
Toughening Effect of 2,5-Furandicaboxylate Polyesters on Polylactide-Based Renewable Fibers

 ,
,  ,
,  ,
,  , and
, and 
Abstract
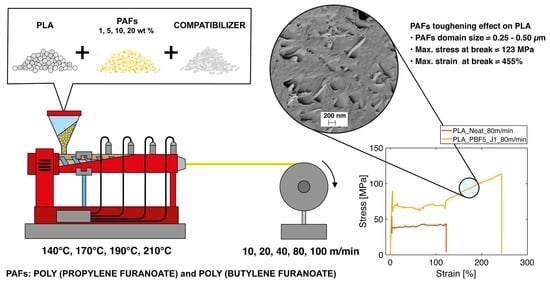
1. Introduction
2. Results
2.1. Characterization of the Bulk (Sheet) Samples
2.1.1. Rheological Properties
2.1.2. Microstructural Properties of the Bulk (Sheet) Samples
2.1.3. Thermal Properties
2.1.4. Mechanical Properties
2.2. Characterization of the Fiber Samples
2.2.1. Microstructural Characterization
2.2.2. Thermal Characterization
2.2.3. Mechanical Characterization
3. Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.2.1. Bulk Samples
3.2.2. Fiber Samples
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Meraldo, A. 4—Introduction to Bio-Based Polymers. In Multilayer Flexible Packaging, 2nd ed.; Wagner, J.R., Ed.; William Andrew Publishing: Norwich, NY, USA, 2016; pp. 47–52. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, 1700782. [Google Scholar] [CrossRef]
- White, E.; Bassilakis, R.; Nogués, S. From the Plastics Present to a Sustainable Future: The Bioplastics Innovation Landscape, Players and Market Opportunities; ClarivateTM: London, UK, 2020. [Google Scholar]
- Fredi, G.; Dorigato, A. Recycling of bioplastic waste: A review. Adv. Ind. Eng. Polym. Res. 2021, 4, 159–177. [Google Scholar] [CrossRef]
- Konstantopoulou, M.; Terzopoulou, Z.; Nerantzaki, M.; Tsagkalias, J.; Achilias, D.S.; Bikiaris, D.N.; Exarhopoulos, S.; Papageorgiou, D.G.; Papageorgiou, G.Z. Poly(ethylene furanoate-co-ethylene terephthalate) biobased copolymers: Synthesis, thermal properties and cocrystallization behavior. Eur. Polym. J. 2017, 89, 349–366. [Google Scholar] [CrossRef]
- Niaounakis, M. Biopolymers Reuse, Recycling, and Disposal; Elsevier: Oxford, UK, 2013. [Google Scholar]
- Yang, Y.; Zhang, M.; Ju, Z.; Tam, P.Y.; Hua, T.; Younas, M.W.; Kamrul, H.; Hu, H. Poly(lactic acid) fibers, yarns and fabrics: Manufacturing, properties and applications. Text. Res. J. 2020, 91, 1641–1669. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Wu, B.; Cui, C.; Guo, Y.; Yan, C. A critical review of fused deposition modeling 3D printing technology in manufacturing polylactic acid parts. Int. J. Adv. Manuf. Technol. 2019, 102, 2877–2889. [Google Scholar] [CrossRef]
- Pegoretti, A.; Fambri, L.; Migliaresi, C. In Vitro degradation of poly(L-lactic acid) fibers produced by melt spinning. J. Appl. Polym. Sci. 1997, 64, 213–223. [Google Scholar] [CrossRef]
- Tait, M.; Pegoretti, A.; Dorigato, A.; Kaladzidou, K. The effect of filler type and content and the manufacturing process on the performance of multifunctional carbon/poly-lactide composites. Carbon 2011, 49, 4280–4290. [Google Scholar] [CrossRef]
