
Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
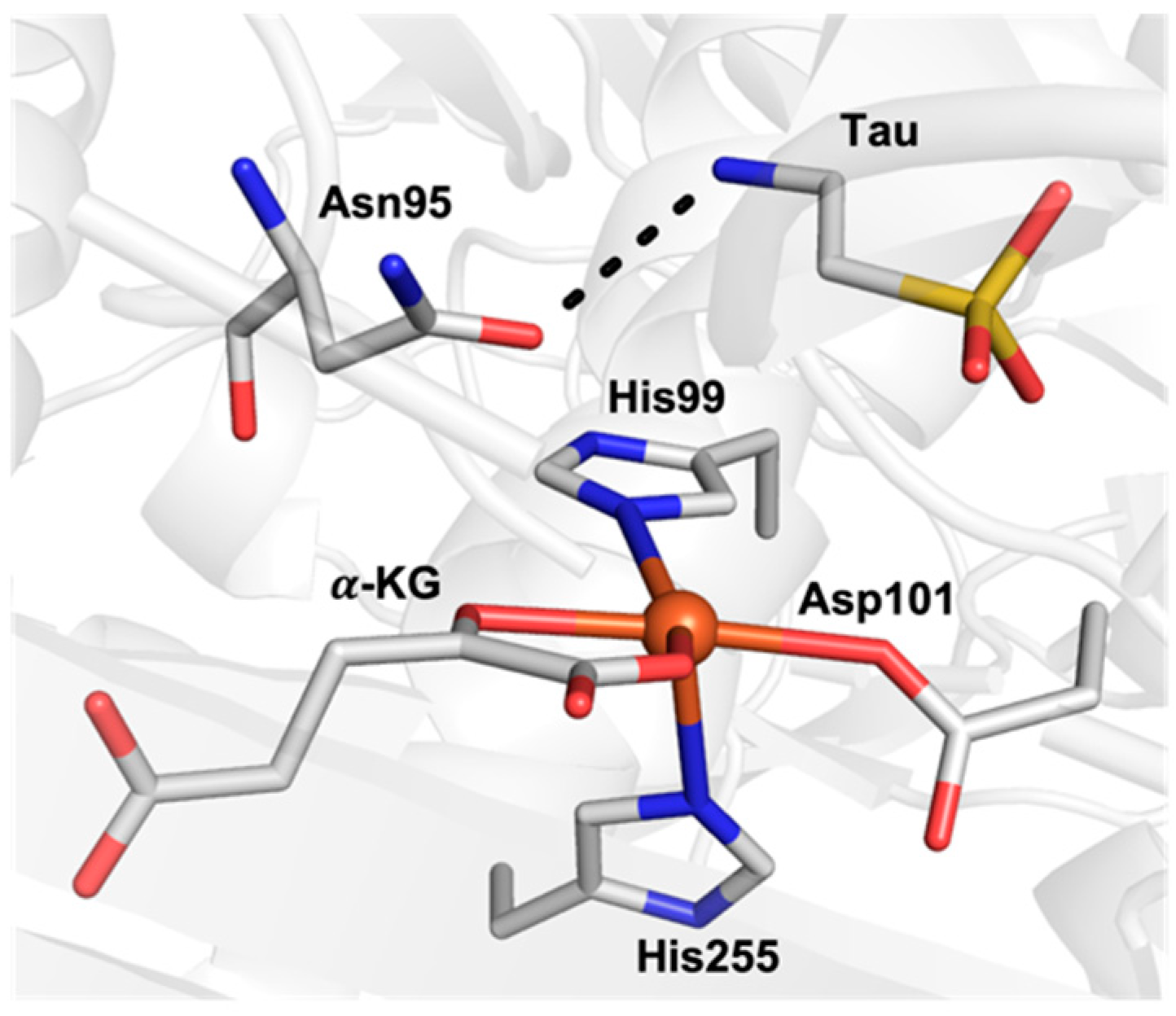
