%) (20 min) | FE Ratio for H2 (%) (20 min) | FE Ratio for CH4 (%) (20 min) | FE Ratio for CO (%) (80min) | FE Ratio for H2 (%) (80min) | FE Ratio for CH4 (%) (80min) | FE Ratio for CO (%) (Hetero) | FE Ratio for H2 (%) (Hetero) | FE Ratio for CH4 (%) (Hetero) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| dmp-A | −0.039 | 0.68 | 14.6 | 15.7 | 69.7 | 5.8 | 15.8 | 78.3 | 8.3 | 3.5 | 88.2 |
| hp | −0.089 | 0.63 | 50.2 | 8.2 | 41.6 | 24.6 | 27.0 | 48.5 | 39.2 | 5.8 | 55.0 |
| hbpy | −0.161 | 0.56 | 57.7 | 5.9 | 36.4 | 50.0 | 6.6 | 43.5 | 22.6 | 4.0 | 73.4 |
| htpy | −0.54 | 0.18 | 30.0 | 17.8 | 52.2 | 13.2 | 32.3 | 54.6 | 23.2 | 7.3 | 69.5 |
| cq | −1.27 | −0.55 | 17.1 | 53.1 | 29.8 | 29.0 | 33.8 | 37.2 | 42.1 | 7.2 | 50.7 |
| dmp-B | −0.039 | 0.68 | 17.1 | 10.5 | 72.4 | 3.4 | 14.5 | 82.1 | 23.0 | 8.8 | 68.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Wagner, P.; Wagner, K.; Mozer, A.J. Effect of the Cu2+/1+ Redox Potential of Non-Macrocyclic Cu Complexes on Electrochemical CO2 Reduction. Molecules 2023, 28, 5179. https://doi.org/10.3390/molecules28135179
Kim K, Wagner P, Wagner K, Mozer AJ. Effect of the Cu2+/1+ Redox Potential of Non-Macrocyclic Cu Complexes on Electrochemical CO2 Reduction. Molecules. 2023; 28(13):5179. https://doi.org/10.3390/molecules28135179
Chicago/Turabian StyleKim, Kyuman, Pawel Wagner, Klaudia Wagner, and Attila J. Mozer. 2023. "Effect of the Cu2+/1+ Redox Potential of Non-Macrocyclic Cu Complexes on Electrochemical CO2 Reduction" Molecules 28, no. 13: 5179. https://doi.org/10.3390/molecules28135179
APA StyleKim, K., Wagner, P., Wagner, K., & Mozer, A. J. (2023). Effect of the Cu2+/1+ Redox Potential of Non-Macrocyclic Cu Complexes on Electrochemical CO2 Reduction. Molecules, 28(13), 5179. https://doi.org/10.3390/molecules28135179