Abstract
Symmetrical and dissymmetrical bolaforms were prepared with good to high yields from unsaturated L-rhamnosides and phenolic esters (ferulic, phloretic, coumaric, sinapic and caffeic) using two eco-compatible synthetic strategies involving glycosylation, enzymatic synthesis and cross-metathesis under microwave activation. The plant-eliciting activity of these new compounds was investigated in Arabidopsis model plants. We found that the monocatenar rhamnosides and bolaforms activate the plant immune system with a response depending on the carbon chain length and the nature of the hydrophilic heads. Their respective antioxidant activities were also evaluated, as well as their cytotoxic properties on dermal cells for cosmetic uses. We showed that phenolic ester-based compounds present good antioxidant activities and that their cytotoxicity is low. These properties are also dependent on the carbon chains used.
1. Introduction
Surfactants are natural or synthetic amphiphilic compounds presenting specific polar and non-polar domains with typical solubility in water. They are able to reduce the interfacial tension of mixtures (oil and water) by adsorbing at the interfaces. Surfactants are classified into three large families according to the nature of their hydrophilic part [1]: ionic (cationic or anionic), zwitterionic (or amphoteric) and neutral (Figure 1) [2,3,4,5,6,7]. The malleability of their structure and the diversity of their properties allow them to be used in many products, particularly in household and industrial detergents, cosmetic formulations and as plant defense inducers [8,9,10,11]. Anionic surfactants (low molecular weight cation associated with sulphates, sulfonates or carboxylates) are the most widely used industrially, particularly in the field of detergents, thanks to their foaming properties [12,13,14,15,16]; cationic surfactants (trimethylated quaternary ammonium salts or pyridinium salts) [17,18,19] are active products in softeners (used to reduce static electricity) or shampoos.
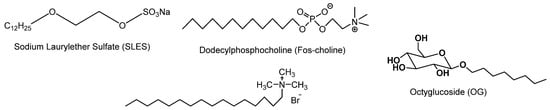
Figure 1.
Examples of surfactants.
Surfactants can also be classified into several categories according to their structure (number and arrangement of hydrophilic and hydrophobic poles within the molecule). The most common structure (in derivatives called monocatenar surfactants) displays a hydrophilic head and a hydrophobic chain. There are surfactants with several hydrophobic chains grafted onto the same hydrophilic head (double-stranded and three-stranded surfactants), but several hydrophilic heads can also be linked to one or more hydrophobic chains (called bolaform or twinned surfactants) [20].
In the current context of sustainable development, the lower availability of raw materials of petrochemical origin and consumers’ acceptance of a “bio-sourced surfactant” have increased interest in new surfactant technologies worldwide. Amphiphilic molecules from agro-resources are an innovative and interesting alternative for the substitution of petroleum-derived surfactants because they present relatively good biodegradability and low toxicity [21]. However, their production cost remains a limiting factor for their development [22]. Among the bio-based surfactants recently developed with these criteria, we can exemplify a sugar head containing alkyl polyglycosides (APG) [23,24,25].
Partially or completely bio-based surfactants are often anionic or non-ionic, including glycolipid, lipopeptide, phospholipid or even fatty acid types [26,27]. The hydrophilic head of the molecule can be a sugar, a carboxylic acid, an amino acid, an alcohol or peptides, while the lipophilic part is often a fatty acid or a fatty alcohol [28,29,30]. Bio-surfactants from agro-resource origins or plant bio-based surfactants are compounds that, in general, have one of the two hydrophilic or hydrophobic moieties of plant origin.
The synthesis of bio-sourced surfactants based on sugars has previously been described in the literature, particularly xylosides and rhamnosides [28,29,30,31,32,33,34]. Indeed, in 1993, 1,12-digluconamidododecane was synthetized from D-gluconolactone [35]. In 2004, Satgé and al. prepared a bolaform surfactant from D-galactose by a microwave-assisted glycosylation of a D-galactose protected by an unsaturated long-chain alcohol, followed by metathesis in the presence of the Grubbs I catalyst; hydrogenation in the presence of (Rh/Al2O3, H2) led to the saturated bolaform [36]. In 2010, K. Dzulkefly et al. synthesized symmetrical bolaform amphiphiles from an acid chloride with D-glucose as the hydrophilic head [37].
Sugar based-bolaamphiphiles have also been prepared, as exemplified by D-xyloside or L-rhamnosides-based bolaamphiphiles [31,32]. In a previous work [38], we described fatty ester-based bolaamphiphiles derived from phloretic acid and dissymmetric bolaamphiphiles with rhamnose and phloretic moieties as polar heads. The first step consisted of glycosylation of the sugar, followed by the enzymatic esterification of the phloretic acid with lipase B from Candida antarctica (CALB). Finally, a classical cross metathesis reaction using a Grubbs I catalyst led to the development of bolaform compounds. However, the kinetics of these reactions was low for an industrial application and the purification requires several chromatographies on silica columns, which are time-consuming and solvent-intensive processes.
Herein, we discuss our newly developed approaches to obtain these bio-based compounds using two eco-compatible and fast synthetic methods coupling either glycosylation or enzymatic synthesis and activation by microwaves [39], which can reduce the reaction times and enhance the selectivity (few degradation products and appropriate quantities with no excess in one or the other substrates). Furthermore, greener solvents have been employed and demonstrated success as a reacting medium. For the processing and purification of the compounds, automated flash chromatography was used to considerably reduce the purification times. In these experiments, we compared old methodologies with the new approaches that we developed. Subsequently, we designed a panel of bio-based bolaforms from L-rhamnose (sugar from pectin) and other phenolic acid derivatives of lignin, such as phloretic, para-coumaric, ferulic, caffeic and sinapic acids, which could present better antioxidant properties (Figure 2).
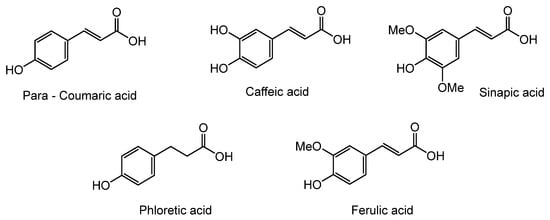
Figure 2.
Structures of derivatives of hydroxycinnamic acids.
The antioxidant properties of the monocatenar and bolaform adducts were then evaluated. Because rhamnose-based surfactants, classified as rhamnolipid mimetics, are well known for their antifungal properties and their ability to trigger an innate immune response in Arabidopsis thaliana [40], we also investigated the plant-eliciting properties of the monocatenar and bolaform adducts. Finally, cytotoxicity on dermal fibroblasts was also tested for a potential use in cosmetics.
2. Results and Discussion
2.1. Synthesis
The glycosylation of L-rhamnose (Scheme 1) was initially performed with p-toluenesulfonic acid (PTSA) as a catalyst under conventional heating conditions with or without a solvent (Table 1). Low yields were obtained at 80 °C for 48 h (Table 1, entries 1 and 2). The time and temperatures were decreased to limit sugar degradation but similar results were obtained (Table 1, entries 3–6); however, the yields increased in the absence of a solvent, as the alcohol itself could be used as a solvent [2,3,4,5,6,7].
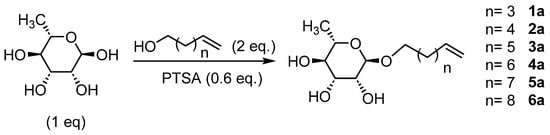
Scheme 1.
Glycosylation of L-rhamnose.

