
Computational Insights into Novel Inhibitor N-(3-(tert-Butylcarbamoyl)-4-methoxyphenyl)-indole and Ingliforib Specific against GP Isoenzyme Dimers Interaction Mechanism


Abstract
1. Introduction
2. Results and Discussion
2.1. Binding Model Studies by Molecular Docking
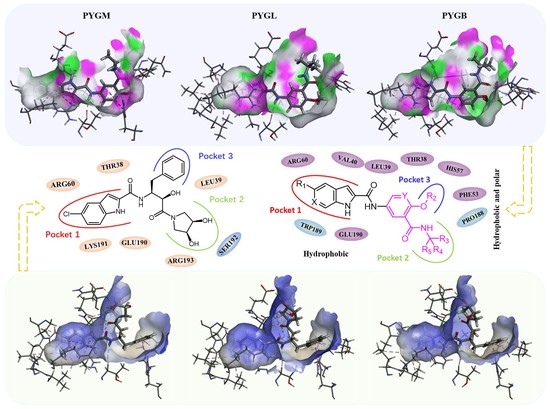
2.1.1. Spatial Position and Orientation
2.1.2. Binding Mode
2.2. Molecular Dynamics Simulations
2.3. Calculation of Binding Free Energy Based on MM-PBSA
2.4. Pharmacophore Modeling
3. Materials and Methods
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation
3.3. Calculation of Binding Free Energy Based on MM-PBSA Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qurtam, A.A.; Mechchate, H.; Es-Safi, I.; Al-Zharani, M.; Nasr, F.A.; Noman, O.M.; Aleissa, M.; Imtara, H.; Aleissa, A.M.; Bouhrim, M.; et al. Citrus Flavanone Narirutin, In Vitro and In Silico Mechanistic Antidiabetic Potential. Pharmaceutics 2021, 13, 1818. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.; Wang, Y.; Yan, Z.; Guo, Y.; Zhang, L. A Novel 5-Chloro-N-phenyl-1H-indole-2-carboxamide Derivative as Brain-Type Glycogen Phosphorylase Inhibitor: Validation of Target PYGB. Molecules 2023, 28, 1697. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Li, S.; Wang, Y.; Li, J.; Ma, C.; Guo, Y.; Zhang, L. Discovery of novel heterocyclic derivatives as potential glycogen phosphorylase inhibitors with a cardioprotective effect. Bioorg. Chem. 2022, 129, 106120. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155. [Google Scholar] [CrossRef]
- Cai, Y.; Guo, H.; Fan, Z.; Zhang, X.; Wu, D.; Tang, W.; Gu, T.; Wang, S.; Yin, A.; Tao, L.; et al. Glycogenolysis Is Crucial for Astrocytic Glycogen Accumulation and Brain Damage after Reperfusion in Ischemic Stroke. iScience 2020, 23, 101136. [Google Scholar] [CrossRef]
- Agius, L. New hepatic targets for glycaemic control in diabetes. Best. Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 587–605. [Google Scholar] [CrossRef]
- Guan, T.; Qian, Y.; Tang, X.; Huang, M.; Huang, L.; Li, Y.; Sun, H. Maslinic acid, a natural inhibitor of glycogen phosphorylase, reduces cerebral ischemic injury in hyperglycemic rats by GLT-1 up-regulation. J. Neurosci. Res. 2011, 89, 1829–1839. [Google Scholar] [CrossRef]
- Migocka-Patrzałek, M.; Elias, M. Muscle Glycogen Phosphorylase and Its Functional Partners in Health and Disease. Cells 2021, 10, 883. [Google Scholar] [CrossRef]
- Chrysina, E.D. The prototype of glycogen phosphorylase. Mini-Rev. Med. Chem. 2010, 10, 1093–1101. [Google Scholar] [CrossRef]
- Aiston, S.; Hampson, L.; Gómez-Foix, A.M.; Guinovart, J.J.; Agius, L. Hepatic Glycogen Synthesis Is Highly Sensitive to Phosphorylase Activity. J. Biol. Chem. 2001, 276, 23858–23866. [Google Scholar] [CrossRef]
- Fletterick, R.J.; Sygusch, J.; Semple, H.; Madsen, N.B. Structure of glycogen phosphorylase a at 3.0 A resolution and its ligand binding sites at 6 A. J. Biol. Chem. 1976, 251, 6142–6146. [Google Scholar] [CrossRef] [PubMed]
