
Scalable Preparation of the Masked Acyl Cyanide TBS-MAC



Abstract
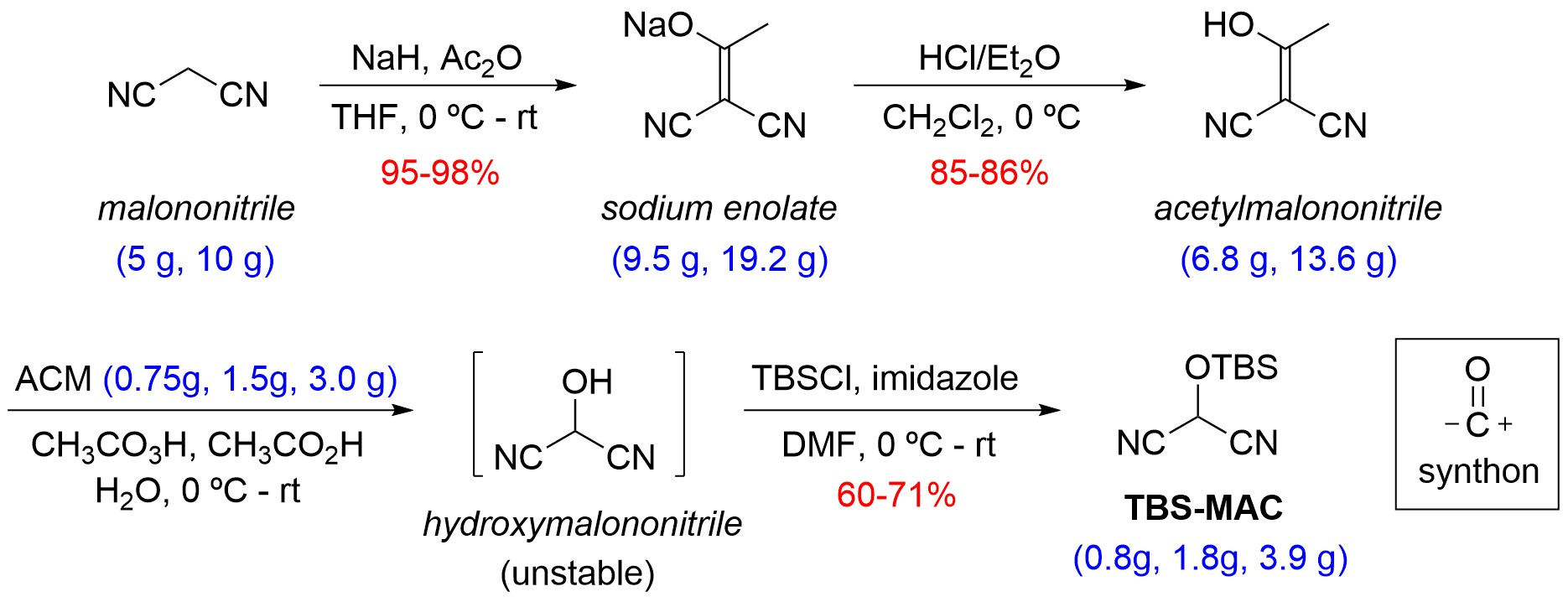
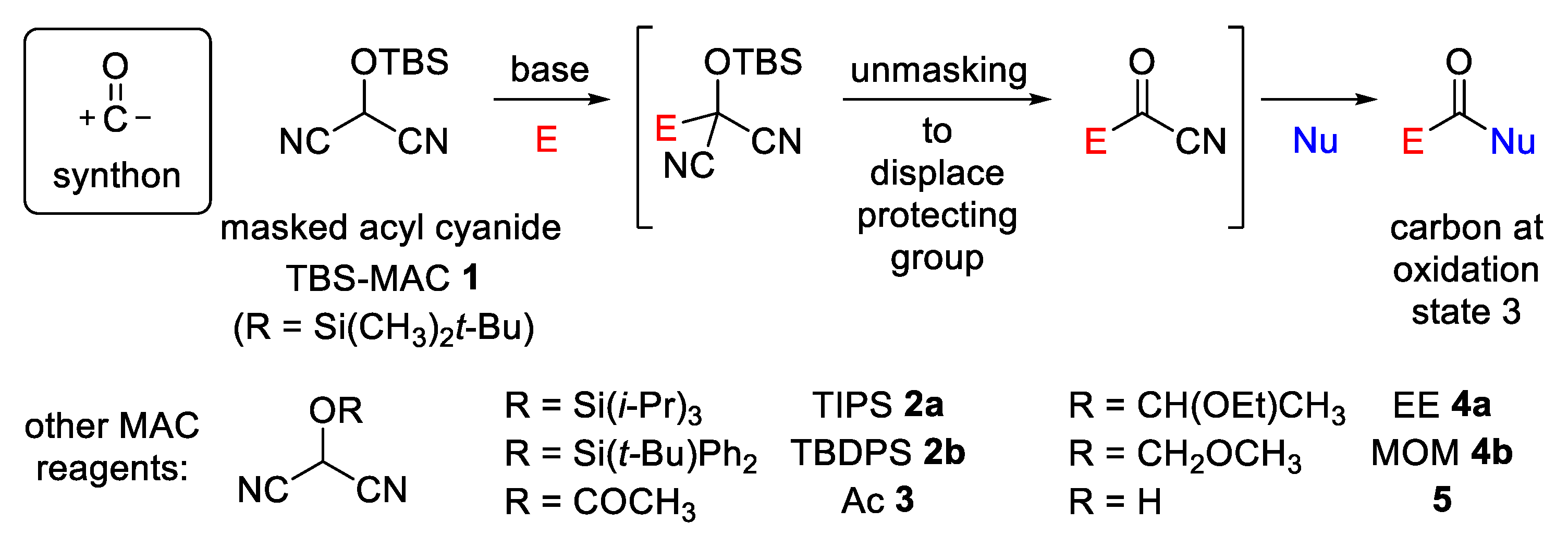
1. Introduction
2. Results and Discussion
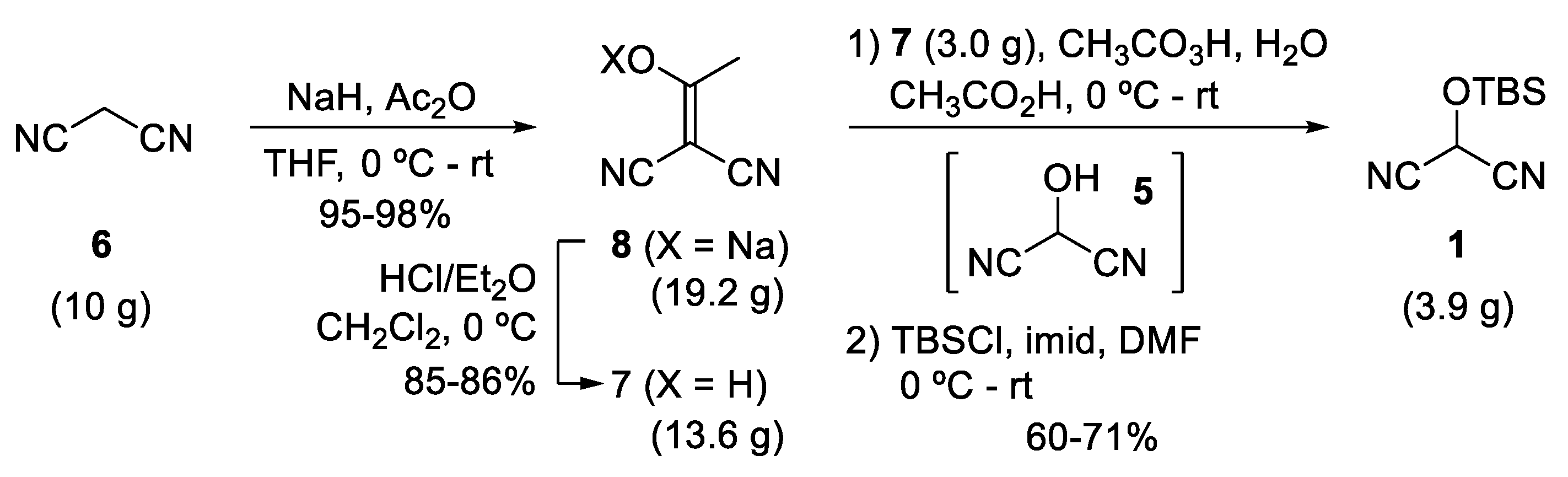
2.1. Synthetic Considerations
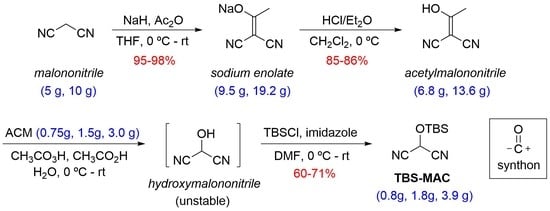
2.2. Step 1—Sodium Enolate 8 (Sodium 1,1-dicyanoprop-1-en-2-olate)
2.3. Step 2—Acetylmalononitrile 6 (2-(1-Hydroxyethylidene)malononitrile)
2.4. Step 3—TBS-MAC 1 (2-((tert-Butyldimethylsilyl)oxy)malononitrile)
3. Materials and Methods
3.1. General Experimental
3.2. Synthetic Procedures and Analytical Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Nemoto, H.; Kubota, Y.; Yamamoto, Y. Development of a New Acyl Anion Equivalent for the Preparation of Masked Activated Esters and Their Use To Prepare a Dipeptide. J. Org. Chem. 1990, 55, 4515–4516. [Google Scholar]
- He, X.; Buchotte, M.; Guillot, R.; Deloisy, S.; Aitken, D.J. A case study of the MAC (masked acyl cyanide) oxyhomologation of N,N-dibenzyl-Lphenylalaninal with anti diastereoselectivity: Preparation of (2S,3S)-allophenylnorstatin esters. Org. Biomol. Chem. 2022, 20, 1761–1781. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H. Protected Dicyanomethanol. In e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Seebach, D. Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. 1979, 18, 239–258. [Google Scholar] [CrossRef]
- Hase, T.A.; Koskimies, J.K. A Compilation of References on Formyl and Acyl Anion Synthons. Aldrichimica Acta 1981, 14, 73–77. [Google Scholar]