Table 1.
Preliminary results for L-rhamnose glycosylation under classic thermic conditions.
In order to increase the yield while avoiding the degradation of the starting reagents, we induced the reaction by microwaves [36,39,41]. First, the reaction conditions were optimized with a combination of L-rhamnose/Hex-5-enol using various conditions of time, temperature and power (Table 2).
Table 2.
Glycosylation of L-rhamnose with hex-5-enol in THF (5 mL) under microwave conditions.
The best results were obtained with an excess of four equivalents of 5-hexen-1-ol at a temperature of 60 °C for 2 h with a power of 60 W (Table 2, entry 8). The more homogeneous heating achieved by the microwaves improved the yield from 19% by conventional heating over 48 h to nearly 64% in only 2 h. It should also be noted that under these conditions, no degradation of rhamnose was observed. These same conditions were then used for the synthesis of L-rhamnosides of different chain lengths. The results obtained are summarized in Table 3.
Table 3.
Glycosylation of L-rhamnose under microwaves (60 °C, 120 min, 60 W).
Rhamnosides were obtained with yields between 47 and 64%, which are inversely proportional to the chain length of the alkyl groups, as observed in the literature with sugars such as D-glucose, D-galactose and D-mannose [42]. The use of microwaves considerably reduces the reaction time and also improves the selectivity (few degradation products). Moreover, 2-methyltetrahydrofuran and γ-valerolactone were used as greener solvents [43] and they led to better yields, especially γ-valerolactone (Table 2, entries 2 and 8 vs. entries 1 and 7, respectively). This can be explained by the more polar and less viscous character of these solvents, which are favorable to the activation of the reagents under microwaves [44]. The formation of the rhamnosides was confirmed by IR (bands at 2926 cm−1 and 1640 cm−1, respectively, for the CH3 group of the rhamnose and the terminal alkene function on the unsaturated alcohols) and 1H NMR, with a shift at 1.25 ppm relative to the CH3 in the C6 position of rhamnose and signals between 4.52–4.72 ppm and 3.17–3.58 ppm for all the protons of rhamnose and at 4.98 ppm and 5.80 ppm for the terminal alkene function. The 13C NMR also confirmed the formation of rhamnosides with a terminal insaturation at 115.2 ppm and 139.2 ppm. In order to obtain bolaform compounds from these monocatenars, a cross-metathesis reaction (Scheme 2) was performed from rhamnosides using two types of Grubbs catalyst (Grubbs I and Grubbs II).
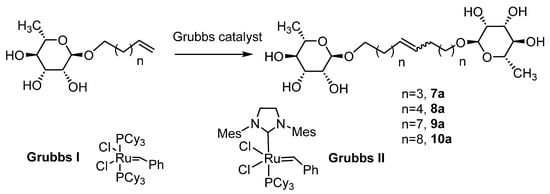
Scheme 2.
Metathesis of L-rhamnoside.
In the first step, we used classical conditions in the presence of Grubbs I with a conventional thermal activation. After several purification steps by silica chromatography, and with the addition of activated carbon and filtration of the celite, we obtained very low yields, lower than 20% (Table 4), regardless of the carbon chain length (Table 4, entries 1–3). These low yields could be explained not only by the purification steps or by the nature of the Grubbs I catalyst, which is less reactive compared to other generations, but also by the solvent used (the dichloromethane in which rhamnosides are not very soluble). The activation through microwaves also induced higher yields (37 and 52%, respectively, for carbon chains of 12 or 10 carbons; Table 4, entries 4 and 5) with a Z/E ratio of 20/80 as confirmed by NMR. These yields were again improved to 60 and 77% by using a Grubbs II catalyst and a mixture of CH2Cl2/MeOH (9/1) (Table 4, entries 7 and 8). Similar results have been also observed concerning the C18 chain (Table 4, entries 3, 6 and 9) and optimal conditions were used to prepare compound 10a involving a C20 chain (Table 4, entry 10).
Table 4.
Cross metathesis of rhamnosides.
This synthetic methodology was extended for the preparation of bolaforms derived from hydroxycinnamic acids, which are widely used in cosmetics and are known for their antioxidant properties [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. We used a method previously described in the literature [38] but improved the yields by using the CombiFlash technique for the purification. These bolaforms derived from cinnamic acids were prepared in two steps. The first step consists of synthesizing monocatenar esters from hydroxycinnamic acids and unsaturated fatty alcohols of different chain lengths. These monocatenar compounds were synthesized by a chemoenzymatic method in the presence of a lipase (lipase Novozym 435, lipase B from Candida antarctica “CAL-B”). According to the results of the literature [38], we chose 2-methyl-2-butanol as a solvent because of the stability and reactivity of the “CAL-B” enzyme. Polar alcohol allows for better chemical and thermal stability of the enzyme, but also avoids the possibility of having a competitive esterification with the starting phenolic acids.
Initial experiments with phloretic acid (used in excess amounts to obtain better yields) and alcohols of different unsaturated chain lengths (5-hexen-1-ol, 6-hepten-1-ol, 9-decen-1-ol 10-undecen-1-ol respectively) under the conditions described by Obounou et al. [38] showed low yields of phloretic acid esters despite good conversion (Table 5). Therefore, we modified the purification method by using a semiautomatic method with CombiFlash coupled with a UV detector at the output (UV1 = 280 nm and UV2 = 360 nm). After several trials, the elution program retained for all the products was as follows: a flow of 15 mL/min, 3 min eluting with 92% petroleum ether and 8% ethyl acetate, 27 min with 80% petroleum ether and 20% ethyl acetate and 32 min with 60% petroleum ether and 40% ethyl acetate. This eluting program allowed the recovery of the different pure monocatenars in a single step as opposed to a classical purification by flash chromatography, which requires several purification steps. This new purification method was very efficient in reducing product losses because the yields in the esters were closed to the conversions (Table 5).
Table 5.
Enzymatic catalyzed esterification of phloretic acids.
After purification, these esters were characterized by infrared spectroscopy (IR), nuclear magnetic resonance (NMR) and mass spectrometry. In FTIR, we observed an elongation band at 1215 cm−1, characteristic of the phenol function; a band at 1640 cm−1, characteristic of the alkene function; and a band at 1704 cm−1 corresponding to the ester (C=O) function. The 1H NMR shows two doublets between 7.01 and 6.72 ppm (integrating for 8 H) corresponding to the aromatic protons, a multiplet for the protons of the alkene function between 5.82 and 4.95 ppm and a triplet at 4.02 ppm, which corresponds to the protons in alpha position of the ester function. The 13C NMR also confirms the presence of the aromatic ring (between 130 and 115 ppm), the carbonyl ring (173 ppm) and the alkene ring (131 ppm).
This methodology was extended to other phenolic acids (paracoumaric, ferulic, caffeic and sinapic). The first experiments carried out with these different acids in 2-methyl-2-butanol (2M2B) did not give any result for some of them (ferulic and sinapic acids) because of their very low solubility in 2M2B. Therefore, we used a mixture of 2M2B/THF (1/2) for caffeic acid and acetone for sinapic acid, respectively, as the solvents. However, the yields obtained from these acids were lower compared to those obtained with phloretic acid (Table 6). This result can be explained by the presence of an unsaturation in the α-position of the acid function, which reduces their reactivity towards enzymatic transformations [50,51]; this is not the case with phloretic acid.
Table 6.
Esterification of paracoumaric, ferulic, caffeic and sinapic acids (conditions: acid (6 eq.) and alcohol (1 eq., lipase 2.5 g per 100 g of reaction mixture, molecular sieves (50 g·L−1), 60 °C, 48 h).
In the second step, the synthesis of phloretic ester bolaforms was first performed by a metathesis in the presence of a Grubbs I catalyst added in 6 portions for 40 min at 45 °C and under microwave activation. These conditions led to low yields of C10 (20%), C12 (16%) and C18 (24%) bolaform compounds, respectively. We subsequently used the more reactive Grubbs II catalyst; after 40 min under microwave conditions, the yields were improved (Table 7). The same experiment was extended using the para-coumaric esters 5 and 6b; good yields were obtained with a short carbon chain, while the yields remained relatively low with a longer chain (Table 7).
Table 7.
Metathesis of phenolic esters.
The compounds were classically characterized by IR, NMR, UV and elemental analysis. In IR, we identified the band at 1704 cm−1 of the carbonyl group, the band between 1640–1690 cm−1 corresponding to the double bond and the elongation band at 1215 cm−1, characteristic of the phenol function. In NMR, the singlet at 5.37 ppm integrating the two protons was also significant for the alkene function.
In order to obtain a system with both a good hydrophilic character (necessary, for example, for a good interaction with phospholipid membranes) and good antioxidant activity, we considered dissymmetric bolaform surfactants with a glycosylated head (L-rhamnose) and the other based on a hydroxycinnamic acid derivative. These dissymmetric bolaform surfactants were prepared by cross-metathesis. Different methods were tested according to the protocols described in the literature using different Ru-based catalysts, different ratios of rhamnosides/fatty acid esters or reaction conditions (time or temperature) [52]. Dissymmetric bolaamphiphiles were always obtained in a minority compared to the symmetric ones. However, inspired by the work of Blechert and Obounou et al. [38,53], four dissymmetric bolaamphiphiles were obtained under classical heating conditions and in the presence of Grubbs II (Scheme 3).
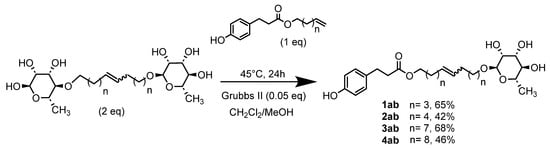
Scheme 3.
Cross-metathesis between rhamnosides and phenolic esters.
These dissymmetrical bolaforms were thus prepared by cross-metathesis of the symmetrical L-Rhamnose-based bolaform and the derived unsaturated phloretic esters. The use of a CH2Cl2/MeOH solvent mixture significantly improved the solubility of the reactants in contrast to CH2Cl2. TLC monitoring showed that the predominant compound formed was the dissymmetrical bolaform with a small amount of symmetrical bolaform derived from the unsaturated phloretic esters. After rapid purification (35 min) by automated flash chromatography, bolaforms 1ab–4ab were obtained in yields up to 70% in 24 h.
These dissymmetric bolaforms were characterized by classical spectroscopic and spectrometric methods: IR (1615, 1732 and 3353 cm−1, respectively, for the C=C, C=O and OH bonds), NMR (5.80 ppm for the protons of the C=C, 2 doublets between 6.99 and 6.65 ppm for the aromatic protons and signals between 3.12 and 3.54 ppm for the hydrogens of the rhamnose, except the methyl group), UV and elemental analysis.
2.2. Antioxidant Properties
To evaluate the antioxidant activity, several methods [54,55] exist in the literature demonstrating the use of 2,2’-azinobis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS) [56], β-carotene [57], Trolox (ORAC method) [58] and 2,2-diphenyl-1-picrylhydrazyl DPPH [59] (Scheme 4), respectively. The latter is based on the reduction of DPPH●, which is accompanied by a significant color change from purple to yellow, monitored by UV-visible spectroscopy at an absorption wavelength of 517 nm. We used this method to determine the antioxidant activity of our surfactants.
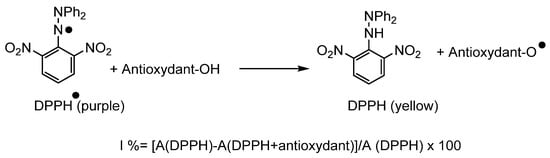
Scheme 4.
Reaction of an antioxidant with the DPPH● radical.
The antioxidant activity of phenolic ester and the derived bolaforms were evaluated in methanol by measuring their ability to reduce the DPPH● at a concentration of 10−4 mol·L−1. These activities were expressed as percent of inhibition according to the formula given in Scheme 4; vitamin C was used as a positive control. The antioxidant tests were performed at 45 min and in triplicate to confirm the repeatability of the results.
Preliminary observations showed that the monocatenar phloretic esters 1b and 3b have no antioxidant activity, but corresponding bolaforms 12b and 14b presented rather interesting antioxidant activity (Table 8, entries 8 and 9). In addition, the antioxidant activity of ferulic ester is greater than that of the phloretic ester containing the same carbon chain (Table 8, entries 5 and 7). This difference could be explained by the low stability of the phenoxy radical resulting from phloretic ester compared to the radical derived from ferulic ester, which could be more stabilized through the methoxy function in the ortho position. These results show that the antioxidant activity depends on both the stability of the resulting radical and the number of phenolic moieties.
Table 8.
Antioxidant properties of phenolic (di)esters.
In order to better understand the structure/antioxidant activity relationship of monocatenar esters, we have extended our measurements to para-coumaric, caffeic and sinapic esters; the values of IC50 are summarized in Table 8.
With the same carbon chain length, we observed that the monocatenar ester of caffeic acid presents the best antioxidant activity before the sinapic, the coumaric and the ferulic esters, respectively (Table 8, entries 2–5). We can therefore deduce that the number of OH groups on the aromatic ring has a great influence on the antioxidant power, as well as the double bond in the α position of the ester (compared to the phloretic ester, which shows no antioxidant activity). This ranking of antioxidant activities corresponds fairly well to one of the acids alone, except for the derivatives of sinapic acid because it has higher antioxidant activities alone than all the other acids [60]. The length of the carbon chain also seems to play an important role because we can note that the antioxidant powers are higher for a chain in C10 than for a chain in C6 (Table 8, entries 6 and 7 vs. entries 2 and 5), which could be explained by a more important inductive effect of a chain in C10, which stabilizes the radical.
Moreover, the antioxidant activity of dissymmetric bolaforms 1ab and 3ab was evaluated, but the study revealed no antioxidant activity, which is not very surprising considering that the phenolic part of the bolaform is constituted of phoretic acid. The solubility, on the other hand, is enhanced by the presence of the rhamnoside entity. Thus, it would be interesting to develop dissymmetrical bolaforms based on caffeic or sinapic esters and rhamnosides in a future study.
In conclusion, we have five compounds (7b, 6b, 9b, 11b and 15b) that present antioxidant activity superior to that of ascorbic acid (Table 8, entry 1). Furthermore, some of these compounds (7b and 15b) have a very interesting hydrophilic character, which may demonstrate them as good candidates for cosmetic applications.
2.3. Plant-Eliciting Properties
As rhamnolipid- and xylolipid-based bolaforms have previously been characterized as plant defense inducers [40], we evaluated the eliciting properties of our new compounds in the plant model Arabidopsis thaliana. Rhamnolipids bolaforms were found to differentially activate early and late immunity-related plant defense responses, depending on the carbon chain length [40]. In this previous study, the 1′,14′-bis-tetradec-7′-enyl-L-rhamnopyranoside was proven to be the best candidate by combining the right chain length, an unsaturation and rhamnose as sugar.
Here, we aimed to compare the eliciting activities of monocatenar and bolaform rhamnosides (1–10a) to the dissymmetric bolaform 4ab in order to observe a potential synergy between phenolic and rhamnose moieties. To investigate the immune response of compounds 1–10a and 4ab in Arabidopsis thaliana, we monitored the extracellular reactive oxygen species (ROS) production [40].
Our results show that only monocatenar rhamnosides with carbon chains containing 10 or 11 atoms are active on Arabidopsis thaliana, as compounds 5a and 6a triggered significant ROS production in planta (Figure 3).
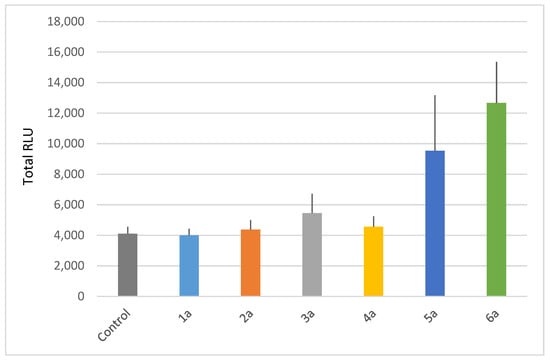
Figure 3.
Plant-eliciting activities of monocatenar rhamnosides. Extracellular reactive oxygen species (ROS) production following treatment of Arabidopsis with C6 to C11 rhamnosides. Production of ROS was measured in leaf disks following treatment with the synthetic glycolipids at 100 µM. MeOH (0.5%) was used as a control. ROS production was measured using the chemiluminescence of luminol, and photon counts were expressed as relative luminescence units (RLUs). Histograms were calculated as the total RLUs over 12 h of monitoring. Data are mean (n = 6). Experiments were independently realized twice with similar results.
We also compared the activity of the unsaturated and saturated compounds, respectively, 5a and 6a towards 5a′ and 6a′ obtained from 5a and 6a through classical Pd-catalyzed hydrogenation [61]. Our results show that the unsaturation of lipid chains leads to a stronger ROS response (Figure 4). Therefore, these data highlight the importance of combined long chains and unsaturation to enhance a monocatenar rhamnoside-triggered immune response in Arabidopsis.
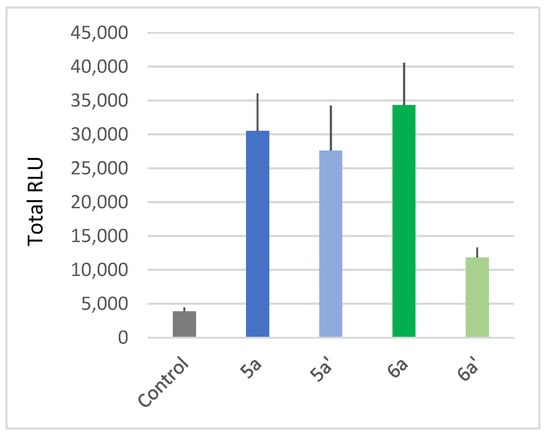
Figure 4.
Plant-eliciting activities comparison between saturated and unsaturated monocatenar rhamnosides. Production of ROS was measured in leaf disks following treatment with the synthetic glycolipids at 100 µM. MeOH (0.5%) was used as a control. ROS production was measured using the chemiluminescence of luminol, and photon counts were expressed as relative luminescence units (RLUs). Histograms were calculated as the total RLUs over 12 h of monitoring. Data are mean ± SEM (n = 6). Experiments were independently realized twice with similar results.
In addition, we found that the dissymmetrical C20 bolaform 4ab induces ROS production close to the level measured for its monocatenar analogue 6a (Figure 5). The symmetrical C20 bolaform 10a was inactive at a concentration of 100 µM. In our previous study, we showed that symmetrical bolaform C18 (9a) was inactive and that the C10 bolaform (7a) was only slightly active at this concentration [40]. This robust immune response obtained with the dissymmetric bolaform may be explained by the presence of the phloretic acid, a relatively close analogue to salicylic acid, a well-known plant signal [62,63].
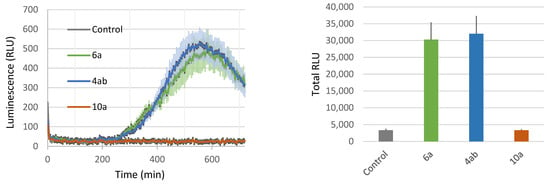
Figure 5.
Plant-eliciting activities comparison between C11 rhamnoside, C20 dissymmetrical and symmetrical bolaforms. Production of ROS was measured in leaf disks following treatment with the synthetic glycolipids at 100 µM. MeOH (0.5%) was used as a control. ROS production was measured using the chemiluminescence of luminol and photon counts were expressed as relative luminescence units (RLUs). Kinetics of production (left panel) and sum of RLU from the same samples (right panel) are shown. Data are mean ± SEM (n = 6). Experiments were independently realized twice with similar results.
Altogether, our results demonstrate that monocatenar rhamnosides 5a and 6a and the dissymmetrical bolaform 4ab can trigger a strong plant defense response in Arabidopsis, and therefore could be promising as plant-elicitor compounds.
2.4. Cytotoxicity Evaluation
As surfactants can be used in cosmetics, we completed our study with cytotoxicity tests on dermal fibroblasts. The toxicity of monocatenar and bolaform rhamnosides was evaluated on human dermal fibroblasts according to the protocol described in the experimental section. A fibroblast model is the best suited to the desired applications in cosmetics, specifically in the field of dermatology; we used the WST1 assay [64] to measure cell viability.
We first incubated dermal fibroblast cells for 48 h in the presence of our rhamnose-based compounds at increasing concentrations (1 to 1000 µg/mL). A dose-response presented by the residual dehydrogenase activity as a function of the surfactant concentration represented in mg/mL allowed us to determine the cytotoxic effect for each compound tested.
In the concentration range tested, we observed a slight cytotoxicity of monocatenar rhamnosides 1–4a; in contrast, 5a (chain length of 10 carbons) showed a lower cell growth of about 46% starting from 50 µg/mL (Figure 6). This cytotoxicity could be due to the hydrophobic character of 5a, which enables it to form micelles in the intracellular medium that can induce a disruption of the cell cycle and causing cell death by either apoptosis or necrosis [65].
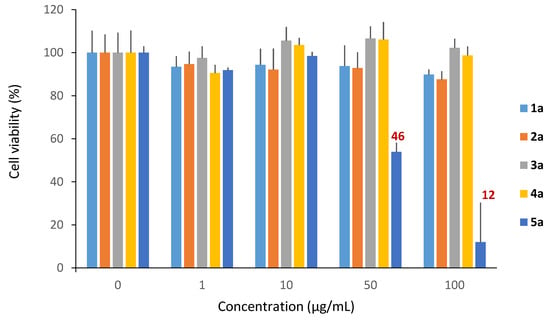
Figure 6.
Cytotoxicity of monocatenar unsaturated rhamnosides.
This low toxicity of rhamnosides is in accordance with the use of rhamnose or rhamnose derivatives, which are known as promising biocompatible molecules for biomedical applications, e.g., in the internalization of nanoparticles with a therapeutic aim [66].
We subsequently studied the cytotoxicity of the unsaturated bolaform rhamnosides 7a and 9a (Figure 7a). As discussed previously, the length of the carbon chain has a strong influence on the cytotoxic character; indeed, the results show that 9a is five times more cytotoxic than 7a (cytotoxicity at 200 µg/mL and 1000 µg/mL, respectively). Nevertheless, when we compared monocatenar rhamnosides and bolaform rhamnosides, i.e., 5a and 9a, the bolaamphiphilic structures clearly decrease the cytotoxicity compared to the monocatenar compounds (Figure 7b).
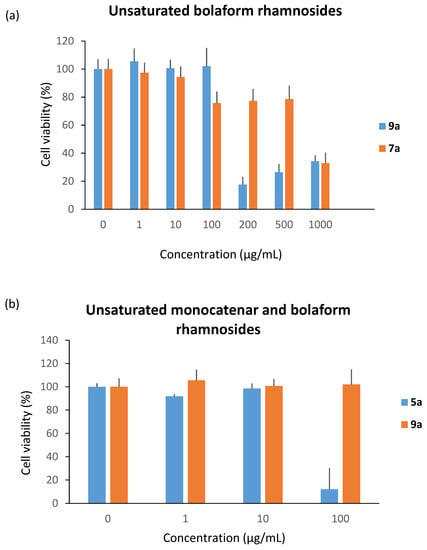
Figure 7.
Cytotoxicity of (a) unsaturated bolaform rhamnosides and (b) monocatenar and bolaform rhamnosides.
Next, the cytotoxicity of unsaturated phloretic esters was evaluated under the same conditions at concentrations between 1 and 1000 µg/mL. In contrast to the monocatenar rhamnosides, all the unsaturated phloretic esters present cytotoxicity starting from 50 µg/mL (Figure 8a). This result can be explained by the intrinsic cytotoxicity of phloretic acid on specific cells, as previously described in the literature [67].
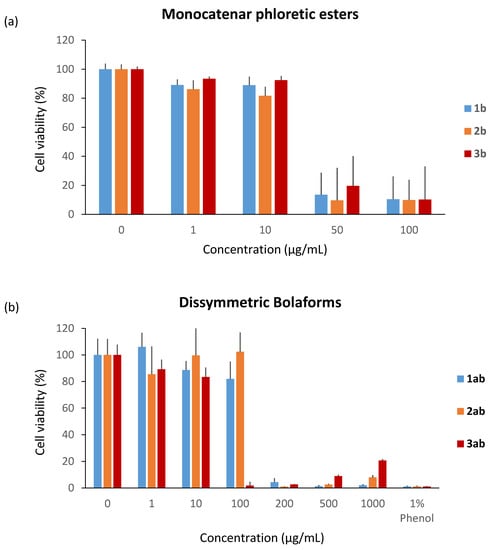
Figure 8.
Cytotoxicity of (a) unsaturated monocatenar phloretic esters and (b) dissymmetric bolaforms.
Regarding the boloform compounds, only the cytotoxicity of the dissymmetrical rhamnose/phloretic ester bolaforms could be evaluated because of the low solubility of the phloretic ester bolaforms in the culture medium. We also observed a decrease of the cytotoxicity when the phloretic esters are coupled with a monocatenar rhamnoside through a cross-metathesis step. In Figure 8b, we can observe low cytotoxicity for compounds 1ab and 2ab at 100 μg/mL, but high cytotoxicity for higher concentrations or for compound 3ab at 100 μg/mL. This cytotoxicity decrease is most likely due to the presence of rhamnose, which is biocompatible with dermal cells and more hydrophilic, improving the solubility of the bolaform.
An in vitro study on dermal cells showed, first, a large difference concerning the cytotoxicity between the monocatenar compounds derived from L-rhamnose and those derived from phloretic acid and, secondly, the importance of the carbon chain length. Moreover, the bolaform structures (always depending on the carbon chain length) contribute to a reduction of the cytotoxic effect on dermal cells (even with bolaforms phloretic esters).
3. Experimental
All the reagents were commercially available and used as received. The solvents were dried and distilled under argon before use (CH2Cl2 over CaCl2 and THF over sodium/benzophenone) and stored over molecular sieves. The NMR spectra were recorded on a spectroscopic apparatus of the Bruker Avance Neo AC type (Wissembourg, France) (500 MHz for 1H, 126 MHz for 13C). The multiplicity of signals is cited according to s: singlet; sl: large singlet; d: doublet; t: triplet; q: quadruplet; m: multiplet; dt: doublet of triplets; dd: doublet of doublets; ddd: doublet of doublets of doublets; and td: triplet of doublets. The coupling constants (J) are expressed in Hertz (Hz). The chemical shifts (in ppm) for the 1H and 13C NMR spectra were referenced to residual protic solvent peaks. Elemental analyses (C, H and N) were carried out on a PerkinElmer 2400 C, H and N element analyzer. The IR spectra of the liquid and solid compounds were recorded on a Bruker Alpha-T FTIR (Wissembourg, France) spectrometer at room temperature. The 1H and 13C NMR spectra were recorded at room temperature with a Bruker AC 500 spectrometer (Wissembourg, France) (500 MHz for 1H, 62.5 MHz for 13C). The specific optical rotation ([α) of rhamnosides and rhamnoside-based boloamphiphiles or fatty ester and rhamnoside-based asymmetric bolaamphiphiles were measured using a polarimeter Anton Paar MCP 5100 (Les Ulis, France) at room temperature. Chromatography was carried out on SDS Silica 60 (40–63 nm), Art 2050044 (flash-chromatography) using the Reveleris® X2 Flash Chromatography System from BUCHI. The microwave oven is a monomode CEM DISCOVERS-CLASS.
3.1. Cytotoxicity and Plant-Eliciting Activities
Normal human dermal fibroblasts were purchased from Promocell (Heidelberg, Germany). They were grown in DMEM supplemented with 10% fetal bovine serum (FBS), according to the manufacturer’s specifications, in Nunclon® 75 cm2 flasks (Dutscher Brumath, France) at 37 °C in a humid atmosphere containing 5% CO2.
For the WST-1 assay, the cells were seeded in sterile 96-well microtiter plates (1 × 10,000 cells/well) and were allowed to settle for 24 h. Effectors were added to the cells at final concentrations ranging from 0 to 1000 µg/mL in DMEM supplemented with 0.5% FBS. The cells were incubated for 48 h and the medium was replaced by fresh medium containing 10% Wst-1 reagent. The absorbance was measured at 450 nm using a microplate spectrophotometer (SPECTROstar® Nano, BMG Labtech, Champigny-sur-Marne, France) and the cell viability was calculated. The results are expressed as the mean ± standard deviation (n = 4). The plant-eliciting properties were monitored through the production of reactive oxygen species (ROS) in planta, as described in [22].
3.2. General Procedure for the Preparation of Rhamnosides under Microwave Activation with Tetrahydrofuran (THF) or 2-Methyltetrahydrofuran (2-MeTHF) as a Solvent
In a microwave tube, a solution of L-rhamnose and unsaturated alcohol in THF or 2-MeTHF (5 mL) was added at 60 °C with p-toluene sulfonic acid (PTSA). The mixture was stirred (600 rpm) under microwave irradiation for 2 h at a power of 60 W and a temperature of 60 °C. After 2 h of reaction, the reaction medium was neutralized with the addition of a 0.5 M MeONa solution (≈26 mL). After evaporation under reduced pressure, the purification of the rhamnosides was realized through a Reveleris® X2 Flash Chromatography System by gradient elution of a CH2Cl2/MeOH mixture (8/2).
- Hex-5′-enyl-α-L-rhamnopyranoside (1a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (2 g; 10.35 mmol; 1 eq.), 5-Hexen-1-ol (5.27 mL; 41.4 mmol; 4 eq.) and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 1a is obtained as a thick yellow liquid with a yield of 64% (with THF) and 63% (with 2-MeTHF).