- Balla, E.; Daniilidis, V.; Karlioti, G.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Vlachopoulos, A.; Koumentakou, I.; Bikiaris, D.N. Poly(lactic Acid): A Versatile Biobased Polymer for the Future with Multifunctional Properties—From Monomer Synthesis, Polymerization Techniques and Molecular Weight Increase to PLA Applications. Polymers 2021, 13, 1822. [Google Scholar] [CrossRef]
- Fambri, L.; Pegoretti, A.; Fenner, R.; Incardona, S.D.; Migliaresi, C. Biodegradable fibres of poly(L-lactic acid) produced by melt spinning. Polymer 1997, 38, 79–85. [Google Scholar] [CrossRef]
- Fambri, L.; Bragagna, S.; Migliaresi, C. Biodegradable Fibers of Poly-L, DL-lactide 70/30 Produced by Melt Spinning. Macromol. Symp. 2006, 234, 20–25. [Google Scholar] [CrossRef]
- Sousa, A.F.; Patrício, R.; Terzopoulou, Z.; Bikiaris, D.N.; Stern, T.; Wenger, J.; Loos, K.; Lotti, N.; Siracusa, V.; Szymczyk, A.; et al. Recommendations for replacing PET on packaging, fiber, and film materials with biobased counterparts. Green Chem. 2021, 23, 8795–8820. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, J.; Yu, S.; Wei, L.; Hu, Z.; Xiang, H.; Zhu, M. Melt-spun bio-based PLA-co-PET copolyester fibers with tunable properties: Synergistic effects of chemical structure and drawing process. Int. J. Biol. Macromol. 2023, 226, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Perego, G.; Cella, G.D.; Bastioli, C. Effect of molecular weight and crystallinity on poly(lactic acid) mechanical properties. J. Appl. Polym. Sci. 1996, 59, 37–43. [Google Scholar] [CrossRef]
- Labrecque, L.V.; Kumar, R.A.; Dave, V.; Gross, R.A.; McCarthy, S.P. Citrate esters as plasticizers for poly(lactic acid). J. Appl. Polym. Sci. 1997, 66, 1507–1513. [Google Scholar] [CrossRef]
- Portes dos Santos, T.; Dias, K.B.; Bischoff, E.; Santos Mauler, R. Synthesis of esters derived from 2,5-furandicarboxylic acid and study of its plasticizing effects on poly(lactic acid). J. Polym. Res. 2022, 29, 58. [Google Scholar] [CrossRef]
- Cabedo, L.; Feijoo, J.L.; Villanueva, M.P.; Lagaron, J.M.; Gimenez, E. Optimization of Biodegradable Nanocomposites Based on aPLA/PCL Blends for Food Packaging Applications. Macromol. Symp. 2006, 233, 191–197. [Google Scholar] [CrossRef]
- Wang, L.; Ma, W.; Gross, R.A.; McCarthy, S.P. Reactive compatibilization of biodegradable blends of poly(lactic acid) and poly(ε-caprolactone). Polym. Degrad. Stab. 1998, 59, 161–168. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Odelius, K.; Guo, Z.; Guan, X.; Hakkarainen, M. Nanostructured Phase Morphology of a Biobased Copolymer for Tough and UV-Resistant Polylactide. ACS Appl. Polym. Mater. 2021, 3, 1973–1982. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Smyrnioti, D.; Nikolaidis, G.N.; Tsitsimaka, I.; Christodoulou, E.; Bikiaris, D.N.; Charitopoulou, M.A.; Achilias, D.S.; Kapnisti, M.; Papageorgiou, G.Z. Sustainable Plastics from Biomass: Blends of Polyesters Based on 2,5-Furandicarboxylic Acid. Polymers 2020, 12, 225. [Google Scholar] [CrossRef]