- Kagawa, N.; Nibbs, A.E.; Rawal, V.H. One-Carbon Homologation of Primary Alcohols to Carboxylic Acids, Esters, and Amides via Mitsunobu Reactions with MAC Reagents. Org. Lett. 2016, 18, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Hethcox, J.C.; Shockley, S.E.; Stoltz, B.M. Enantioselective Iridium-Catalyzed Allylic Alkylation Reactions of Masked Acyl Cyanide Equivalents. Org. Lett. 2017, 19, 1527–1529. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kubota, Y.; Honda, Y.; Fukui, H.; Asao, N.; Nemoto, H. Transition Metal Catalyzed Addition of Certain Nucleophiles to Imines. J. Am. Chem. Soc. 1994, 116, 3161–3162. [Google Scholar] [CrossRef]
- Nemoto, H.; Ma, R.; Suzuki, I.; Shibuya, M. A New One-Pot Method for the Synthesis of α-Siloxyamides from Aldehydes or Ketones and Its Application to the Synthesis of (-)-Bestatin. Org. Lett. 2000, 2, 4245–4247. [Google Scholar] [CrossRef]
- Nemoto, H.; Kawamura, T.; Miyoshi, N. A Highly Efficient Carbon-Carbon Bond Formation Reaction via Nucleophilic Addition to N-Alkylaldimines without Acids or Metallic Species. J. Am. Chem. Soc. 2005, 127, 14546–14547. [Google Scholar]
- Nemoto, H.; Ma, R.; Kawamura, T.; Kamiya, M.; Shibuya, M. Synthesis of α-Amino Acid Precursors Directly from Aldehydes Using Masked Acyl Cyanide Reagents and N,O-Dialkylated Hydroxylamines. J. Org. Chem. 2006, 71, 6038–6043. [Google Scholar]
- Nemoto, H.; Moriguchi, H.; Ma, R.; Kawamura, T.; Kamiya, M.; Shibuya, M. Highly diastereoselective nucleophilic addition reactions of masked acyl cyanide reagents to tert-butanesulfiminides. Tetrahedron Asymm. 2007, 18, 383–389. [Google Scholar] [CrossRef]
- Yang, K.S.; Rawal, V.H. Synthesis of α-Amino Acid Derivatives and Peptides via Enantioselective Addition of Masked Acyl Cyanides to Imines. J. Am. Chem. Soc. 2014, 136, 16148–16151. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Matsumoto, N.; Kibe, M.; Abe, K.; Takehara, T.; Suzuki, T. Enantiodivergent Reaction of Ketimines with Malononitriles Using Single Cinchona Alkaloid Sulfonamide Catalysts. Adv. Syn. Catal. 2022, 364, 781–786. [Google Scholar] [CrossRef]