νmax (ATR) cm−1: 3363 (OH), 2926–2856 (C–H), 1641 (C=C), 1384 (CH3), 1265–1231 (C–OHTert.). [α (589 nm, MeOH) = −52.501. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.25 (3H, d, J 7.5 Hz, H6′), 1.50 (2H, m, H3), 1.63 (2H, m, H2), 2.13 (2H, m, H4), 3.17–3.58 (5H, m, H1, H2′, H3′, H4′, H5′), 4.52–4.72 (3H, s, 3 OH), 4.98 (2H, d, J 7.2 Hz, H6), 5.80 (1H, m, H5). 13C NMR (125 MHz; DMSO-d6, ppm) δ 18.4 (C6′), 25.4 (C3), 29.1, 33.4 (C2, C4), 66.6 (C1), 68.9–73.9 (C2′, C3′, C4′, C5′), 100.6 (C1′), 115.3 (C6), 139.1 (C5). Analysis (%): calculated for: C12H22O5: C 58.52, H 9.02. Found: C 58.97, H 8.88.
- Hept-6′-enyl-α-L-rhamnopyranoside (2a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (0.76 g; 3.93 mmol; 1 eq.), 6-hepten-1-ol (3 mL; 15.72 mmol; 4 eq.) and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 2a is obtained as a thick yellow liquid with a yield of 60%.

νmax (ATR) cm−1: 3368 (OH), 2926–2856 (C–H), 1641 (C=C), 1382 (CH3),1227 (C–OHTert.). [α (589 nm, MeOH) = −49.101. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.13 (3H, d, J 7.5 Hz, H6′), 1.36 (4H, m, H3, H4), 1.50 (2H, m, H2), 2.02 (2H, m, H5), 3.17–3.56 (5H, m, H1, H2′, H3′, H4′, H5′), 4.52–4.73 (3H, s, 3 OH), 4.98 (2H, d, J 7.2 Hz, H7), 5.80 (1H, m, H6). 13C NMR (125 MHz; DMSO-d6, ppm) δ 18.38 (C6′), 25.7 (C4), 28.5, 29.3, 33.6 (C3, C2, C5), 66.8 (C1), 68.9–72.5 (C2′, C3′, C4′, C5′), 100.4 (C1′), 115.2 (C7), 139.2 (C6). Analysis (%): calculated for: C13H24O5: C 59.98, H 9.29. Found: C 59.71, H 9.14.
- Oct-7′-enyl-α-L-rhamnopyranoside (3a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (0.6 g; 3.11 mmol; 1 eq.), 7-octen-1-ol (2.52 mL; 12.4 mmol; 4 eq.) and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 3a is obtained as a thick yellow liquid with a yield of 50%.

νmax (ATR) cm−1: 3371 (OH), 2926–2856 (C–H), 1641 (C=C), 1381 (CH3),1231 (C–OHTert.). [α (589 nm, MeOH) = −46.501. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.13 (3H, d, J 7.5 Hz, H6′), 1.30 (6H, m, (H4, H3, H5), 2.02 (2H, m, H2), 3.18 (2H, m, H6), 3.36–3.58 (5H, m, H1, H2′, H3′, H4′, H5′), 4.53–5.02 (3H, s, 3 OH), 5.18 (2H, d, J 7.2 Hz, H8), 5.80 (1H, m, H7). 13C NMR (125 MHz; DMSO-d6) 18.4 (C6′), 26.0 (C4), 28.7, 28.8 29.5, 33.6 (C3, C5, C2, C6), 66.78 (C1), 68.9–72.5 (C2′, C3′, C4′, C5′), 100.4 (C1), 115.2 (C8), 139.3 (C7). Analysis (%): calculated for: C14H26O5: C 61.29, H 9.55. Found: C 60.83, H 9.41.
- Non-8′-enyl-α-L-rhamnopyranoside (4a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (0.57 g; 2.95 mmol; 1 eq.) and 8-nonen-1-ol (2.5 mL; 11.80 mmol; 4 eq.), and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 4a is obtained as a thick yellow liquid with a yield of 53%.

νmax (ATR) cm−1: 3367 (OH), 2926–2856 (C–H), 1641 (C=C), 1382 (CH3),1230 (C–OHTert.). [α (589 nm, MeOH) = −45.001. 1H NMR (500, 1 MHz; CD3OD) δ 1.13 (3H, d, J 7.5 Hz, H6′), 1.32 (8H, m, H5, H6, H4, H3), 1.50 (2H, m, H2), 2.02 (2H, m, H7), 3.15–3.54 (5H, m, H1, H2′, H3′, H4′, H5′), 4.51–5.06 (3H, s, 3 OH), 5.27 (2H, d, J 7.2 Hz, H9), 5.80 (1H, m, H8). 13C NMR (125 MHz; DMSO-d6) δ 18.4 (C6′), 26.0 (C5), 26.1, 28.8, 29.1, 29.6, 33.6 (C6, C4, C3, C2, C7), 66.8 (C1), 68.8–73.2 (C2′, C3′, C4′, C5′), 100.5 (C1), 115.1 (C9), 139.3 (C8). Analysis (%): calculated for: C15H28O5: C 62.47, H 9.79. Found: C 62.35, H 9.65.
- Dec-9′-enyl-α-L-rhamno-pyranoside (5a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (0.53 g; 2.74 mmol; 1 eq.), 9-decen-1-ol (2.5 mL; 10.96 mmol; 4 eq.) and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 5a is obtained as a thick yellow liquid with a yield of 48% (with THF) and 55% (with 2-MeTHF).