- Sousa, A.F.; Silvestre, A.J.D. Plastics from renewable sources as green and sustainable alternatives. Curr. Opin. Green Sustain. Chem. 2022, 33, 100557. [Google Scholar] [CrossRef]
- Eerhart, A.J.J.E.; Faaij, A.P.C.; Patel, M.K. Replacing fossil based PET with biobased PEF; Process analysis, energy and GHG balance. Energy Environ. Sci. 2012, 5, 6407. [Google Scholar] [CrossRef]
- Paszkiewicz, S.; Walkowiak, K.; Irska, I.; Mechowska, S.; Stankiewicz, K.; Zubkiewicz, A.; Piesowicz, E.; Miadlicki, P. Influence of the Multiple Injection Moulding and Composting Time on the Properties of Selected Packaging and Furan-Based Polyesters. J. Polym. Environ. 2022, 31, 722–742. [Google Scholar] [CrossRef]
- Nguyen, H.T.H.; Qi, P.; Rostagno, M.; Feteha, A.; Miller, S.A. The quest for high glass transition temperature bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Poulopoulou, N.; Kantoutsis, G.; Bikiaris, D.N.; Achilias, D.S.; Kapnisti, M.; Papageorgiou, G.Z. Biobased Engineering Thermoplastics: Poly(butylene 2,5-furandicarboxylate) Blends. Polymers 2019, 11, 937. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, R.Y.; Huang, J.C.; Wang, J.G.; Jiang, Y.H.; Hu, G.H.; Yang, J.; Zhu, J. Tensile Property Balanced and Gas Barrier Improved Poly(lactic acid) by Blending with Biobased Poly(butylene 2,5-furan dicarboxylate). ACS Sustain. Chem. Eng. 2017, 5, 9244–9253. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Zamboulis, A.; Papadopoulos, L.; Grigora, M.E.; Tsongas, K.; Tzetzis, D.; Bikiaris, D.N.; Papageorgiou, G.Z. Blending PLA with Polyesters Based on 2,5-Furan Dicarboxylic Acid: Evaluation of Physicochemical and Nanomechanical Properties. Polymers 2022, 14, 4725. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, L.; Wang, J.; Hao, X.; Dong, Y.; Sun, R. Ductile polylactic acid-based blend derived from bio-based poly(butylene adipate-co-butylene furandicarboxylate). Polym. Bull. 2022. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Bortolotti, M.; Pegoretti, A.; Bikiaris, D.N. Mechanical and Functional Properties of Novel Biobased Poly(decylene-2,5-furanoate)/Carbon Nanotubes Nanocomposite Films. Polymers 2020, 12, 2459. [Google Scholar] [CrossRef]
- Rigotti, D.; Soccio, M.; Dorigato, A.; Gazzano, M.; Siracusa, V.; Fredi, G.; Lotti, N. Novel biobased polylactic acid/poly(pentamethylene 2,5-furanoate) blends for sustainable food packaging. ACS Sustain. Chem. Eng. 2021, 9, 13742–13750. [Google Scholar] [CrossRef]
- Fredi, G.; Jafari, M.K.; Dorigato, A.; Bikiaris, D.N.; Pegoretti, A. Improving the Thermomechanical Properties of Poly(lactic acid) via Reduced Graphene Oxide and Bioderived Poly(decamethylene 2,5-furandicarboxylate). Materials 2022, 15, 1316. [Google Scholar] [CrossRef]