- Nemoto, H.; Ma, R.; Moriguchi, H.; Kawamura, T.; Kamiya, M.; Shibuya, M. Direct One-Pot Synthesis of α-Siloxy-Weinreb Amides from Aldehydes. J. Org. Chem. 2007, 72, 9850–9853. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Ma, R.; Kawamura, T.; Yatsuzuka, K.; Kamiya, M.; Shibuya, M. One-Pot Synthesis of α-Siloxy Esters Using a Silylated Masked Acyl Cyanide. Synthesis 2008, 23, 3819–3827. [Google Scholar] [CrossRef]
- Nemoto, H.; Kawamura, T.; Kitasaki, K.; Yatsuzuka, K.; Kamiya, M.; Yoshioka, Y. One-Portion Synthesis of 2-Acetoxy Carbonyl Compounds from Aldehydes by Using an Acetylated Masked Acyl Cyanide. Synthesis 2009, 10, 1694–1702. [Google Scholar] [CrossRef]
- Nomura, Y.; Thuaud, F.; Sekine, D.; Ito, A.; Maeda, S.; Koshino, H.; Hashizume, D.; Muranaka, A.; Cruchter, T.; Uchiyama, M.; et al. Synthesis of All Stereoisomers of Monomeric Spectomycin A1/A2 and Evaluation of Their Protein SUMOylation-Inhibitory Activity. Chem. Eur. J. 2019, 25, 8387–8392. [Google Scholar] [CrossRef]
- Esgulian, M.; Buchotte, M.; Guillot, R.; Deloisy, S.; Aitken, D.J. Reversal of Diastereoselectivity in a Masked Acyl Cyanide (MAC) Reaction: Synthesis of Protected erythro-β-Hydroxyaspartate Derivatives. Org. Lett. 2019, 21, 2378–2382. [Google Scholar] [CrossRef]
- Jentsch, N.G.; Hume, J.D.; Crull, E.B.; Beauti, S.M.; Pham, A.H.; Pigza, J.A.; Kessl, J.; Donahue, M.G. Quinolines from the cyclocondensation of isatoic anhydride with ethyl acetoacetate: Preparation of ethyl 4-hydroxy-2-methylquinoline-3-carboxylate and derivatives. Beilstein J. Org. Chem. 2018, 14, 2529–2536. [Google Scholar] [CrossRef]