- Oikonomakos, N.G.; Skamnaki, V.T.; E Tsitsanou, K.; Gavalas, N.G.; Johnson, L.N. A new allosteric site in glycogen phosphorylase b as a target for drug interactions. Structure 2000, 8, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; de la Sierra-Gallay, I.L.; Duval, R.; Xu, X.; Cocaign, A.; Léger, T.; Woffendin, G.; Camadro, J.-M.; Etchebest, C.; Haouz, A.; et al. Insights into Brain Glycogen Metabolism. J. Biol. Chem. 2016, 291, 18072–18083. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Dupret, J.-M.; Rodrigues-Lima, F. The structure of brain glycogen phosphorylase-from allosteric regulation mechanisms to clinical perspectives. FEBS J. 2016, 284, 546–554. [Google Scholar] [CrossRef]
- Freeman, S.; Bartlett, J.B.; Convey, G.; Hardern, I.; Teague, J.L.; Loxham, S.J.G.; Allen, J.M.; Poucher, S.M.; Charles, A.D. Sensitivity of glycogen phosphorylase isoforms to indole site inhibitors is markedly dependent on the activation state of the enzyme. Br. J. Pharmacol. 2006, 149, 775–785. [Google Scholar] [CrossRef]
- Konkimalla, V. An Improved Comparative Docking Approach for Developing Specific Glycogen Phosphorylase Inhibitors Using Pentacyclic Triterpenes. Curr. Top. Med. Chem. 2017, 17, 1640–1645. [Google Scholar] [CrossRef]
- Tracey, W.R.; Treadway, J.L.; Magee, W.P.; Sutt, J.C.; McPherson, R.K.; Levy, C.B.; Wilder, D.E.; Yu, L.J.; Chen, Y.; Shanker, R.M.; et al. Cardioprotective effects of ingliforib, a novel glycogen phosphorylase inhibitor. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1177–H1184. [Google Scholar] [CrossRef]
- Chehardoli, G.; Bahmani, A. Synthetic strategies, SAR studies, and computer modeling of indole 2 and 3-carboxamides as the strong enzyme inhibitors: A review. Mol. Divers. 2020, 25, 535–550. [Google Scholar] [CrossRef]
- Henke, B.R.; Sparks, S.M. Glycogen phosphorylase inhibitors. Mini Rev. Med. Chem. 2006, 6, 845–857. [Google Scholar] [CrossRef]
- Rath, V.L.; Ammirati, M.; Danley, D.E.; Ekstrom, J.L.; Gibbs, E.M.; Hynes, T.R.; Mathiowetz, A.M.; McPherson, R.K.; Olson, T.V.; Treadway, J.L.; et al. Human liver glycogen phosphorylase inhibitors bind at a new allosteric site. Chem. Biol. 2000, 7, 677–682. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Liu, J.; Zhu, Q.; Leng, Y.; Luo, X.; Jiang, H.; Liu, H. Discovery of novel dual-action antidiabetic agents that inhibit glycogen phosphorylase and activate glucokinase. Eur. J. Med. Chem. 2012, 58, 624–639. [Google Scholar] [CrossRef] [PubMed]
- PYGB—Glycogen Phosphorylase, Brain Form—Homo Sapiens (Human) UnitProtKB|Uniprot. Available online: https://www.uniprot.org/uniprot/P11216 (accessed on 1 August 2022).
- Onda, K.; Suzuki, T.; Shiraki, R.; Yonetoku, Y.; Negoro, K.; Momose, K.; Katayama, N.; Orita, M.; Yamaguchi, T.; Ohta, M.; et al. Synthesis of 5-chloro-N-aryl-1H-indole-2-carboxamide derivatives as inhibitors of human liver glycogen phosphorylase a. Bioorg. Med. Chem. 2008, 16, 5452–5464. [Google Scholar] [CrossRef] [PubMed]
- PYGL—Glycogen Phosphorylase, Liver Form—Homo Sapiens (Human) UnitProtKB|Uniprot. Available online: https://www.uniprot.org/uniprot/P06737 (accessed on 1 August 2022).