νmax (ATR) cm−1: 3374 (OH), 2926–2856 (C–H), 1641 (C=C), 1382 (CH3),1229 (C–OHTert.). [α (589 nm, MeOH) = −43.701. 1H NMR (500, 1 MHz; CD3OD) δ 1.13 (3H, d, J 7.5 Hz, H6′), 1.32 (10H, m, H5, H6, H4, H7, H3), 1.50 (2H, m, H2), 2.02 (2H, m, H8), 3.15–3.54 (5H, m, H1, H2′, H3′, H4′, H5′), 4.15–4.54 (3H, s, 3 OH), 4.97 (2H, d, J 7.2 Hz, H10), 5.80 (1H, m, H9). 13C NMR (125 MHz; DMSO-d6) δ 18.4 (C6′), 26.1 (C5), 28.7, 28.9, 29.2, 29.3, 29.5, 33.6 (C6, C4, C7, C3, C2, C8), 66.8 (C1), 68.9–73.5 (C2′, C3′, C4′, C5′), 100.4 (C1′), 115.1 (C10), 139.3 (C9). Analysis (%): calculated for: C16H30O5: C 63.55, H 10.03. Found: C 63.16, H 9.82.
- Undec-10′-enyl-α-L-rhamnopyranoside (6a)
The general procedure for the preparation of the rhamnosides under microwave activation with L-rhamnose (0.5 g; 2.59 mmol; 1 eq.), 10-undecen-1-ol (2.5 mL; 10.36 mmol; 4 eq.) and PTSA (1.25 g; 6.21 mmol; 0.6 eq.) is as follows. Compound 6a is obtained as a thick yellow liquid with a yield of 47%.

νmax (ATR) cm−1: 3366 (OH), 2926–2856 (C–H), 1641 (C=C), 1382 (CH3),1229 (C–OHTert.). [α (589 nm, MeOH) = −37.001. 1H NMR (500, 1 MHz; CD3OD) δ 1.13 (3H, d, J 7.5 Hz, H6′), 1.31 (12H, H6, H7, H5, H4, H8, H3), 1.50 (2H, m, H2), 2.01 (2H, m,H9), 3.08–3.54 (5H, m, H1, H2′, H3′, H4′, H5′), 4.52–4.72 (3H, s, 3 OH), 4.97 (2H, d, J 7.2 Hz, H11), 5.80 (1H, m, H10). 13C NMR (125 MHz; DMSO-d6) δ 18.4 (C6′), 26.1 (C6), 28.7, 28.9, 29.2, 29.3, 29.4, 29.5, 33.6 (C5, C7, C4, C8, C3, C2, C9), 66.80 (C1), 68.9–72.5 (C2′, C3′, C4′, C5′),100.6 (C1′),115.1 (C11), 139.3 (C10). Analysis (%): calculated for C17H32O5: C 64.53, H 10.19. Found: C 63.91, H 10.14.
3.3. General Procedure for the Preparation of Rhamnosides under Microwave Activation with γ-Valerolactone as Solvent
The solution of L-rhamnose and unsaturated alcohol (5-Hexen-1-ol or 9-decen-1-ol) in γ-valerolactone was stirred at 60 °C with PTSA (p-toluenesulfonic acid) in a microwave tube. The mixture was irradiated under microwaves for 2 h at a power of 60 W and a temperature of 60 °C. After 2 h, the crude was extracted with diethylether (60 mL) after the addition of a NaCl-saturated solution (50 mL). The organic phase was dried over MgSO4 and evaporated under reduced pressure. The purification of the rhamnosides was then realized through the Reveleris® X2 Flash Chromatography System by gradient elution of a CH2Cl2/MeOH mixture.
Compound 1a was obtained as a thick yellow liquid with a yield of 66% from L-rhamnose (2 g; 10.35 mmol; 1 eq.), 5-Hexen-1-ol (5.27 mL; 41.4 mmol; 4 eq.), 1.25 g of PTSA (6.21 mmol; 0.6 eq.) and γ-valerolactone (5 mL).
Compound 5a was obtained as a thick yellow liquid with a yield of 73% from L-rhamnose (1 g; 5.18 mmol; 1 eq.), 9-decen-1-ol (3.75 mL; 20.7 mmol; 4 eq.), 0.62 g of PTSA (3.11 mmol; 0.6 eq.) and γ-valerolactone (2.5 mL).
3.4. General Procedure for the Preparation of Rhamnoside-Based Boloamphiphiles under Microwave Activation
The rhamnoside was dissolved in a mixture of CH2Cl2/MeOH (8/2 mL) in a microwave tube under argon and the Grubbs II catalyst was added in three portions over the whole reaction time. The mixture was irradiated under microwaves for 40 min at a power of 60 W and a temperature of 60 °C. After 40 min of reaction, the reaction medium was treated with activated charcoal to remove the residual Grubbs catalyst or derivatives and was then filtered through celite. After evaporation, the residue was purified by the Reveleris® X2 Flash Chromatography System with an elution mixture of CH2Cl2/MeOH (9/1) during 45 min.
- 1′,10′-bis-dec-5′-eny-L-rhamnopyranoside (7a)
The general procedure for the preparation of the rhamnoside-based bolaamphiphiles with compound 1a (1.5 g, 6.09 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.5 g, 0.6 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2/MeOH is as follows. Compound 7a is obtained as a brown paste with a yield of 77%.

νmax (ATR) cm−1: 3347 (OH), 2971–2901 (C–H), 1634 (C=C), 1384 (CH3),1128–1048 (C-O-C). [α (589 nm, MeOH) = −34.801. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.24 (6H, d, J 7.5 Hz, H6′), 1.48 (4H, m, H3), 1.61 (4H, m, H2), 2.17(4H, m, H4), 3.56–3.12 (10H, m, H1, H2′, H3′, H4′, H5′), 4.70–4.48 (6H, s, 6 OH), 5.78 (2H, d, J 7.2 Hz, H5). 13C NMR (125 MHz; DMSO-d6, ppm) δ 18.3 (2 C6′), 23.6 (2 C3), 30.1, 33.2 (2 C2, 2 C4), 64.3 (2 C1), 67.6–71.9 (2 C2′, 2 C3′, 2 C4′, 2 C5′), 102.1 (2 C1′), 130.1 (CH2CH(Z)=CH(Z)CH2), 130.6 (CH2CH(E)=CH(E)CH2). Analysis (%): calculated for C22H40O10: C 56.88, H 8.68. Found: C 56.47, H 8.96.
- 1′,12′-bis-dodec-6′-enyl-l-rhamnopyranoside (8a)
The general procedure for the preparation of the rhamnoside-based bolaamphiphiles with compound 2a (0.7 g, 2.69 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.2 g, 0.27 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2/MeOH is as follows. Compound 8a is obtained as a brown paste with a yield of 60%.

νmax (ATR) cm−1: 3342 (OH), 2970–2913 (C–H), 1632 (C=C), 1386 (CH3), 1124–1051 (C–O–C). [α (589 nm, MeOH) = −41.301. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.14 (6H, d, J 7.5 Hz, H6′), 1.38 (8H, m, H3, H4), 1.51 (4H, m, H2), 2.07 (4H, m, H5), 3.36–3.18 (10H, m, H1, H2′, H3′, H4′, H5′), 4.71–4.58 (6H, s, 6 OH), 5.78 (2H, d, J 7.2 Hz, H5). 13C NMR (125 MHz; DMSO-d6, ppm) δ 17.3 (2 C6′), 25.6 (2 C3), 29.0, 29.4, 33.6 (2 C3, 2 C2, 2 C5), 65.1 (2 C1), 68.6–72.6 (2 C2′, 2 C3′, 2 C4′, 2 C5′), 100.2 (2 C1′), 138.0 (CH2CH(Z)=CH(Z)CH2), 138.1 (CH2CH(E)=CH(E)CH2). Analysis (%): calculated for: C24H44O10: C 58.52, H 9.00. Found: C 58.87, H 9.19.
- 1′,18′-bis-octadec-9′-enyl-l-rhamnopyranoside (9a)
The general procedure for the preparation of the rhamnoside-based bolaamphiphiles with compound 5a (1.45 g, 4.80 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.4 g, 0.48 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2/MeOH is as follows. Compound 9a is obtained as a brown paste with a yield of 56%.

νmax (ATR) cm−1: 3345 (OH), 2971–2922 (C–H), 1634 (C=C), 1382 (CH3), 1127–1051 (C–O–C). [α (589 nm, MeOH) = −32.601. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.13 (6H, d, J 7.5 Hz, H6′), 1.34 (20H, m, H3, H4, H5, H6, H7), 1.51 (4H, m, H2), 2.09 (4H, m, H8), 3.16–3.58 (10H, m, H1, H2′, H3′, H4′, H5′), 4.31–4.58 (6H, s, 6 OH), 5.70 (2H, d, J 7.2 Hz, H9). 13C NMR (125 MHz; DMSO-d6, ppm) δ 18.3 (2 C6′), 26.2 (2 C5), 28.6, 28.9, 29.4, 29.7, 33.8 (2 C3, 2 C2, 2 C4, 2 C5, 2 C6, 2 C7, 2 C8), 66.8 (2 C1), 68.7–72.9 (2 C2′, 2 C3′, 2 C4′, 2 C5′), 100.4 (2 C1′), 130.1 (CH2CH(Z)=CH(Z)CH2), 130.5 (CH2CH(E)=CH(E)CH2). Analysis (%): calculated for C30H56O10: C 62.47, H 9.79. Found: C 62.11, H 9.29.
- 1′,20′-bis-eicosa-10′-enyl-l-rhamnopyranoside (10a)
The general procedure for the preparation of the rhamnoside-based bolaamphiphiles with compound 6a (1.09 g, 3.60 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.3 g, 0.36 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2/MeOH is as follows. Compound 10a is obtained as a brown paste with a yield of 32%.

νmax (ATR) cm−1: 3338 (OH), 2968–2919 (C–H), 1632 (C=C), 1381 (CH3), 1124–1053 (C–O–C). [α (589 nm, MeOH) = −35.001. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.18 (6H, d, J 7.5 Hz, H6′), 1.39 (24H, m, H3, H4, H5, H6, H7, H8), 1.57 (4H, m, H2), 2.15 (4H, m, H9), 3.19–3.61 (10H, m, H1, H2′, H3′, H4′, H5′), 4.28–4.47 (6H, s, 6 OH), 5.74 (2H, d, J 7.2 Hz, H10). 13C NMR (125 MHz; DMSO-d6, ppm) δ 17.5 (2 C6′), 24.9 (2 C5), 27.4, 28.8, 29.1, 29.8, 32.5, 35.0 (2 C3, 2 C2, 2 C4, 2 C5, 2 C6, 2 C7, 2 C8, 2 C9), 66.5 (2 C1), 70.1–73.0 (2 C2′, 2 C3′, 2 C4′, 2 C5′), 107.3 (2 C1′), 131.3 (CH2CH(Z)=CH(Z)CH2), 133.1 (CH2CH(E)=CH(E)CH2). Analysis (%): calculated for C32H60O10: C 63.55, H 10.00. Found: C 63.41, H 9.89.
3.5. General Procedure for the Hydrogenation of Monocatenar Unsaturated Rhamnosides
The unsaturated rhamnoside (1 eq.) was dissolved in 4 mL of ethanol under an argon atmosphere. After 10 min of stirring, palladium on activated charcoal (Pd/C, 10% w/w, 0.02 eq.) was added and the solution was stirred for an additional 10 min under an argon atmosphere before being submitted to a H2 flow until completion (24 h at room temperature). Once the reaction was completed, the reaction mixture was filtered through celite. The obtained solution was then evaporated under reduced pressure. The saturated rhamnoside was obtained with quantitative yields.
- Decyl-α-l-rhamno-pyranoside (5a’)
The general procedure for the hydrogenation of monocatenar unsaturated rhamnosides with unsaturated l-rhamnoside 5a (0.4 g; 2.04 mmol; 1 eq.), palladium on activated charcoal (4.6 mg; 0.02 eq.) and 4 mL ethanol is as follows. Compound 5a′ is obtained as a thick yellow liquid with a quantitative yield.

νmax (ATR) cm−1: 3373 (OH), 2928–2854 (C–H), 1381 (CH3), 1227 (C–OHTert). [α (589 nm, MeOH) = −43.501. 1H NMR (500, 1 MHz; CD3OD) δ 0.86 (3H, t, J = 7.5 Hz, H10); 1.14 (3H, d, J 7.5 Hz, H6′), 1.32 (10H, m, H3, H4, H5, H6, H7, H8, H9), 1.51 (2H, m, H2), 3.14–3.56 (5H, m, H1, H2′, H3′, H4′, H5′), 4.12–4.57 (3H, s, 3 OH). 13C NMR (125 MHz; DMSO-d6) δ 14.1 (C10), 18.4 (C6′), 28.7, 28.9, 29.2, 29.3, 29.5, 33.6 (C9, C8, C7, C6, C5, C4, C3), 46.2 (C2), 66.8 (C1), 68.9–73.5 (C2′, C3′, C4′, C5′), 100.4 (C1′). Analysis (%): calculated for: C16H32O5: C 63.13, H 10.60. Found: C 63.32, H 10.72.
- Undecyl-α-L-rhamnopyranoside (6a′)
The general procedure for the hydrogenation of monocatenar unsaturated rhamnosides with unsaturated l-rhamnoside 6a (4 g; 2.34 mmol; 1 eq.), palladium on activated charcoal (4.4 mg; Pd/C 10% w/w; 0.02 eq.) is as follows. Compound 6a’ was obtained as a thick yellow liquid with a quantitative yield.