- Fredi, G.; Jafari, M.K.; Dorigato, A.; Bikiaris, D.N.; Checchetto, R.; Favaro, M.; Brusa, R.S.; Pegoretti, A. Multifunctionality of reduced graphene oxide in bioderived polylactide/poly(dodecylene furanoate) nanocomposite films. Molecules 2021, 26, 2398. [Google Scholar] [CrossRef]
- Fredi, G.; Rigotti, D.; Bikiaris, D.N.; Dorigato, A. Tuning thermo-mechanical properties of poly(lactic acid) films through blending with bioderived poly(alkylene furanoate)s with different alkyl chain length for sustainable packaging. Polymer 2021, 218, 123527. [Google Scholar] [CrossRef]
- Perin, D.; Rigotti, D.; Fredi, G.; Papageorgiou, G.Z.; Bikiaris, D.N.; Dorigato, A. Innovative bio-based poly(lactic acid)/poly(alkylene furanoate) fiber blends for sustainable textile applications. J. Polym. Environ. 2021, 29, 3948–3963. [Google Scholar] [CrossRef]
- Perin, D.; Fredi, G.; Rigotti, D.; Lotti, N.; Dorigato, A. Sustainable textile fibers made of bioderived polylactide/poly(pentamethylene 2,5-furanoate) blends. J. Appl. Polym. Sci. 2022, 139, 51740. [Google Scholar] [CrossRef]
- Rigotti, D.; Fredi, G.; Perin, D.; Bikiaris, D.N.; Pegoretti, A.; Dorigato, A. Statistical Modeling and Optimization of the Drawing Process of Bioderived Polylactide/Poly(Dodecylene Furanoate) Wet-Spun Fibers. Polymers 2022, 14, 396. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Dussin, A.; Xanthopoulou, E.; Bikiaris, D.N.; Botta, L.; Fiore, V.; Pegoretti, A. Compatibilization of polylactide/poly(ethylene furanoate) (PLA/PEF) blends for sustainable and bioderived packaging. Molecules 2022, 27, 6371. [Google Scholar] [CrossRef]
- Zhu, X.; Ren, Q.; Li, W.; Wu, M.; Weng, Z.; Wang, J.; Zheng, W.; Wang, L. In situ nanofibrillar fully-biobased poly (lactic acid)/poly (ethylene 2,5-furandicarboxylate) composites with promoted crystallization kinetics, mechanical properties, and heat resistance. Polym. Degrad. Stab. 2022, 206, 110172. [Google Scholar] [CrossRef]
- Mysiukiewicz, O.; Barczewski, M.; Skorczewska, K.; Matykiewicz, D. Correlation between Processing Parameters and Degradation of Different Polylactide Grades during Twin-Screw Extrusion. Polymers 2020, 12, 1333. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.; Castell, P.; Garcia, L.; Fernandez, A. Effect of Extrusion on the Mechanical and Rheological Properties of a Reinforced Poly(Lactic Acid): Reprocessing and Recycling of Biobased Materials. Materials 2015, 8, 7106–7117. [Google Scholar] [CrossRef]
- Yahyaee, N.; Javadi, A.; Garmabi, H.; Khaki, A. Effect of Two-Step Chain Extension using Joncryl and PMDA on the Rheological Properties of Poly (lactic acid). Macromol. Mater. Eng. 2019, 305, 1900423. [Google Scholar] [CrossRef]