- Lukacs, C.M.; Oikonomakos, N.G.; Crowther, R.L.; Hong, L.N.; Kammlott, R.U.; Levin, W.; Li, S.; Liu, C.M.; Lucas-McGady, D.; Pietranico, S.; et al. The crystal structure of human muscle glycogen phosphorylase a with bound glucose and AMP: An intermediate conformation with T-state and R-state features. Proteins 2006, 63, 1123–1126. [Google Scholar] [CrossRef]
- PYGM—Glycogen Phosphorylase, Muscle Form—Homo Sapiens (Human)|UnitProtKB|Uniprot. Available online: https://www.uniprot.org/uniprot/P11217 (accessed on 1 August 2022).
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D., Jr.; Feig, M.; Brooks, C.L., 3rd. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Beglov, D.; Roux, B. Finite representation of an infinite bulk system: Solvent boundary potential for computer simulations. J. Chem. Phys. 1994, 100, 9050–9063. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Acceptor | Donor | Occupancy (%) | Distance (Å) | Angle (°) |
|---|---|---|---|---|---|
| PYGB–compound 1 | Glu190@O | MOL@H1N1 | 97.16 | 2.938 | 148.2 |
| Thr38′@O | MOL@H3N2 | 3.82 | 2.949 | 148.5 | |
| MOL@O2 | His57′@HE2NE2 | 3.31 | 3.088 | 147.5 | |
| PYGL–compound 1 | Glu190@O | MOL@H4N1 | 99.74 | 2.893 | 157.6 |
| Thr38′@O | MOL@H6N2 | 94.15 | 2.955 | 146.6 | |
| MOL@O2 | His57′@HE2NE2 | 75.24 | 3.074 | 148.9 | |
| MOL@O3 | Arg60′@HH12NH1 | 20.00 | 2.858 | 158.0 | |
| MOL@O3 | Arg60′@HH22NH2 | 9.31 | 3.173 | 137.8 | |
| MOL@O3 | His57′@HE2NE2 | 8.16 | 3.183 | 131.5 | |
| PYGM–compound 1 | His57@ND1 | MOL@H1N1 | 99.85 | 2.936 | 155.3 |
| MOL@O2 | His57′@HE2NE2 | 23.01 | 3.019 | 141.0 | |
| MOL@O3 | Lys191@HZ2NZ | 22.91 | 2.818 | 149.7 | |
| Thr38′@O | MOL@H3N2 | 19.77 | 3.149 | 135.0 | |
| MOL@O3 | Lys191@HZ1NZ | 13.89 | 2.811 | 148.0 | |
| MOL@O3 | Lys191@HZ3NZ | 10.98 | 2.837 | 149.6 | |
| PYGB–ingliforib | Glu190@O | MOL@H4N1 | 96.86 | 2.929 | 149.8 |
| MOL@O5 | Lys191@HZ3NZ | 37.16 | 2.804 | 153.2 | |
| MOL@O5 | Lys191@HZ1NZ | 29.79 | 2.792 | 154.7 | |
| MOL@O5 | Lys191@HZ2NZ | 25.25 | 2.791 | 154.4 | |
| PYGL–ingliforib | Thr38′@O | MOL@H21O2 | 99.43 | 2.761 | 159.3 |
| Glu190@O | MOL@H4N1 | 99.03 | 2.857 | 153.7 | |
| Thr38′@O | MOL@H6N2 | 93.73 | 3.129 | 139.1 | |
| Ser192@O | MOL@H22O3 | 76.07 | 2.679 | 160.3 | |
| PYGM–ingliforib | Glu190@O | MOL@H4N1 | 99.91 | 2.863 | 154.5 |
| Thr38′@O | MOL@H21O2 | 97.39 | 2.835 | 151.9 | |
| Thr38′@OG1 | MOL@H6N2 | 67.59 | 3.151 | 149.5 | |