νmax (ATR) cm−1: 3375 (OH), 2927–2854 (C–H), 1383 (CH3),1228 (C–OHTert). [α (589 nm, MeOH) = −37.03. 1H NMR (500, 1 MHz; CD3OD) δ 0.85 (3H, t, J = 7.5 Hz, H11); 1.17 (3H, d, J 7.5 Hz, H6′), 1.34 (10H, m, H3, H4, H5, H6, H7, H8, H9, H10), 1.54 (2H, m, H2), 3.16–3.67 (5H, m, H1, H2′, H3′, H4′, H5′), 4.14–4.55 (3H, s, 3 OH). 13C NMR (125 MHz; DMSO-d6) δ 13.6 (C11), 18.3 (C6′), 27.5, 28.6, 28.9, 29.7, 30.5, 33.9 (C10, C9, C8, C7, C6, C5, C4, C3), 48.1 (C2), 67.1 (C1), 69.8–75.5 (C2′, C3′, C4′, C5′), 103.4 (C1′). Analysis (%): calculated for: C17H34O5: C 64.12, H 10.76. Found: C 64.25, H 10.64.
3.6. General Procedure for the Preparation of Monocatenar Esters Derived from Phenolic Acids
In a 250 mL flask, bicol was dissolved in phenolic acid (6 eq.) and unsaturated alcohol (1 eq.) in a solvent (2-methyl-2-butanol, or 2-methyl-2-butanol/THF or acetone, depending on the nature of the phenolic acid) under magnetic stirring. The resulting mixture was heated at 60 °C in the presence of 3 Ǻ molecular sieves (50 g·L−1). After adding an appropriate amount of the enzyme (2.5 g per 100 g of reaction mixture), the mixture was stirred for 48 h. Once the reaction was completed, the reaction mixture was filtered through celite to remove the enzyme; the resulting solution was evaporated under reduced pressure and the residue was purified on silica gel by the Reveleris® X2 Flash Chromatography System with elution of a gradient petroleum ether/ethyl acetate.
- Hex-5′-enyl-3-(4-hydroxyphenyl)propionic (1b)
The general procedure for the preparation of the monocatenar ester with 5-hexen-1-ol (0.7 mL; 6.53 mmol; 1 eq.) and 3-(4-hydroxyphenyl)propionic acid (6.50 g; 39.18 mmol; 6 eq.) in 60 mL of 2-methyl-2-butanol is as follows.
Compound 1b was obtained as a clear oil with a yield of 95%.

νmax (ATR) cm−1: 3397 (OH), 2932 (C–H), 1732 (C=O), 1614 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.36 (2H, broad, H3), 1.55 (2H, broad, H4), 2.03 (2H, broad, H2), 2.54 (2H, t, J 7.2 Hz, H2′), 2.77 (2H, t, J 7.2 Hz, H1′), 4.01 (2H, t, J 7.2 Hz, H1), 4.98 (2H, broad, H6), 5.78 (1H, broad, H5), 6.73 (2H, d, J 10 Hz, H4′, H8′), 7.00 (2H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.0 (C3), 28.1 (C4), 30.0 (C2), 33.2 (C2′), 36.0 (C1′), 64.0 (C1),115.4 (C4′, C8′), 115.5 (C5′, C7′), 129.5 (C6), 131.0 (C5), 138.9 (C3′), 156.1 (C6′), 172.8 (C=O). Analysis (%): calculated for C15H20O3: C 72.55, H 8.12. Found: C 72.93, H 8.46.
- Hept-6′-enyl-3-(4-hydroxyphenyl)propionic (2b)
The general procedure for the preparation of the monocatenar ester with 5-hepten-1-ol (0.47 mL; 3.01 mmol; 1 eq.) and 3-(4-hydroxyphenyl)propionic acid (3 g; 18.05 mmol; 6 eq.) in 60 mL of 2-methyl-2-butanol is as follows.
Compound 2b was obtained as a clear oil with a yield of 88%.

νmax (ATR) cm−1: 3392 (OH), 2930 (C–H), 1732 (C=O), 1614 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.29 (2H, m, H4), 1.38 (2H, m, H3), 1.56 (2H, m, H5), 2.03 (2H, m, H2), 2.55 (2H, t, J 7.2 Hz, H2′), 2.74 (2H, t, J 7.2 Hz, H1′), 3.98 (2H, t, J 7.2 Hz, H1), 5.03 (2H, m, H7), 5.83 (1H, m, H6), 6.66 (2H, d, J 10 Hz, H5′, H7′), 7.02 (2H, d, J 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.3 (C4), 28.3 (C3), 28.4 (C5), 30.0 (C2), 33.5 (C2′), 36.0 (C1′), 64.1 (C1),115.2 (C7), 115.5 (C4′, C8′), 129.5 (C5′, C7′), 131.0 (C3′), 139.1 (C6), 156.1 (C6′), 172.8 (C=O). Analysis (%): calculated for C16 H22O3: C 73.25, H 8.45. Found: C 72.93, H 8.24.
- Dec-9′-enyl-3-(4-hydroxyphenyl)propionic (3b)
The general procedure for the preparation of the monocatenar ester with 9-decen-1-ol (1.2 mL; 3.01 mmol; 1 eq.) and 3-(4-hydroxyphenyl)propionic acid (6,51 g; 18.06 mmol; 6 eq.) in 60 mL of 2-methyl-2-butanol is as follows.
Compound 3b was obtained as a clear oil with a yield of 96%.

νmax (ATR) cm−1: 3397 (OH), 2925 (C–H), 1733 (C=O), 1614 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.26 (8H, m, H4, H5, H6, H7), 1.36 (2H, m, H3), 1.55 (2H, m, H8), 2.04 (2H, m, H2), 2.54 (2H, t, J 7.2 Hz, H2′), 2.77 (2H, t, J 7.2 Hz, H1′), 4.00 (2H, t, J 7.2 Hz, H1), 5.00 (2H, m, H10), 5.83 (1H, m, H9), 6.71 (2H, d, J 10 Hz, H5′, H7′), 6.99 (2H, d, J 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.8 (C5), 28.6 (C6), 28.8 (C4), 28.9 (C7), 29.1 (C3), 29.2 (C8), 30.1 (C2), 33.7 (C2′), 36.1 (C1′), 64.2 (C1), 115.0 (C10), 115.5 (C4′, C8′), 129.5 (C5′, C7′), 130.9 (C3′), 139.2 (C9), 156.1 (C6′), 172.7 (C=O). Analysis (%): calculated for C19 H28O3: C 74.96, H 9.27. Found: C 75.13, H 9.03.
- Undec-10′-enyl-3-(4-hydroxyphenyl)propionic (4b)
The general procedure for the preparation of the fatty esters with 36.83 mmol of 3-(4-hydroxyphenyl)propionic acid (6.12 g; 6 eq.) and 6.14 mmol of 10-undecen-1-ol (1.045 g; 1 eq.) is as follow. Compound 4b was obtained as a bright oil with a yield of 79%.

νmax (ATR) cm−1: 3387 (OH), 2931 (C–H), 1732 (C=O), 1613 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.24 (8H, m, H4, H5, H6, H7, H8), 1.34 (2H, m, H3), 1.58 (2H, m, H9), 2.07 (2H, m, H2), 2.51 (2H, t, J 7.2 Hz, H2′), 2.74 (2H, t, J 7.2 Hz, C1′), 3.97 (2H, t, J 7.2 Hz, H1), 5.21 (2H, m, C11), 5.81 (1H, m, H10), 6.65 (2H, d, J 10 Hz, H5′, H7′), 7.09 (2H, d, J 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 24.8 (C5), 28.1 (C6), 28.5 (C4), 28.8 (C7), 29.1 (C3), 29.2 (C8), 30.0 (C9), 30.1 (C2), 33.5 (C2′), 35.3 (C1′), 65.1 (C1), 114.0 (C11), 115.6 (C4′, C8′), 130.0 (C5′, C7′), 130.7 (C3′), 140.0 (C10), 154.6 (C6′), 172.1 (C=O). Analysis (%): calculated for C20 H30O3: C 75.43, H 9.50. Found: C 75.37, H 9.49.
- Hex-5′-enyl-3-(4-hydroxyphenyl)prop-2-enoic (5b)
The general procedure for the preparation of the monocatenar ester with 5-hexen-1-ol (0.36 mL; 3.04 mmol; 1 eq.) and p-coumaric acid (3.28 g; 18.21 mmol; 6 eq.) in 40 mL of 2-methyl-2-butanol is as follows. Compound 5b was obtained as a pale yellow oil with a yield of 70%.

νmax (ATR) cm−1: 3397 (OH), 2932 (C–H), 1732 (C=O), 1614 (–CH=CH2–), 1668 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.51 (2H, m, H3), 1.75 (2H, m, H4), 2.15 (2H, m, H2), 4.25 (2H, t, J 7.2 Hz, H1), 5.01 (2H, m, H6), 5.80 (1H, m, H5), 6.35 (1H, d, J 7.2 Hz, H1′), 6.80 (2H, d, J 7.2 Hz, H7′, H5′), 7.40 (1H, d, J 7.2 Hz, H1′), 7.65 (2H, d, J 7.2 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 23.2 (C3), 25.2 (C4), 34.2 (C2), 68.3 (C1), 114.2 (C6), 115.0 (C1′), 127.3 (C5), 130.1 (C7′, C5′), 140.1 (C8′, C4′), 145.0 (C3′), 158.0 (C6′), 168.4 (C=O). Analysis (%): calculated for C15H18O3: C 73.15, H 7.37. Found: C 72.77, H 7.66.
- Dec-9′-enyl-3-(4-hydroxyphenyl)prop-2-enoïc (6b)
The general procedure for the preparation of the monocatenar ester with 9-decen-1-ol (0.57 mL; 3.2 mmol; 1 eq.) and para-coumaric acid (3.15 g; 19.2 mmol; 6 eq.) in 40 mL of 2-methyl-2-butanol is as follows. Compound 6b was obtained as a yellow oil with a yield of 74%.

νmax (ATR) cm−1: 3397 (OH), 2932 (C–H), 1732 (C=O), 1612 (–CH=CH2–), 1665 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 11.22 (8H, m, H4, H5, H6, H7), 1.70 (2H, m, H3), 2.10 (2H, m, H8), 3.75 (2H, m, H2), 4.20 (2H, t, J 7.2 Hz, H1), 5.01 (2H, d, J 7.2 Hz, H10), 5.81 (1H, m, H9), 6.32 (1H, d, J 7.2 Hz, H1′), 6.80 (2H, d, J 10 Hz, H7′, H5′), 7.43 (1H, d, J 7.2 Hz, H2′), 7.65 (2H, d, J 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.1 (C5), 27.5 (C6), 28.4 (C4), 28.9 (C7), 29.1 (C3), 29.5 (C8), 34.0 (C2), 77.2 (C1), 114.1 (C10), 115.2 (C1′), 116.0 (C9), 127.1 (C5′, C7′), 130.3 (C4′, C8′), 140.2 (C9), 145.1 (C2′), 158.7 (C6′), 168.9 (C=O). Analysis (%): calculated for C19 H26O3: C 75.46, H 8.67. Found: C 75.67, H 9.21.
- Hex-5′-enyl-3-(3,5-dihydroxyphenyl)prop-2-enoic (7b)
The general procedure for the preparation of the monocatenar ester with 5-hexen-1-ol (0.36 mL; 3.04 mmol; 1 eq.) and caffeic acid (3.28 g; 18.24 mmol; 6 eq.) in 30 mL of 2-methyl-2-butanol/THF (10/20) is as follows. Compound 7b was obtained as a pale yellow oil with a yield of 52%.

νmax (ATR) cm−1: 3390 (OH), 2930 (C–H), 1734 (C=O), 1615 (–CH=CH2–), 1664 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.59 (2H, m, H3), 1.61 (2H, m, H2), 2.21 (2H, m, H4), 4.22 (2H, t, J 7.2 Hz, H1), 5.05 (2H, d, J 7.2 Hz, H6), 5.12 (1H, d, J 7.2 Hz, H1′), 5.85 (1H, m, H5), 6.36 (2H, d, J 7.2 Hz, H2′), 6.79 (1H, d, J 7.2 Hz, H8′), 6.94 (1H, d, J 7.2 Hz, H7′), 7.18 (1H, s, C4′), 7.50 (1H, d, J 7.2 Hz, C2′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 24.9 (C3), 25.4 (C4), 33.9 (C2), 65.1 (C1), 115.2 (C6), 116.4 (C1′), 117.1 (C7′), 122.2 (C8′), 128.1 (C3′), 139.0 (C5), 144.2 (C4′), 147.2 (C5′, C6′), 167.4 (C=O). Analysis (%): calculated for C15H18O4: C 68.69, H 6.92. Found: C 68.24, H 7.98.
- Dec-9′-enyl-3-(4-hydroxyphenyl)prop-2-enoic (8b)
The general procedure for the preparation of the monocatenar ester with 9-decen-1-ol (0.74 mL; 3.7 mmol; 1 eq.) and caffeic acid (4 g; 19.20 mmol; 6 eq.) in 30 mL of 2-methyl-2-butanol/THF (10/20) is as follows. Compound 8b was obtained as a pale yellow oil with a yield of 45%.