- Kahraman, Y.; Alkan Goksu, Y.; Özdemir, B.; Eker Gümüş, B.; Nofar, M. Composition design of PLA/TPU emulsion blends compatibilized with multifunctional epoxy-based chain extender to tackle high impact resistant ductile structures. J. Appl. Polym. Sci. 2021, 139, 51833. [Google Scholar] [CrossRef]
- Mofokeng, J.P.; Luyt, A.S.; Tábi, T.; Kovács, J. Comparison of injection moulded, natural fibre-reinforced composites with PP and PLA as matrices. J. Thermoplast. Compos. Mater. 2011, 25, 927–948. [Google Scholar] [CrossRef]
- Xie, H.; Wu, L.; Li, B.-G.; Dubois, P. Poly(ethylene 2,5-furandicarboxylate-mb-poly(tetramethylene glycol)) multiblock copolymers: From high tough thermoplastics to elastomers. Polymer 2018, 155, 89–98. [Google Scholar] [CrossRef]
- Sanusi, O.M.; Papadopoulos, L.; Klonos, P.A.; Terzopoulou, Z.; Hocine, N.A.; Benelfellah, A.; Papageorgiou, G.Z.; Kyritsis, A.; Bikiaris, D.N. Calorimetric and Dielectric Study of Renewable Poly(hexylene 2,5-furan-dicarboxylate)-Based Nanocomposites In Situ Filled with Small Amounts of Graphene Platelets and Silica Nanoparticles. Polymers 2020, 12, 1239. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Sousa, A.F.; Andrade, M.; Silva, N.H.C.S.; Freire, C.S.R.; Mendes, A.; Silvestre, A.J.D. Furanoate-Based Nanocomposites: A Case Study Using Poly(Butylene 2,5-Furanoate) and Poly(Butylene 2,5-Furanoate)-co-(Butylene Diglycolate) and Bacterial Cellulose. Polymers 2018, 10, 810. [Google Scholar] [CrossRef]
- Gomes, M.; Gandini, A.; Silvestre, A.J.D.; Reis, B. Synthesis and characterization of poly(2,5-furan dicarboxylate)s based on a variety of diols. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 3759–3768. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, R.; Shi, L.; Ying, W.B.; Wang, J.; Zhu, J. Modification of Poly(butylene 2,5-furandicarboxylate) with Lactic Acid for Biodegradable Copolyesters with Good Mechanical and Barrier Properties. Ind. Eng. Chem. Res. 2018, 57, 11020–11030. [Google Scholar] [CrossRef]
- Paszkiewicz, S.; Janowska, I.; Pawlikowska, D.; Szymczyk, A.; Irska, I.; Lisiecki, S.; Stanik, R.; Gude, M.; Piesowicz, E. New functional nanocomposites based on poly(trimethylene 2,5-furanoate) and few layer graphene prepared by in situ polymerization. Express Polym. Lett. 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Guigo, N.; Tsanaktsis, V.; Exarhopoulos, S.; Bikiaris, D.N.; Sbirrazzuoli, N.; Papageorgiou, G.Z. Fast Crystallization and Melting Behavior of a Long-Spaced Aliphatic Furandicarboxylate Biobased Polyester, Poly(dodecylene 2,5-furanoate). Ind. Eng. Chem. Res. 2016, 55, 5315–5326. [Google Scholar] [CrossRef]