| Thr38′@O | MOL@H6N2 | 58.24 | 3.090 | 136.0 | |
| MOL@O5 | Lys191@HZ1NZ | 32.18 | 2.816 | 149.7 | |
| MOL@O5 | Lys191@HZ3NZ | 30.03 | 2.813 | 149.5 | |
| MOL@O5 | Lys191@HZ2NZ | 24.19 | 2.807 | 150.5 |
| Energy Component (kcal·mol−1) | Compound 1 | ||
| PYGB | PYGL | PYGM | |
| ELE | −31.72 ± 4.15 | −21.51 ± 4.97 | −32.07 ± 4.36 |
| VDW | −46.75 ± 2.05 | −47.85 ± 3.04 | −47.56 ± 2.1 |
| GAS | −78.47 ± 3.34 | −69.36 ± 2.78 | −79.63 ± 3.68 |
| GBSUR | −5.86 ± 0.11 | −6.26 ± 0.19 | −6.4 ± 0.14 |
| GB | 46.08 ± 3.15 | 40.53 ± 2.77 | 48.42 ± 2.89 |
| GBSOL | 40.21 ± 3.19 | 34.27 ± 2.89 | 42.02 ± 2.82 |
| GBELE | 14.35 ± 1.83 | 19.02 ± 3.22 | 16.35 ± 2.59 |
| GBTOT | −38.26 ± 1.61 | −35.10 ± 2.57 | −37.61 ± 2.08 |
| Energy Component (kcal·mol−1) | Ingliforib | ||
| PYGB | PYGL | PYGM | |
| ELE | −54.05 ± 6 | −50 ± 5.64 | −48.15 ± 2.56 |
| VDW | −50.9 ± 1.14 | −51.97 ± 4.01 | −46.4 ± 5.64 |
| GAS | −104.95 ± 6.74 | −101.97 ± 3.51 | −94.55 ± 7.39 |
| GBSUR | −6.45 ± 0.25 | −6.98 ± 0.1 | −6.52 ± 0.33 |
| GB | 70.65 ± 8.84 | 66.72 ± 4.53 | 62.45 ± 3.27 |
| GBSOL | 64.2 ± 8.69 | 59.74 ± 4.45 | 55.94 ± 2.94 |
| GBELE | 16.6 ± 2.91 | 16.72 ± 2.18 | 14.3 ± 1.62 |
| GBTOT | −40.75 ± 1.96 | −42.23 ± 1.91 | −38.62 ± 4.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, S.; Yan, Z.; Zhang, L. Computational Insights into Novel Inhibitor N-(3-(tert-Butylcarbamoyl)-4-methoxyphenyl)-indole and Ingliforib Specific against GP Isoenzyme Dimers Interaction Mechanism. Molecules 2023, 28, 4909. https://doi.org/10.3390/molecules28134909
Wang Y, Li S, Yan Z, Zhang L. Computational Insights into Novel Inhibitor N-(3-(tert-Butylcarbamoyl)-4-methoxyphenyl)-indole and Ingliforib Specific against GP Isoenzyme Dimers Interaction Mechanism. Molecules. 2023; 28(13):4909. https://doi.org/10.3390/molecules28134909
Chicago/Turabian StyleWang, Youde, Shuai Li, Zhiwei Yan, and Liying Zhang. 2023. "Computational Insights into Novel Inhibitor N-(3-(tert-Butylcarbamoyl)-4-methoxyphenyl)-indole and Ingliforib Specific against GP Isoenzyme Dimers Interaction Mechanism" Molecules 28, no. 13: 4909. https://doi.org/10.3390/molecules28134909
APA StyleWang, Y., Li, S., Yan, Z., & Zhang, L. (2023). Computational Insights into Novel Inhibitor N-(3-(tert-Butylcarbamoyl)-4-methoxyphenyl)-indole and Ingliforib Specific against GP Isoenzyme Dimers Interaction Mechanism. Molecules, 28(13), 4909. https://doi.org/10.3390/molecules28134909