νmax (ATR) cm−1: 3375(OH), 2926 (C–H), 1731 (C=O), 1614 (–CH=CH2–), 1661 (Phenyl-CH=CH–COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.28 (8H, m, H4, H5, H6, H7), 1.44 (2H, m, H3), 1.62 (2H, m, H2), 2.18 (2H, m, H8), 3.99 (2H, t, J 7.2 Hz, H1), 5.02 (2H, d, J 7.2 Hz, H10), 5.17 (1H, d, J 7.2 Hz, H1′), 5.83 (1H, m, H9), 6.30 (1H, d, J 10 Hz, H8′), 6.79 (1H, d, J 7.2 Hz, H7′), 6.95 (1H, d, J 7.2 Hz, H4′), 7.49 (1H, d, J 7.2 Hz, H2′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.52 (C5), 29.32, 29.57, 31.16, 32.48 (C6, C4, C7, C3), 32.97 (C2), 33.93 (C8), 65.31 (C1), 115.25 (C10), 116.48 (C1′), 117.16 (C3′), 122.12 (C8′), 128.17 (C7′), 129.18 (C4′), 139.13 (C9), 144.95 (C1′), 147.23 (C5′, C6′), 167.13 (C=O). Analysis (%): calculated for C19H26O4: C 71.67, H 8.23. Found: C 71.91, H 8.66.
- Hex-5′-enyl-3-(3,5-dimethoxy-4-hydroxyphenyl)prop-2-enoic (9b)
The general procedure for the preparation of the monocatenar ester with 5-hexen-1-ol (1 mL; 0.74 mmol; 1 eq.) and sinapic acid (1 g; 4.44 mmol; 6 eq.) in 30 mL of acetone is as follows. Compound 9b was obtained as an orange-yellow oil with a yield of 40%.

νmax (ATR) cm−1: 3392 (OH), 2934 (C–H), 1732 (C=O), 1612 (–CH=CH2–), 1665 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 0.58 (2H, m, H3), 1.62 (2H, m, H2), 2.19 (2H, m, H4), 3.83 (6H, t, J 7.2 Hz, (CH3-O)2-Phenyl), 3.99 (2H, d, J 7.2 Hz, H1), 5.12 (1 H, d, J 7.2 Hz, H6), 5.82 (1H, m, H1′), 6.32 (1H, d, J 7.2 Hz, H5), 6.72 (2H, s, H4′, H8′), 7.48 (1H, d, J 7.2 Hz, H2′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.2 (C3), 25.6 (C2), 33.7 (C4), 56.3 ((CH3-O)2-Phenyl), 65.1 (C1), 108.2 (C6), 115.2 (C1′), 116.1 (C3′), 126.1 (C4′, C8′), 136.0 (C6′),139.2 (C5), 146.3 (C2′), 148.1 (C5′, C7′), 167.9 (C=O). Analysis (%): calculated for C17H22O5: C 66.65, H 7.24. Found: C 66.39, H 7.62.
- Hex-5′-enyl-3-(4-hydroxy-3-méthoxyphényl)prop-2-enoïc (10b)
The general procedure for the preparation of the monocatenar ester with 5-hexen-1-ol (0.7 mL; 6.53 mmol; 1 eq.) and ferulic acid or 3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic (7.61 mg; 39.2 mmol; 6 eq.) in 60 mL of 2-methyl-2-butanol is as follows. Compound 10b was obtained as a clear oil with a yield of 52%.

νmax (ATR) cm−1: 3392 (OH), 2927 (C–H), 1713 (C=O), 1659–1640 (CH=CH2), 1688–1664 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.48 (2H, m, H3), 1.67 (2H, m, H4), 2.09 (2H, m, H2), 3.82 (3H, s, CH3-O-Phenyl), 4.14 (2H, t, J 7.2 Hz, H1), 5.05 (2H, m, H6), 5.86 (1H, m, H5), 6.49 (1H, d, J 7.2 Hz, H7′), 6.80 (1H, d, J 7.2 Hz, H1′), 7.13 (1H, d, J 7.2 Hz, H2′), 7.33 (1H, s, H4′), 7.56 (1H, d, J 7.2, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.2 (C3), 28.3 (C4), 33.2 (C2), 56.2 (CH3-O-Phenyl), 64.0 (C1), 111.6 (C7′), 114.9 (C1′), 115.5 (C6), 115.9 (C4′), 123.6 (C2′), 126.0 (C3′), 138.9 (C5), 145.4 (C8′), 149.8 (C6′), 148.4 (C5′), 167.2 (C=O). Analysis (%): calculated for C16H20O4: C 69.55, H 7.30. Found: C 69.73, H 7.52.
- Dec-9′-enyl-3-enyl-3-(4-hydroxy-3-méthoxyphényl)prop-2-enoïc (11b)
The general procedure for the preparation of the monocatenar ester with 9-decen-1-ol (1.2 mL; 3.01 mmol; 1 eq.) and 3-(4-hydroxy-3-methoxyphenyl)prop-2-enoic acid (3.51 mg; 18.06 mmol; 6 eq.) in 60 mL of 2-methyl-2-butanol is as follows. Compound 11b was obtained as a clear oil with a yield of 52%.

νmax (ATR) cm−1: 3394 (OH), 2926 (C–H), 1724 (C=O), 1660–1640 (CH=CH2), 1690–1664 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.33 (8H, m, H4, H5, H6, H7), 1.55 (2H, m, H3), 1.63 (2H, m, H8), 2.02 (2H, m, H2), 3.82 (3H, s, CH3-O-Phenyl), 4.12 (2H, t, J 7.2 Hz, H1), 4.99 (2H, m, H10), 5.82 (1H, m, H9), 6.47 (1H, d, J 7.2 Hz, H7′), 6.80 (1H, d, J 7.2 Hz, H1′), 7.11 (1H, d, J 7.2 Hz, H2′), 7.31 (1H, s, H4′), 7.56 (1H, d, J 7.2, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 28.9 (C5), 28.9 (C6), 29.0 (C4), 29.1 (C7), 29.2 (C3), 29.3 (C8), 33.6 (C2), 56.1 (CH3-O-Phenyl), 64.2 (C1), 111.5 (C7′), 114.9 (C1′), 115.0 (C6), 115.9 (C4′), 123.5 (C2′), 126.0 (C3′), 139.2 (C5), 145.4 (C8′), 148.4 (C6′), 149.8 (C5′), 167.1 (C=O). Analysis (%): calculated for C20H28O4: C 72.26, H 8.49. Found: C 71.92, H 8.64.
3.7. General Procedure for the Preparation of the Fatty Ester-Based Bolaamphiphiles under Microwave Activation
In a microwave tube, the fatty ester in CH2Cl2 (10 mL) was dissolved under argon and the Grubbs II catalyst was then added in three portions over the whole reaction time. The mixture was irradiated for 40 min at a power of 60 W and a temperature of 60 °C. After 40 min of reaction, the reaction medium was treated with activated charcoal to remove the Grubbs catalyst and filtered through celite. After evaporation of the solvent under reduced pressure, the residue was purified by a flash chromatography system coupled with a UV detector with elution mixture CH2Cl2/MeOH (9/1) over 35 min.
- 1′,10′-bis-dec-5′-enyl-3-(4-hydroxyphenyl)propionic ester (12b)
The general procedure for the preparation of the fatty ester-based bolaamphiphiles with compound 1b (1 g, 4.03 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.34 g, 0.403 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2 is as follows. Compound 12b is obtained as a brown paste with a yield of 56%.

νmax (ATR) cm−1: 3385 (OH), 2927–2855 (C–H), 1730 (C=O), 1614 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.27–1.36 (12H, m, H3, H4, H2), 2.51 (4H, t, J 7.2 Hz, H2′), 2.74 (4H, t, J 7.2 Hz, H1′), 3.98 (4H, t, J 7.2 Hz, H1), 5.74 (2H, d, J 7.2 Hz, H5), 6.71 (4H, d, J 10 Hz, H4′, H8′), 6.96 (4H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.9 (2 C3), 28.2 (2 C4), 30.1 (2 C2), 33.4 (2 C2′), 36.4 (2 C1′), 64.2 (2 C1),116.2 (2 C4′, 2 C8′), 129.5 (2 C5′, 2 C7′), 130.1 (2 C3′), 130.5 (CH2CH(Z)=CH(Z)CH2), 130.9 (CH2CH(E)=CH(E)CH2), 156.1 (2 C6′), 172.8 (C=O). Analysis (%): calculated for: C28H36O6: C 71.77, H 7.74. Found: C 72.04, H 7.44%.
- 1′,12′-bis-dodec-6′-enyl-3-(4-hydroxyphenyl)propionic ester (13b)
The general procedure for the preparation of the fatty ester-based bolaamphiphiles with compound 2b (0.45 g, 1.72 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.15 g, 0.172 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2 is as follows. Compound 13b is obtained as a brown paste with a yield of 48%.

νmax (ATR) cm−1: 3374 (OH), 2923–2835 (C–H), 1732 (C=O), 1621 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.28–1.32 (12H, m, H3, H4, H5, H2), 2.53 (4H, t, J 7.2 Hz, H2′), 2.72 (4H, t, J 7.2 Hz, H1′), 4.08 (4H, t, J 7.2 Hz, H1), 5.84 (2H, d, J 7.2 Hz, H6), 6.73 (4H, d, J 10 Hz, H4′, H8′), 6.92 (4H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 25.9 (2C3), 28.3 (2 C4), 28.6 (2 C5), 32.2 (2 C2), 33.58 (2 C2′), 36.04 (2 C1′), 64.25 (2 C1),115.18 (2 C4′, 2 C8′), 129.4 (2 C5′, 2 C7′), 130.2 (2C3′), 130.5 (CH2CH(Z)=CH(Z)CH2), 130.9 (CH2CH(E)=CH(E)CH2), 156.2 (2 C6′), 173.0 (C=O). Analysis (%): calculated for C30H40O6: C 72.55, H 8.12. Found: C 72.06, H 7.93.
- 1′,18′-bis-octadec-9′-enyl-3-(4-hydroxyphenyl)propionic ester (14b)
The general procedure for the preparation of the fatty ester-based bolaamphiphiles with compound 3b (1 g, 3.29 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.28 g, 0.329 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2 is as follows. Compound 14b is obtained as a brown paste with a yield of 60%.

νmax (ATR) cm−1: 3383 (OH), 2927–2854 (C–H), 1731 (C=O), 1614 (C=C). 1H NMR (500 MHz; CD3OD, ppm) δ 1.29 (20H, m, H3, H4, H5, H6, H7), 1.51 (4H, m, J 7.2 Hz, H2), 1.94 (4H, t, J 7.2 Hz, H8), 2.53 (4H, t, J 7.2 Hz, H1′), 2.72 (4H, t, J 7.2 Hz, H2′), 3.97 (4H, t, J 10 Hz, H1), 5.36 (2H, t, J 7.2 Hz, H9), 6.65 (4H, d, J 10 Hz, H5′, H7′), 6.90 (4H, d, J 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 28.9 (2 C5), 29.1 (2 C6), 29.2 (2 C4), 29.4 (2 C7), 29.4 (2 C3), 30.0 (2 C8), 32.4 (2 C2), 32.4 (2 C2′), 36.0 (2 C1′), 64.2 (2 C1), 115.5 (2 C4′, 2 C8′), 129.5 (2 C5′, 2 C7′), 130.1 (2 C3′), 130.5 (CH2CH(Z)=CH(Z)CH2), 130.9 (CH2CH(E)=CH(E)CH2), 156.1 (C6′), 172.8 (C=O). Analysis (%): calculated for C36H52O6: C 74.45, H 9.02. Found: C 74.07, H 8.78.
- 1′,10′-bis-dec-5′-enyl-3-(4-hydroxyphenyl)prop-2-enoic (15b)
The general procedure for the preparation of the fatty ester-based bolaamphiphiles with compound 4b (0.1 g, 4.06 mmol, 1 eq.) under microwave activation with Grubbs II catalyst (0.35 g, 0.406 mmol; 0.1 eq.) dissolved in 10 mL CH2Cl2 is as follows. Compound 14b is obtained as a brown paste with a yield of 42%.

νmax (ATR) cm−1: 3395 (OH), 2937 (C–H), 1731 (C=O), 1614 (C=C), 1668 (Phenyl-CH=CH-COO). 1H NMR (500 MHz; CD3OD, ppm) δ 1.25–1.51 (12H, m, H3, H4, H2), 4.25 (4H, t, J 7.2 Hz, H1), 5.48 (2H, t, J 7.2 Hz, H5), 6.30 (2H, d, J 7.2 Hz, H2′), 6.81 (4H, d, J 10 Hz, H5′, H7′), 7.30 (2H, d, J 7.2 Hz, H1′), 7.74 (4, d, 10 Hz, H4′, H8′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 24.1 (2 C3), 25.1 (2 C4), 31.3 (2 C2), 32.2 (2 C2′), 65.3 (2 C1), 114.1 (2 C2′), 1115.0 (2 C5′, 2 C7′), 127.2 (2 C3′), 129.6 (2 C4′, 2 C8′), 130.1 (CH2CH(Z)=CH(Z)CH2), 130.3 (CH2CH(E)=CH(E)CH2), 145.4 (2 C6′), 158.2 (2 C1′), 170.5 (C=O). Analysis (%): calculated for C28H32O6: C 72.39, H 6.94. Found: C 72.82, H 7.21.
3.8. General Procedure for the Preparation of the Unsymmetrical Fatty Ester- and Rhamnoside-Based Bolaamphiphiles by Classic Heating
The rhamnoside-based bolaamphiphile and the fatty ester were dissolved in CH2Cl2 (35 mL)/MeOH (5 mL) in a Schlenk tube under argon and the Grubbs II catalyst was added in three portions over the whole reaction time. The mixture was irradiated at a temperature of 45 °C. After 24 h of reaction, the reaction medium was treated with activated charcoal to remove the Grubbs catalyst and filtered through celite. After evaporation, the residue was purified by a flash chromatography system coupled with a UV detector with elution mixture CH2Cl2/MeOH (9/1) over 45 min.
- 1′,10′-bis-dec-5′-enyl-3-(4-hydroxyphenyl)propionicacid–α–L-rhamnopyranoside (1ab)
The general procedure for the preparation of the unsymmetrical fatty ester- and rhamnoside-based bolaamphiphiles with compound 6a (1 g, 2.15 mmol, 2 eq.) and compound 1b (0.267 g, 1.080 mmol) under argon with Grubbs II catalyst (0.09 g, 0.108 mmol; 0.05 eq.) dissolved in 40 mL CH2Cl2/MeOH (35/5) is as follows. Compound 1ab is obtained as a brown paste with a yield of 65%.