- Tsanaktsis, V.; Terzopoulou, Z.; Nerantzaki, M.; Papageorgiou, G.Z.; Bikiaris, D.N. New poly(pentylene furanoate) and poly(heptylene furanoate) sustainable polyesters from diols with odd methylene groups. Mater. Lett. 2016, 178, 64–67. [Google Scholar] [CrossRef]
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. A biocatalytic approach towards sustainable furanic–aliphatic polyesters. Polym. Chem. 2015, 6, 5198–5211. [Google Scholar] [CrossRef]
- Fambri, L.; Dabrowska, I.; Ceccato, R.; Pegoretti, A. Effects of Fumed Silica and Draw Ratio on Nanocomposite Polypropylene Fibers. Polymers 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Papageorgiou, D.G.; Exarhopoulos, S.; Papageorgiou, M.; Bikiaris, D.N. Evaluation of polyesters from renewable resources as alternatives to the current fossil-based polymers. Phase transitions of poly(butylene 2,5-furan-dicarboxylate). Polymer 2014, 55, 3846–3858. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Tsanaktsis, V.; Bikiaris, D.N. Synthesis of the bio-based polyester poly(propylene 2,5-furan dicarboxylate). Comparison of thermal behavior and solid state structure with its terephthalate and naphthalate homologues. Polymer 2015, 62, 28–38. [Google Scholar] [CrossRef]
- Bruckmoser, K.; Resch, K. Effect of processing conditions on crystallization behavior and mechanical properties of poly(lactic acid) staple fibers. J. Appl. Polym. Sci. 2015, 132, 42432. [Google Scholar] [CrossRef]
- Pivsa-Art, W.; Pivsa-Art, S. Multifilament yarns of polyoxymethylene/poly(lactic acid) blends produced by a melt-spinning method. Text. Res. J. 2020, 90, 294–301. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Garlotta, D. A Literature Review of Poly(Lactic Acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Terzopoulou, Z.; Bikiaris, D.N. Production of bio-based 2,5-furan dicarboxylate polyesters: Recent progress and critical aspects in their synthesis and thermal properties. Eur. Polym. J. 2016, 83, 202–229. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAF wt% | PPF Domain Size (µm) | PBF Domain Size (µm) |
|---|---|---|
| 1 | 0.25 + 0.10 | 0.44 + 0.11 |
| 5 | 0.31 + 0.12 | 0.43 + 0.14 |
| 10 | 0.30 + 0.10 | 0.50 + 0.10 |
| 20 | 0.31 + 0.14 | 0.51 + 0.15 |
| Assignment | Label | PLA | PPF | PLA-PPF20-J1 | PBF | PLA-PBF20-J1 |
|---|---|---|---|---|---|---|
| CH (Fu) | A | / | 3159 | / | 3152 | / |
| CH (Fu) | / | 3126 | / | 3117 | / | |
| CH3 | B | 2996 | / | 2995 | / | 2995 |
| CH3 | 2945 | / | 2945 | / | 2945 | |
| CH | 2919 | 2963 | 2921 | 2964 | 2921 | |
| CH | 2852 | 2850 | / | 2894 | 2851 | |
| C=O | C | 1750 | 1712 | 1750/1712 | 1718 | 1749/1721 |
| C=C (Fu) | D | / | 1581 | / | 1574 | 1582 |
| CH3 | E | 1453 | 1453 | 1453 | 1453 | 1458 |
| CH3 | 1383 | 1389 | 1383 | 1389 | 1383 | |
| C–O | F | 1180 | 1265 | 1181 | 1268/1121 | 1273/1180 |
| Fu ring breathing | G | / | 1018 | / | 1027 | / |