- Kuboto, Y.; Nemoto, H.; Yamamoto, Y. Synthesis of 1,4-Dicarbonyl Compounds via the Conjugate Addition of a Masked Activated Ester, ROCH(CN)2. J. Org. Chem. 1991, 56, 7195–7196. [Google Scholar] [CrossRef]
- Rawal, V.H.; Türkmen, Y.E.; Nibbs, A.E.; Yang, K.S. Squaramide-Catalyzed Enantioselective Michael Addition of Masked Acyl Cyanides to Substituted Enones. J. Am. Chem. Soc. 2013, 135, 16050–16053. [Google Scholar]
- Zhao, K.; Zhi, Y.; Wang, A.; Enders, D. Synthesis of Malononitrile-Substituted Diarylmethines via 1,6-Addition of Masked Acyl Cyanides to para-Quinone Methides. Synthesis 2018, 50, 872–880. [Google Scholar]
- Roche, S.P.; Faure, S.; Aitken, D.J. Total Synthesis of Cyclotheonamide C using a Tandem Backbone-Extension–Coupling Methodology. Angew. Chem. Int. Ed. 2008, 47, 6840–6842. [Google Scholar] [CrossRef] [PubMed]
- Yamatsugu, K.; Yin, L.; Kamijo, S.; Kimura, Y.; Kanai, M.; Shibasaki, M. A Synthesis of Tamiflu by Using a Barium-Catalyzed Asymmetric Diels–Alder-Type Reaction. Angew. Chem. Int. Ed. 2009, 48, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Chu, H.; Stratton, T.P.; Kossler, D.; Eberle, K.J.; Flood, D.T.; Baran, P.S. Total Synthesis of Tagetitoxin. J. Am. Chem. Soc. 2020, 142, 13683–13688. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Xu, H.; Zhang, T.; Zhang, S.; Zhao, X.; Jiang, P.; Li, J.; Ye, B.; Sun, Y.; et al. Elastase Inhibitor Cyclotheonellazole A: Total Synthesis and In Vivo Biological Evaluation for Acute Lung Injury. J. Med. Chem. 2022, 65, 2971–2987. [Google Scholar] [CrossRef]
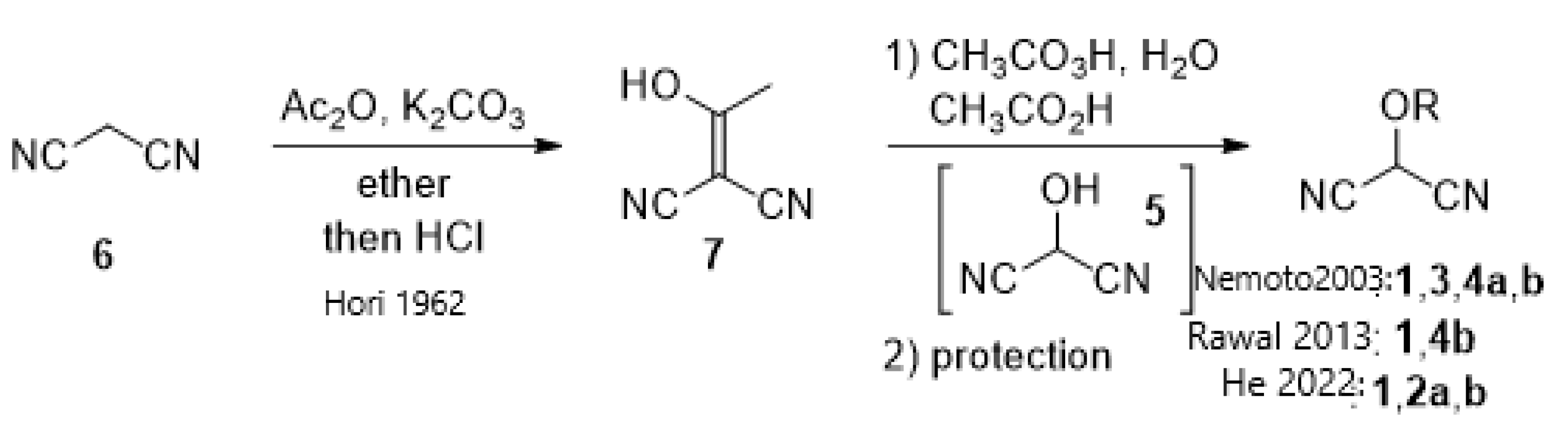