νmax (ATR) cm−1: 3353 (OH), 2928 (C–H), 1732 (C=O), 1615 (C=C). [α]20D (589 nm, MeOH) = −26.601. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.27 (3H, d, J 7.5 Hz, H6′-ose), 1.41 (4H, m, H3), 1.83 (4H, m, H2), 2.15 (4H, m, H4), 2.53 (2H, t, J 7.2 Hz, H2′-acid), 2.72 (2H, t, J 7.2 Hz, H1′-acid), 3.12–3.54 (5H, m, H1′-ose, C2′-ose, C3′-ose, C4′-ose, C5′-ose), 4.01 (2H, t, J 7.2 Hz, H1-acid), 4.52–4.72 (3H, s, 3 OH-ose), 5.76 (2H, t, J 7.2 Hz, 2 H5-C=C-), 6.71 (2H, d, J 10 Hz, H4′, H8′), 6.98 (2H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 17.7 (C6′-ose), 25.8 (2 C3), 28.6, 31.7 (2 C2, 2 C4), 32.9 (C2′-acid), 36.1 (C1′-acid), 63.9 (C1-acid), 66.3 (C1-ose), 68.7–74.1 (C2′-ose, C3′-ose, C4′-ose, C5′-ose), 100.4 (C1′-ose), 115.4 (C4′-acid, C8′-acid), 115.5 (C5′-acid, C7′-acid), 131.0 (CH2CH(Z)=CH(Z)CH2), 131.5 (CH2CH(E)=CH(E)CH2), 136.3 (C3′), 156.1 (C6′), 172.8 (C=O). Analysis (%): calculated for C25H38O8: C 64.36, H 8.21. Found: C 64.09, H 8.57.
- 1′,12′-bis-dodec-6′-enyl-3-(4-hydroxyphenyl)propionicacid–α–l-rhamnopyranoside (2ab)
The general procedure for the preparation of the unsymmetrical fatty ester- and rhamnoside-based bolaamphiphiles with compound 6a ((0,20 g, 0,4 mmol, 2 éq.)) and compound 2b (0.053 g, 0.2 mmol, 1 eq.) under argon with Grubbs II catalyst (0.017 g, 0.02 mmol; 0.05 eq.) dissolved in 40 mL CH2Cl2/MeOH (35/5) is as follows. Compound 1ab is obtained as a brown paste with a yield of 42%.

νmax (ATR) cm−1: 3351 (OH), 2923 (C–H), 1731 (C=O), 1614 (C=C). [α (589 nm, MeOH) = −16.900. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.16 (3H, d, J 7.5 Hz, H6′-ose), 1.28 (4H, m, H4) 1.43 (4H, m, H3), 1.80 (4H, m, H2), 2.12 (4H, m, H4), 2.56 (2H, t, J 7.2 Hz, H2′-acid), 2.70 (2H, t, J 7.2 Hz, H1′-acid), 3.14–3.52 (5H, m, H1-ose, H2′-ose, H3′-ose, H4′-ose, H5′-ose), 4.13 (2H, t, J 7.2 Hz, H1-acid), 4.42–4.82 (3H, s, 3 OH-ose), 5.74 (2H, t, J 7.2 Hz, H5-C=C-), 6.67 (2H, d, J 10 Hz, H4′, H8′), 7.08 (2H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 17.3 (C6′-ose), 25.4 (2 C4), 25.7 (2 C3), 28.6, 31.7 (2 C2, 2 C4), 32.8 (C2′-acid), 36.2 (C1′-acid), 63.9 (C1-acid), 66.1 (C1-ose), 68.5–74.1 (C2′-ose, C3′-ose, C4′-ose, C5′-ose), 100.6 (C1′-ose), 115.1 (C4′-acid, C8′-acid), 114.3 (C5′-acid, C7′-acid), 130.9 (CH2CH(Z)=CH(Z)CH2), 131.6 (CH2CH(E)=CH(E)CH2), 136.3 (C3′), 156.4 (C6′), 172.8 (C=O). Analysis (%): calculated for C27H42O8: C 65.56, H 8.56. Found: C 65.80, H 8.74.
- 1′,18′-bis-dodec-9′-enyl-3-(4-hydroxyphenyl)propionicacid–α–l-rhamnopyranoside (3ab)
The general procedure for the preparation of the unsymmetrical fatty ester- and rhamnoside-based bolaamphiphiles with compound 8a (1 g, 1.73 mmol, 2 eq.) and compound 3b (0.264 g, 0.867 mmol) under argon with Grubbs II catalyst (0.074 g, 0.086 mmol; 0.05 eq.) dissolved in 40 mL CH2Cl2/MeOH (35/5) is as follows. Compound 3ab is obtained as a brown paste with a yield of 68%.

νmax (ATR) cm−1: 3353 (OH), 2925 (C–H), 1732 (C=O), 1614 (C=C). [α (589 nm, MeOH) = −32.601. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.12 (3H, d, J 7.5 Hz, H6′-ose), 1.34 (20H, m, H3, H4, H5, H6, H7), 1.52 (4H, m, H2), 2.03 (4H, m, H8), 2.51 (2H, t, J 7.2 Hz, H2′-acid), 2.74 (2H, t, J 7.2 Hz, H1′-acid), 3.18–3.47 (5H, m, H1-ose, H2′-ose, H3′-ose, H4′-ose, H5′-ose), 4.06 (2H, t, J 7.2 Hz, H1-acide), 4.13–4.51 (3H, s, 3 OH-ose), 5.81 (2H, t, J 7.2 Hz, H9-C=C-), 6.72 (2H, d, J 10 Hz, H4′, H8′), 7.02 (2H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 18.3 (C6′-ose), 26.1, 28.7, 28.9, 29.2, 29.4 (2 C3, 2 C4, 2 C5, 2 C6, 2 C7), 29.4 (2 C2), 31.8 (2 C8), 33.4 (C2′-acid), 36.2 (C1′-acid), 64.2 (C1-acid), 66.8 (C1-ose), 68.8–73.5 (C2′-ose, C3′-ose, C4′-ose, C5′-ose), 100.7 (C1′-ose), 115.5 (C4′-acid, C8′-acid), 128.3 (C5′-acid, C7′-acid), 130.7 (CH2CH(Z)=CH(Z)CH2), 131.1 (CH2CH(E)=CH(E)CH2), 138.5 (C3′-acid),156.2 (C6′), 171.8 (C=O). Analysis (%): calculated for: C33 H54O8: C 68.48, H 9.40. Found: C 67.98, H 9.78.
- 1′,20′-bis-eicosa-10′-enyl-3-(4-hydroxyphenyl)propionicacid–α–l-rhamnopyranoside (4ab)
The general procedure for the preparation of the unsymmetrical fatty ester- and rhamnoside-based bolaamphiphiles with compound 10a (0.5 g, 0.872 mmol, 2 eq.) and compound 4b (0.132 g, 0.437 mmol, 1 eq.) under argon with Grubbs II catalyst (0.017 g, 0.019 mmol; 0.05 eq.) dissolved in 40 mL CH2Cl2/MeOH (35/5) is as follows.
Compound 4ab is obtained as a brown paste with a yield of 46%.