| Fu ring bending | / | 965; 825; 760 | 965; 825; 760 | 965; 822; 763 | 966; 823; 764 |
| PLA | PPF | PBF | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Thermal Cycle | H1 | C | H2 | H1 | C | H2 | H1 | C | H2 |
| Tg [°C] | 71.8 | 50.7 | 60.5 | 62.0 | 45.2 | 51.6 | 50.0 | / | 40.7 |
| Tcc [°C] | / | / | 111.0 | 105.1 | / | / | / | / | / |
| ∆Hcc [J/g] | / | / | 36.9 | 3.3 | / | / | / | / | / |
| Tm [°C] | 186.7 | / | 179.4 | 173.3 | / | 170.6 | 178.4 | / | 170.8 |
| ∆Hm [J/g] | 51.8 | / | 42.8 | 59.3 | / | 0.5 | 61.6 | / | 46.1 |
| Tc [°C] | / | / | / | / | / | / | / | 114.7 | / |
| ∆Hc [J/g] | / | / | / | / | / | / | / | 61.6 | / |
| Xc [%] | 55.2 | / | 6.3 | 39.5 | / | 0.4 | 47.8 | / | 35.7 |
| Sample | Tg (°C) | Tcc (°C) | ∆Hcc (J/g) | Tm (°C) | ∆Hm (J/g) | Xc (PLA) (%) | Tc (°C) | ∆Hc (J/g) |
|---|---|---|---|---|---|---|---|---|
| PLA | 59.9 | 94.7 | 26.3 | 183.2 | 57.3 | 33.1 | 95.8 | 10.9 |
| PLA-J1 | 56.7 | 96.7 | 31.2 | 176.1 | 43.2 | 12.9 | 94.8 | 3.8 |
| PLA-PPF10 | 58.3 | 95.3 | 37.3 | 176.8 | 51 | 16.2 | 98.7 | 12.9 |
| PLA-PPF1-J1 | 61.3 | 97.2 | 32.3 | 175.6 | 45.3 | 14.2 | / | / |
| PLA-PPF5-J1 | 59.3 | 97.4 | 32.1 | 178.2 | 42 | 11.2 | / | / |
| PLA-PPF10-J1 | 54.7 | 96.8 | 28.2 | 176.8 | 37.3 | 10.9 | / | / |
| PLA-PPF20-J1 | 54.7 | 96.8 | 28.2 | 176.8 | 37.3 | 12.3 | / | / |
| PLA-PBF1-J1 | 58.9 | 97 | 31.5 | 178.9 | 43.5 | / | 96.1 | 3.8 |
| PLA-PBF5-J1 | 62.8 | 96 | 31.9 | 177.9 | 45 | / | 97.8 | 17 |
| PLA-PBF10-J1 | 39.3/59.0 | 97.1 | 29.7 | 177.9 | 45.2 | / | 95 | 5 |
| PLA-PBF20-J1 | 39.5/59.2 | 96.7 | 28.3 | 178.5 | 45.3 | / | 99.4 | 9.8 |
| Sample | E (GPa) | UTS (MPa) | εb (%) |
|---|---|---|---|
| PLA | 4.0 ± 0.1 | 56.3 ± 0.4 | 5.2 ± 0.1 |
| PLA-J1 | 3.1 ± 0.1 | 60.7 ± 0.8 | 6.6 ± 0.4 |
| PLA-PPF1-J1 | 2.7 ± 0.1 | 60.2 ± 0.9 | 7.2 ± 0.4 |
| PLA-PPF5-J1 | 2.5 ± 0.1 | 59.8 ± 1.3 | 7.7 ± 0.8 |
| PLA-PPF10 | 3.0 ± 0.1 | 54.7 ± 1.0 | 5.3 ± 0.3 |
| PLA-PPF10-J1 | 3.1 ± 0.1 | 58.5 ± 2.8 | 6.2 ± 0.9 |
| PLA-PPF20-J1 | 2.6 ± 0.1 | 56.1 ± 4.3 | 5.6 ± 0.7 |
| PLA-PBF1-J1 | 3.4 ± 0.1 | 55.5 ± 0.5 | 8.3 ± 1.1 |
| PLA-PBF5-J1 | 3.3 ± 0.1 | 53.4 ± 1.9 | 22.0 ± 1.5 |
| PLA-PBF10-J1 | 3.4 ± 0.2 | 49.7 ± 1.0 | 55.5 ± 24.5 |
| PLA-PBF20-J1 | 2.9 ± 0.1 | 44.8 ± 3.7 | 11.5 ± 3.1 |
| Diameters (µm) | 10 m/min | 20 m/min | 40 m/min | 80 m/min | 100 m/min |
|---|---|---|---|---|---|
| PLA | 569 ± 22 (3.1) | 347 ± 21 (8.3) | 302 ± 14 (11.0) | 207 ± 38 (23.3) | 151 ± 21 (43.9) |
| PLA-J1 | 457 ± 70 (4.8) | / | 219 ± 15 (20.9) | 184 ± 13 (29.5) | / |
| PLA-PPF10 | / | / | 225 ± 15 (19.8) | 134 ± 12 (55.7) | / |
| PLA-PPF1-J1 | 485 ± 25 (4.3) | / | 235 ± 24 (18.1) | / | / |
| PLA-PPF5-J1 | 449 ± 34 (5.0) | / | 232 ± 19 (18.6) | 209 ± 30 (22.9) | / |
| PLA-PPF10-J1 | 497 ± 22 (4.0) | / | 242 ± 18 (17.1) | 179 ± 19 (31.2) | / |