- Nemoto, H.; Li, X.; Ma, R.; Suzuki, I.; Shibuya, M. A three-step preparation of MAC reagents from malononitrile. Tetrahedron Lett. 2003, 44, 73–75. [Google Scholar] [CrossRef]
- Hori, I.; Midorikawa, H. Acylation of cyanoacetic ester by acetic anhydride and potassium carbonate. Sci. Papers Inst. Phys. Chem. Res. 1962, 56, 216–217. [Google Scholar]
- Fleury, J.P.; Libis, B. Acyl-et sulfonylmalodinitriles et leurs sels de triethylamine. Bull. Soc. Chim. Fr. 1964, 413–414. [Google Scholar]
- Jentsch, N.G.; Hart, A.P.; Hume, J.D.; Sun, J.; McNeely, K.A.; Lama, C.; Pigza, J.A.; Donahue, M.G.; Kessl, J.J. Synthesis and Evaluation of Aryl Quinolines as HIV-1 Integrase Multimerization Inhibitors. ACS Med. Chem. Lett. 2018, 9, 1007–1012. [Google Scholar] [CrossRef]
- Sun, J.; Patel, K.; Hume, J.; Pigza, J.A.; Donahue, M.G.; Kessl, J.J. Optimized binding of substituted quinoline ALLINIs within the HIV-1 integrase oligomer. J. Biol. Chem. 2021, 296, 100363. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yin, Y.; He, Z.; Ma, L.; Li, X.; Zhang, Z.; Lin, X.; Ma, R. 2-Hydroxymalonitrile—A Useful Reagent for One-step Synthesis of α-Hydroxy Esters. Chem. Res. Chin. Univ. 2015, 31, 321–324. [Google Scholar] [CrossRef]
- Organic Syntheses Instructions for Authors, Appendix 1. Available online: http://www.orgsyn.org/instructions.aspx (accessed on 27 June 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Scale | Time 1 | Times 2, 3 | Acetone Slurry | Yield |
| 1 | 5 g | 30 min | 15 min, 15 min | 2 × 100 mL | 9.5 g, 97% |
| 2 | 10 g | 1 h | 30 min, 30 min | 2 × 200 mL | 17.3 g, 88% |
| 3 | 10 g | 1 h | 30 min, 30 min | 3 × 100 mL | 13.8 g, 70% |
| 4 | 10 g | 1 h | 30 min, 30 min | 3 × 200 mL | 18.7 g, 95% |