νmax (ATR) cm−1: 3350 (OH), 2921 (C–H), 1729 (C=O), 1612 (C=C). [α (589 nm, MeOH) = −20,300. 1H NMR (500 MHz; DMSO-d6, ppm) δ 1.15 (3H, d, J 7.5 Hz, H6′-ose), 1.32 (24H, m, H3, H4, H5, H6, H7, H8), 1.50 (4H, m, H2), 2.31 (4H, m, H9), 2.58 (2H, t, J 7.2 Hz, H2′-acid), 2.73 (2H, t, J 7.2 Hz, H1′-acid), 3.14–3.52 (5H, m, H1-ose, H2′-ose, H3′-ose, H4′-ose, H5′-ose), 4.09 (2H, t, J 7.2 Hz, H1-acide), 4.15–4.73 (3H, s, 3 OH-ose), 5.95 (2H, t, J 7.2 Hz, H10-C=C-), 6.74 (2H, d, J 10 Hz, H4′, H8′), 7.08 (2H, d, J 10 Hz, H5′, H7′). 13C NMR (125 MHz; DMSO-d6, ppm) δ 17.5 (C6′-ose), 24.8, 27.6, 28.1, 28.9, 29.2, 29.3 (2 C3, 2 C4, 2 C5, 2 C6, 2 C7, 2 C8), 29.7 (2 C2), 32.1 (2 C9), 34.8 (C2′-acid), 36.1 (C1′-acid), 64.5 (C1-acid), 67.0 (C1-ose), 68.1–74.0 (C2′-ose, C3′-ose, C4′-ose, C5′-ose), 105.7 (C1′-ose), 115.0 (C4′-acid, C8′-acid), 129.2 (C5′-acid, C7′-acid), 130.6 (CH2CH(Z)=CH(Z)CH2), 131.1 (CH2CH(E)=CH(E)CH2), 137.7 (C3′-acid),154.5 (C6′), 171.8 (C=O). Analysis (%): calculated for: C35 H58O8: C 69.27, H 9.63. Found: C 69.38, H 9.71.
4. Conclusions
In this study, we developed greener syntheses of six monocatenar and five bolaform rhamnosides, respecting the fundamental principles of green chemistry, which, when coupled with a CombiFlash purification technique, allowed the obtention of compounds with good to very good yields. We have also prepared 10 phenolic acid esters by an esterification reaction, as well as four corresponding bolaforms via a cross-metathesis reaction with good yields. Four dissymmetric bolaforms derived from phloretic esters and rhamnosides have been also easily prepared.
Two monocatenar rhamnosides, as well as a dissymmetrical bolaform (phloretic ester/rhamnoside) trigger a plant defense response in Arabidopsis. Phenolic acid esters have shown good antioxidant activities and we observed that the levels of cytotoxicity depend on the carbon chains we used. The transformation into corresponding bolaforms or into dissymmetric bolaforms associated with rhamnosides significantly decreases their toxicity while retaining their antioxidant properties. These aspects can therefore augur a potential use of some of these compounds in the field of cosmetics. An in-depth study of the mechanisms involved in planta and protection assays will, however, be needed prior to determining the potential use of these compounds in plant protection and pest management strategies. Biophysical studies on their interaction with plant and skin biomimetic plasma membranes would also help to better understand their mode of action at the cell level. An in vitro study on dermal cells proved that L-rhamnose derivatives are less cytotoxic than those from phloretic acid, and that the carbon chain length plays an important role. Moreover, bolaform structures issued from rhamnosides and phloretic esters are less toxic. Further tests on other cell models will be carried out in order to use these compounds in different fields of application.
Author Contributions
Conceptualization, S.B., S.D. and S.B.-P.; methodology, E.K., K.B., S.V., S.B., J.-P.M., S.D., S.B.-P. and J.-C.M.; validation, S.B., J.-P.M., S.D. and S.B.-P.; formal analysis, E.K., K.B., S.V., S.B., J.-P.M., S.D., S.B.-P. and J.-C.M.; validation, S.B., J.-P.M., S.D. and S.B.-P.; investigation, E.K., K.B., S.V., J.-P.M., S.B.-P. and J.-C.M.; validation, S.B., J.-P.M., S.D. and S.B.-P.; writing—original draft preparation, E.K. and J.-P.M.; writing—review and editing, S.D., S.B.-P. and S.B.; supervision, S.B. and J.-P.M. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from the CNRS and the MESRI (Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation) are gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declare no conflict of interest.
Sample Availability
Samples of all synthesized compounds are available from the authors.
References
- Ash, I.; Ash, M. Encyclopedia of Surfactants; Chemical Pub. Co.: New York, NY, USA, 1983. [Google Scholar]
- Gonçalves, R.A.; Holmberg, K.; Lindman, B. Cationic surfactants: A review. J. Mol. Liq. 2023, 375, 121335–121346. [Google Scholar] [CrossRef]
- Sarkar, R.; Pal, A.; Rakshit, A.; Saha, B. Properties and applications of amphoteric surfactant: A concise review. J. Surfactants Deterg. 2021, 24, 709–730. [Google Scholar] [CrossRef]
- Singh, N.; Sharma, L. Synthesis of carbohydrate derived non-ionic gemini surfactants and study of their micellar and reverse micellar behavior—A review. Lett. Org. Chem. 2019, 16, 607–614. [Google Scholar] [CrossRef]
- Zhou, M.; Li, S.; Zhang, Z.; Wang, C.; Luo, G.; Zhao, J. Progress in the Synthesis of Zwitterionic Gemini Surfactants. J. Surfactants Deterg. 2017, 20, 1243–12541. [Google Scholar] [CrossRef]
- Damez, C.; Bouquillon, S.; Harakat, D.; Hénin, F.; Muzart, J.; Pezron, I.; Komunjer, L. Alkenyl and alkenoyl amphiphilic derivatives of D-xylose and their surfactant properties. Carbohydr. Res. 2007, 342, 154–163. [Google Scholar] [CrossRef]
- Zakharova, L.Y.; Pashirova, T.N.; Doktorovova, S.; Fernandes, A.R.; Sanchez-Lopez, E.; Silva, A.M.; Souto, S.B. Cationic surfactants: Self-assembly, structure-activity correlation and their biological applications. Int. J. Mol. Sci. 2019, 20, 5534. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, E.; Badosa, E.; Pla, M.; Montesinos, L.; Bonaterra, A. Biocontrol of Plant Disease: Recent Advances and Prospects in Plant Protection; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 121–147. [Google Scholar]
- Kumar, A.; Singh, S.K.; Kant, C.; Verma, H.; Kumar, D.; Singh, P.P.; Modi, A.; Droby, S.; Kesawat, M.S.; Alavilli, H.; et al. Microbial biosurfactant: A new frontier for sustainable agriculture and pharmaceutical industries. Antioxidants 2021, 10, 1472. [Google Scholar] [CrossRef]
- Bajpai, D.; Tyagi, V.K. Laundry detergents: An overview. J. Oleo Sci. 2007, 56, 327–340. [Google Scholar] [CrossRef]
- Mijaljica, D.; Spada, F.; Harrison, I.P. Skin Cleansing without or with Compromise: Soaps and Syndets. Molecules 2022, 27, 2010. [Google Scholar] [CrossRef]
- Andree, H.; Hessel, J.F.; Meine, G.; Middelhauve, B.; Schmid, K. Alkyl Polyglycosides, Technology, Properties and Applications; Hill, K., Von Rybinski, W., Stoll, G., Eds.; VCH Publishers Inc.: New York, NY, USA, 1996; pp. 99–130. [Google Scholar]
- Naveen, K.; Rashmi, T. Characteristic and Application of Anionic Dimeric Surfactants: A Review. Tenside Surfactants Deterg. 2019, 56, 172–179. [Google Scholar]
- Abe, M.; Schechter, D.; Schechter, R.S.; Wade, H.; Weerasooriya, U.; Yiv, S. Microemulsion formation with branched tail polyoxyethylene sulfonate surfactants. J. Colloid Interface Sci. 1986, 114, 342–356. [Google Scholar] [CrossRef]
- Sunwoo, C.K.; Wade, W.H. Optimal Surfactant Structures for Cosurfactant-Free Microemulston Systems I. C16 and C14 Guerbet Alcohol Hydrophobes. J. Dispersion Sci. Technol. 1992, 13, 491–514. [Google Scholar] [CrossRef]
- Sanchez, J.; del Valle, M. Determination of Anionic Surfactants Employing Potentiometric Sensors-A Review. Crit. Rev. Anal.Chem. 2005, 35, 15–29. [Google Scholar] [CrossRef]
- Kaler, E.W.; Herrington, K.L.; Murthy, A.K.; Zasadzinski, J.A. Phase Behavior and Structures of Mixtures of Anionic and Cationic Surfactants. J. Phys. Chem. 1992, 96, 6698–6707. [Google Scholar] [CrossRef]
- Ahmady, A.R.; Hosseinzadeh, P.; Solouk, A.; Akbari, S.; Szulc, A.M.; Brycki, B.E. Cationic gemini surfactant properties, its potential as a promising bioapplication candidate, and strategies for improving its biocompatibility: A review. Adv. Colloid Interface Sci. 2022, 299, 102581–102609. [Google Scholar] [CrossRef]
- Jesus, C.F.; Alves, A.A.S.; Fiuza, S.M.; Murtinho, D.; Antunes, F.E. Mini-review: Synthetic methods for the production of cationic sugar-based surfactants. J. Mol. Liq. 2021, 342, 117389–117398. [Google Scholar] [CrossRef]
- Holmberg, K. Handbook of Applied Surface and Colloid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Benvegnu, T.; Sassi, J.F. Oligomannuronates from Seaweeds as Renewable Sources for the Development of Green Surfactants. Top. Curr. Chem. 2010, 294, 143–164. [Google Scholar]
- Farn, R.J. Chemistry and Technology of Surfactants; Wiley—Blackwell: Hoboken, NY, USA, 2008. [Google Scholar]
- Hoffmann, B.; Platz, G. Phase and aggregation behaviour of alkylglycosides. Curr. Opin. Colloid Interface Sci. 2010, 6, 171–177. [Google Scholar] [CrossRef]
- Jérôme, F.; Marinkovic, S.; Estrine, B. Transglycosylation: A Key Reaction to Access Alkylpolyglycosides from Lignocellulosic Biomass. ChemSusChem 2018, 11, 1395–1409. [Google Scholar] [CrossRef]
- Geetha, D.; Tyagi, R. Alkyl Poly Glucosides (APGs) Surfactants and Their Properties: A Review. Tenside Surf. Det. 2012, 49, 417–427. [Google Scholar] [CrossRef]
- Roy, S.; Maiti, K.; Moulik, S.P.; Ghosh, R. Synthesis of C14- and C16-carbon chain containing mannitol diester and diether based non-ionic amphiphiles and studies of their Langmuir monolayer films at the air/water interface. Colloids Surf. A Physicochem. Eng. Asp. 2011, 377, 349–355. [Google Scholar] [CrossRef]
- Mulligan, C.N. Recent advances in the environmental applications of biosurfactants. Curr. Opin. Colloid Interface Sci. 2009, 14, 372–378. [Google Scholar] [CrossRef]
- Deleu, M.; Paquot, M. From renewable vegetables resources to microorganisms: New trends in surfactants. C. R. Chim. 2004, 7, 641–646. [Google Scholar] [CrossRef]
- Lin, S.C. Biosurfactants: Recent advances. J. Chem. Technol. Biotechnol. 1996, 66, 109–120. [Google Scholar] [CrossRef]
- Lang, S. Biological amphiphiles (microbial biosurfactants). Curr. Opin. Colloid Interface Sci. 2002, 7, 12–20. [Google Scholar] [CrossRef]
- Gatard, S.; Nasir, M.N.; Deleu, M.; Klai, N.; Legrand, V.; Bouquillon, S. Bolaamphiphiles Derived from Alkenyl L-Rhamnosides and Alkenyl D-Xylosides: Importance of the Hydrophilic Head. Molecules 2013, 18, 6101–6112. [Google Scholar] [CrossRef] [PubMed]
- Nasir, M.N.; Crowet, J.M.; Lins, L.; Obounou Akong, F.; Haudrechy, A.; Bouquillon, S.; Deleu, M. Interactions of sugar-based bolaamphiphiles with biomimetic systems of plasma membranes. Biochimie 2016, 130, 23–32. [Google Scholar] [CrossRef]
- Corma, A.; Hamid, S.B.A.; Iborra, S.; Velty, A. Surfactants from Biomass: A Two-Step Cascade Reaction for the Synthesis of Sorbitol Fatty Acid Esters Using Solid Acid Catalysts. ChemSusChem 2008, 1, 85–90. [Google Scholar] [CrossRef]
- Angyal, S.J. Complexes of Metal Cations with Carbohydrates in Solution. Adv. Carbohydr. Chem. Biochem. 1989, 47, 1–43. [Google Scholar]
- Garelli-Calvet, R.; Brisset, F.; Godefroy, L.; Lattes, A. Alkyldiamides à Double tête de Sucre ou Dérivé de Sucre Comme Composés Amphiphiles. Brevet Européen N° 0 541 467 A2, 7 May 1993. FR 9113851. [Google Scholar]
- Satgé, C.; Granet, R.; Verneuil, B.; Champavier, Y.; Krausz, P. Synthesis and properties of new bolaform and macrocyclic galactose-based surfactants obtained by olefin metathesis. Carbohydr. Res. 2004, 339, 1243–1254. [Google Scholar] [CrossRef]
- Dzulkefly, K.; Khoh, H.F.; Ahmad, F.B.H.; Adlie Ahmad, S.; Lim, W.H. Solvent-free esterification process for the synthesis of glucose bolaform surfactants. Orient. J. Chem. 2010, 26, 747–752. [Google Scholar]
- Obounou Akong, F.; Bouquillon, S. Efficient syntheses of bolaform surfactants from L-rhamnose and/or 3-(4-hydroxyphenyl)propionic acid. Green Chem. 2015, 17, 3290–3300. [Google Scholar] [CrossRef]
- Stuerga, D. Microwave-material interaction and dielectric properties, key ingredients for mastery of chemical microwave process. In Microwave in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; CiteSeer: Princeton, NJ, USA, 2006; pp. 1–61. [Google Scholar]
- Luzuriaga-Loaiza, W.P.; Schellenberger, R.; De Gaetano, Y.; Obounou Akong, F.; Villaume, S.; Crouzet, J.; Haudrechy, A.; Baillieul, F.; Clément, C.; Lins, L.; et al. Synthetic Rhamnolipid Bolaforms trigger an innate immune response in Arabidopsis thaliana. Sci. Rep. 2018, 8, 8534–8547. [Google Scholar] [CrossRef] [PubMed]
- Perreux, L.; Loupy, A. A tentative rationalisation of microwave effects in organic synthesis according to the reaction medium, and mechanistic considerations. Tetrahedron 2001, 57, 9199–9223. [Google Scholar] [CrossRef]
- Bornaghi, F.L.; Poulsen, S.A. Microwave-accelerated Fischer glycosylation. Tetrahedron Lett. 2005, 46, 3485–3488. [Google Scholar] [CrossRef]
- Kerkel, F.; Markiewicz, M.; Stolte, S.; Müller, C.; Kunz, W. The green platform molecule gamma-valerolactone—Ecotoxicity, biodegradability, solvent properties, and potential applications. Green Chem. 2021, 23, 2962–2976. [Google Scholar] [CrossRef]
- Dutta, S.; Yu, I.K.M.; Tsang, D.C.W.; Hau Ng, Y.; Sik Ok, Y.; Sherwood, J.; Clark, J.H. Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts. Chem. Eng. J. 2019, 372, 992–1006. [Google Scholar] [CrossRef]
- Snoussi, M.; Trabelsi, N.; Dehmeni, A.; Benzekri, R.; Bouslama, L.; Hajlaoui, B.; Al-sieni, A.; Papetti, A. Phytochemical analysis, antimicrobial and antioxidant activities of Allium roseum var. odoratissimum (Desf.) Coss extracts. Ind. Crops Prod. 2016, 89, 533–542. [Google Scholar] [CrossRef]
- Selka, A.; Moutombi, F.J.N.; Jean-François, J.; Touaibia, M. Hydroxycinnamic Acids and Their Related Synthetic Analogs: An Update of Pharmacological Activities. Mini Rev. Med. Chem. 2022, 22, 1516–1544. [Google Scholar] [CrossRef]
- Abazari, M.F.; Nasiri, N.; Karizi, S.Z.; Nejati, F.; Haghiaminjan, H.; Norouzi, S.; Piri, P.; Estakhr, L.; Faradonbeh, D.R.; Kohandani, M.; et al. An updated review of various medicinal applications of p-coumaric acid: From antioxidative and anti-inflammatory properties to effects on cell cycle and proliferation. Mini Rev. Med. Chem. 2021, 21, 2187–2201. [Google Scholar] [CrossRef]
- Kaur, J.; Kaur, R. p-Coumaric Acid: A Naturally Occurring Chemical with Potential Therapeutic Applications. Curr. Org. Chem. 2022, 26, 1333–1349. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Ferulic Acid From Plant Biomass: A Phytochemical With Promising Antiviral Properties. Front. Nutr. 2022, 8, 777576. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Escobedo, L.; Gotor-Fernández, V. Solvent role in the lipase-catalysed esterification of cinnamic acid and derivatives. Optimisation of the biotransformation conditions. Tetrahedron 2021, 8112, 131873–131879. [Google Scholar] [CrossRef]
- Chen, H.C.; Twu, Y.K.; Chang, C.M.J.; Liu, Y.C.; Shieh, C.J. Optimized synthesis of lipase-catalyzed octyl caffeate by Novozym® 435. Ind. Crops Prod. 2010, 32, 522–526. [Google Scholar] [CrossRef]
- Kotha, S.; Dipak, M.K. Strategies and tactics in olefin metathesis. Tetrahedron 2012, 68, 397–421. [Google Scholar] [CrossRef]
- Connon, S.J.; Blechert, S. Recent developments in olefin cross-metathesis. Angew. Chem. Int. Ed. 2003, 42, 1900–1923. [Google Scholar] [CrossRef]
- López-Alarcón, C.; Denicola, A. Evaluating the antioxidant capacity of natural products: A review on chemical and cellular-based assays. Anal. Chim. Acta 2013, 763, 1–10. [Google Scholar] [CrossRef]
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22, 3380. [Google Scholar] [CrossRef]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int. J. Mol. Sci. 2020, 21, 1131–1158. [Google Scholar] [CrossRef]
- Moussaoui, A.L.; Jawhari, F.Z.; Almehdi, A.M.; Elmsellem, H.; Benbrahim, K.F.; Bousta, D.; Bari, A. Antibacterial, antifungal and antioxidant activity of total polyphenols of Withania frutescens L. Bioorg. Chem. 2019, 93, 103337–103346. [Google Scholar] [CrossRef]
- Sakurai, S.; Kawakami, Y.; Kuroki, M.; Gotoh, H. Structure/antioxidant activity (oxygen radical absorbance capacity) relationships of phenolic compounds. Struct. Chem. 2022, 33, 1055–1062. [Google Scholar] [CrossRef]
- Zheng, Y.; Karimi-Maleh, H.; Fu, L. Evaluation of Antioxidants Using Electrochemical Sensors: A Bibliometric Analysis. Sensors 2022, 22, 3238. [Google Scholar] [CrossRef] [PubMed]
- Chaillou, L.L.; Nazareno, M.A. New Method to Determine Antioxidant Activity of Polyphenols. J. Agric. Food Chem. 2006, 54, 8397–8402. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Aziz, A.; Kapoor, D. Salicylic Acid: It’s Physiological Role and Interactions. Res. J. Pharm. Technol. 2018, 11, 3171–3177. [Google Scholar] [CrossRef]
- Ullah, C.; Chen, Y.H.; Ortega, M.A.; Tsai, C.J. The diversity of salicylic acid biosynthesis and defense signaling in plants: Knowledge gaps and future opportunities. Curr. Opin. Plant Biol. 2023, 72, 102349. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, I.; Shiga, M.; Sasamoto, K. A new sulfonated tetrazolium salt that produces a highly water-soluble formazan dye. Chem. Pharm. Bull. 1993, 41, 1118–1122. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Paolinia, A.; Porredon Guarch, C.; Ramos-López, D.; de Lapuente, J.; Lascialfari, A.; Guari, Y.; Larionova, J.; Long, J.; Nano, J. Rhamnose-coated superparamagnetic iron-oxide nanoparticles: An evaluation of their in vitro cytotoxicity, genotoxicity and carcinogenicity. J. Appl. Toxicol. 2016, 36, 510–520. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, G.; Wang, H.; Cheng, C.; Zang, G.; Guo, X.; Liu, R.H. Phytochemical Profiles and Antioxidant Activities in Six Species of Ramie Leaves. PLoS ONE 2014, 9, e108140. [Google Scholar] [CrossRef]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).