| PLA-PPF20-J1 | 500 ± 20 (4.0) | / | 250 ± 11 (16.0) | 178 ± 9 (31.6) | / |
| PLA-PBF1-J1 | 497 ± 54 (4.0) | 339 ± 34 (8.7) | 241 ± 17 (17.2) | 163 ± 14 (37.6) | 151 ± 11 (43.9) |
| PLA-PBF5-J1 | 521 ± 38 (3.7) | 326 ± 21 (9.4) | 265 ± 12 (14.2) | 186 ± 18 (28.9) | 87 ± 23 (132.1) |
| PLA-PBF10-J1 | 566 ± 32 (3.1) | 414 ± 18 (5.8) | 303 ± 32 (10.9) | 205 ± 32 (23.8) | 120 ± 32 (69.4) |
| PLA-PBF20-J1 | 557 ± 28 (3.2) | 389 ± 10 (6.6) | 281 ± 11 (12.7) | 202 ± 7 (24.5) | 157 ± 8 (40.6) |
| Titer (tex) | 10 m/min | 20 m/min | 40 m/min | 80 m/min | 100 m/min |
| PLA | 321 ± 3 | 154 ± 9 | 88 ± 4 | 39 ± 8 | 30 ± 2 |
| PLA-J1 | 240 ± 49 | / | 99 ± 13 | 75 ± 12 | / |
| PLA-PPF10 | / | / | 49 ± 5 | 20 ± 2 | / |
| PLA-PPF1-J1 | 219 ± 7 | / | 57 ± 7 | / | / |
| PLA-PPF5-J1 | 196 ± 9 | / | 47 ± 5 | 41 ± 8 | / |
| PLA-PPF10-J1 | 241 ± 6 | / | 58 ± 4 | 46 ± 2 | / |
| PLA-PPF20-J1 | 263 ± 12 | / | 62 ± 6 | 30 ± 1 | / |
| PLA-PBF1-J1 | 259 ± 10 | 140 ± 4 | 62 ± 8 | 31 ± 2 | 24 ± 2 |
| PLA-PBF5-J1 | 256 ± 9 | 110 ± 10 | 69 ± 3 | 35 ± 5 | 5 ± 1 |
| PLA-PBF10-J1 | 319 ± 5 | 177 ± 7 | 84 ± 10 | 44 ± 3 | 23 ± 7 |
| PLA-PBF20-J1 | 311 ± 11 | 155 ± 6 | 83 ± 6 | 42 ± 2 | 25 ± 3 |
| Label | PLA (wt%) | PPF (wt%) | PBF (wt%) | J (phr) |
|---|---|---|---|---|
| PLA | 100 | 0 | 0 | 0 |
| PLA-J1 | 100 | 0 | 0 | 1 |
| PLA_PPF10 | 90 | 10 | 0 | 0 |
| PLA_PPF1_J1 | 99 | 1 | 0 | 1 |
| PLA_PPF5_J1 | 95 | 5 | 0 | 1 |
| PLA_PPF10_J1 | 90 | 10 | 0 | 1 |
| PLA_PPF20_J1 | 80 | 20 | 0 | 1 |
| PLA_PBF1_J1 | 99 | 0 | 1 | 1 |
| PLA_PBF5_J1 | 95 | 0 | 5 | 1 |
| PLA_PBF10_J1 | 90 | 0 | 10 | 1 |
| PLA_PBF20_J1 | 80 | 0 | 20 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fredi, G.; Zonta, E.; Dussin, A.; Bikiaris, D.N.; Papageorgiou, G.Z.; Fambri, L.; Dorigato, A. Toughening Effect of 2,5-Furandicaboxylate Polyesters on Polylactide-Based Renewable Fibers. Molecules 2023, 28, 4811. https://doi.org/10.3390/molecules28124811
Fredi G, Zonta E, Dussin A, Bikiaris DN, Papageorgiou GZ, Fambri L, Dorigato A. Toughening Effect of 2,5-Furandicaboxylate Polyesters on Polylactide-Based Renewable Fibers. Molecules. 2023; 28(12):4811. https://doi.org/10.3390/molecules28124811
Chicago/Turabian StyleFredi, Giulia, Edoardo Zonta, Alessandro Dussin, Dimitrios N. Bikiaris, George Z. Papageorgiou, Luca Fambri, and Andrea Dorigato. 2023. "Toughening Effect of 2,5-Furandicaboxylate Polyesters on Polylactide-Based Renewable Fibers" Molecules 28, no. 12: 4811. https://doi.org/10.3390/molecules28124811
APA StyleFredi, G., Zonta, E., Dussin, A., Bikiaris, D. N., Papageorgiou, G. Z., Fambri, L., & Dorigato, A. (2023). Toughening Effect of 2,5-Furandicaboxylate Polyesters on Polylactide-Based Renewable Fibers. Molecules, 28(12), 4811. https://doi.org/10.3390/molecules28124811