| 5 | 10 g | 1 h | 30 min, 30 min | 4 × 200 mL | 19.2 g, 98% |
 | |||||
|---|---|---|---|---|---|
| Entry | Scale | Time | CH2Cl2 Slurry | Hexanes Slurry | Yield |
| 1 | 9.5 g | 15 min | 1 × 50 mL | 1 × 50 mL | 6.8 g, 86% |
| 2 | 19.2 g | 30 min | 1 × 100 mL | 1 × 100 mL | 13.6 g, 85% |
 | |||||
|---|---|---|---|---|---|
| Entry | Scale | DMF | Time/Temp [a] | Crude (g, Yield) [c] | Purified Yield [d] |
| 1 | 0.75 g | 0.5 M | 30 min 0 °C, 30 min rt | 0.83 g, 61% | --- |
| 2 | 0.75 g | 0.5 M | 30 min 0 °C, 12 h rt | 0.85 g, 63% | --- |
| 3 | 0.75 g | 1 M | 30 min 0 °C, 30 min rt | 0.92 g, 68% | 0.81 g, 60% |
| 4 | 0.75 g | 1 M | 1 h rt | 0.86 g, 63% | --- |
| 5 | 0.75 g | 1 M | 30 min 0 °C, 30 min rt [b] | 0.66 g, 48% | --- |
| 6 | 1.5 g | 1 M | 30 min 0 °C, 30 min rt | 1.94 g, 71% | 1.84 g, 68% |
| 7 | 3.0 g | 1 M | 30 min 0 °C, 30 min rt | 3.91 g, 72% | 3.90 g, 71% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinton, H.; Patterson, J.; Hume, J.; Patel, K.; Pigza, J. Scalable Preparation of the Masked Acyl Cyanide TBS-MAC. Molecules 2023, 28, 5087. https://doi.org/10.3390/molecules28135087
Hinton H, Patterson J, Hume J, Patel K, Pigza J. Scalable Preparation of the Masked Acyl Cyanide TBS-MAC. Molecules. 2023; 28(13):5087. https://doi.org/10.3390/molecules28135087
Chicago/Turabian StyleHinton, Haley, Jack Patterson, Jared Hume, Krunal Patel, and Julie Pigza. 2023. "Scalable Preparation of the Masked Acyl Cyanide TBS-MAC" Molecules 28, no. 13: 5087. https://doi.org/10.3390/molecules28135087
APA StyleHinton, H., Patterson, J., Hume, J., Patel, K., & Pigza, J. (2023). Scalable Preparation of the Masked Acyl Cyanide TBS-MAC. Molecules, 28(13), 5087. https://doi.org/10.3390/molecules28135087