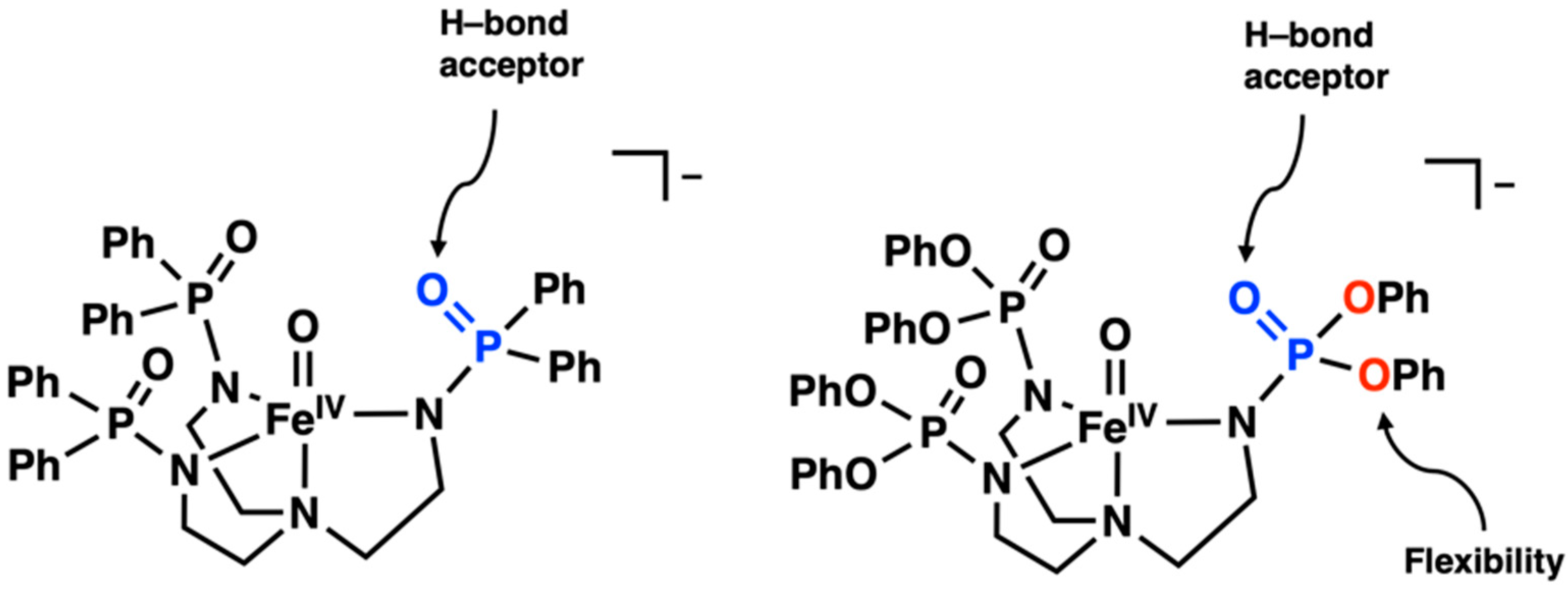
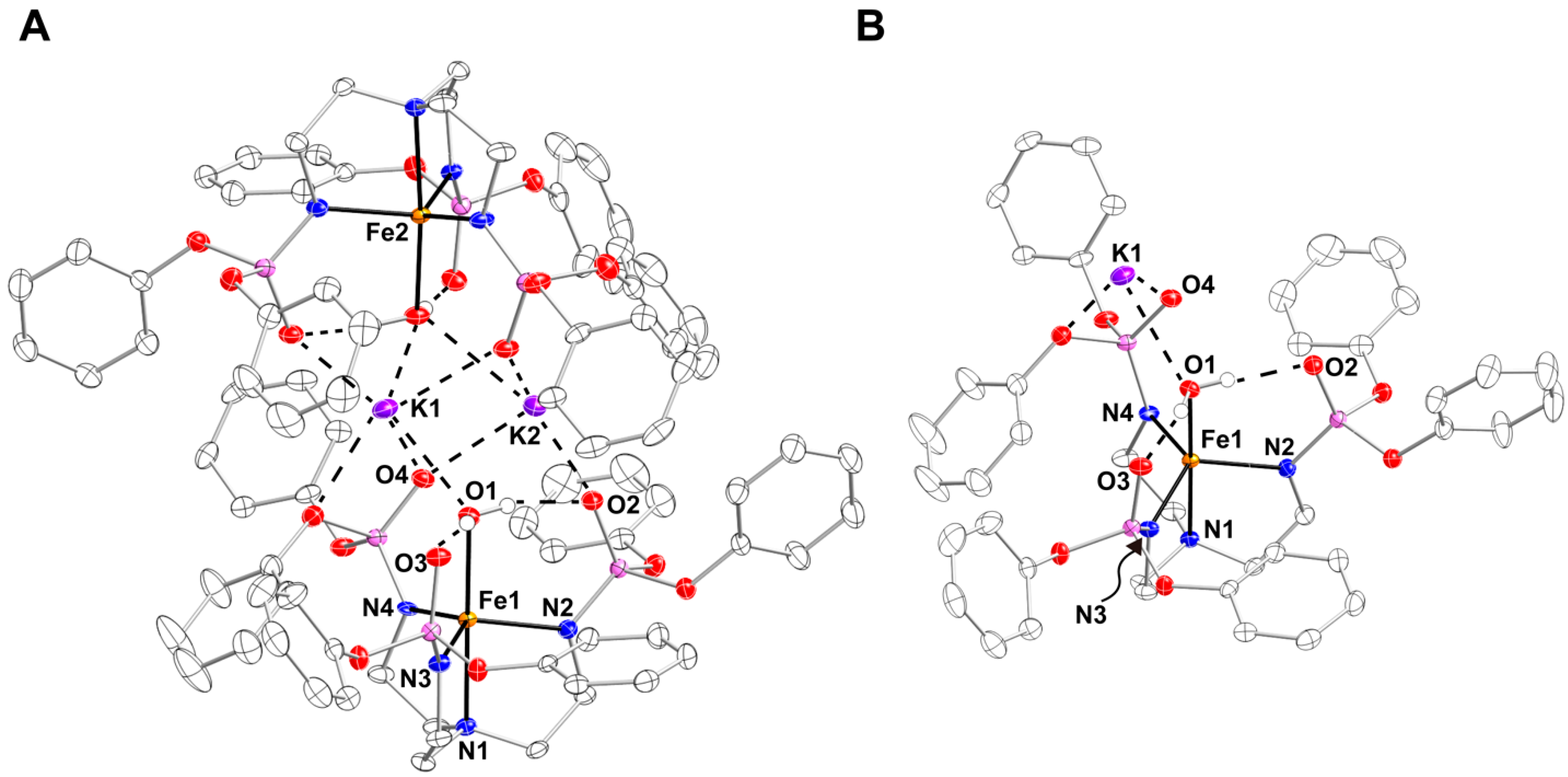
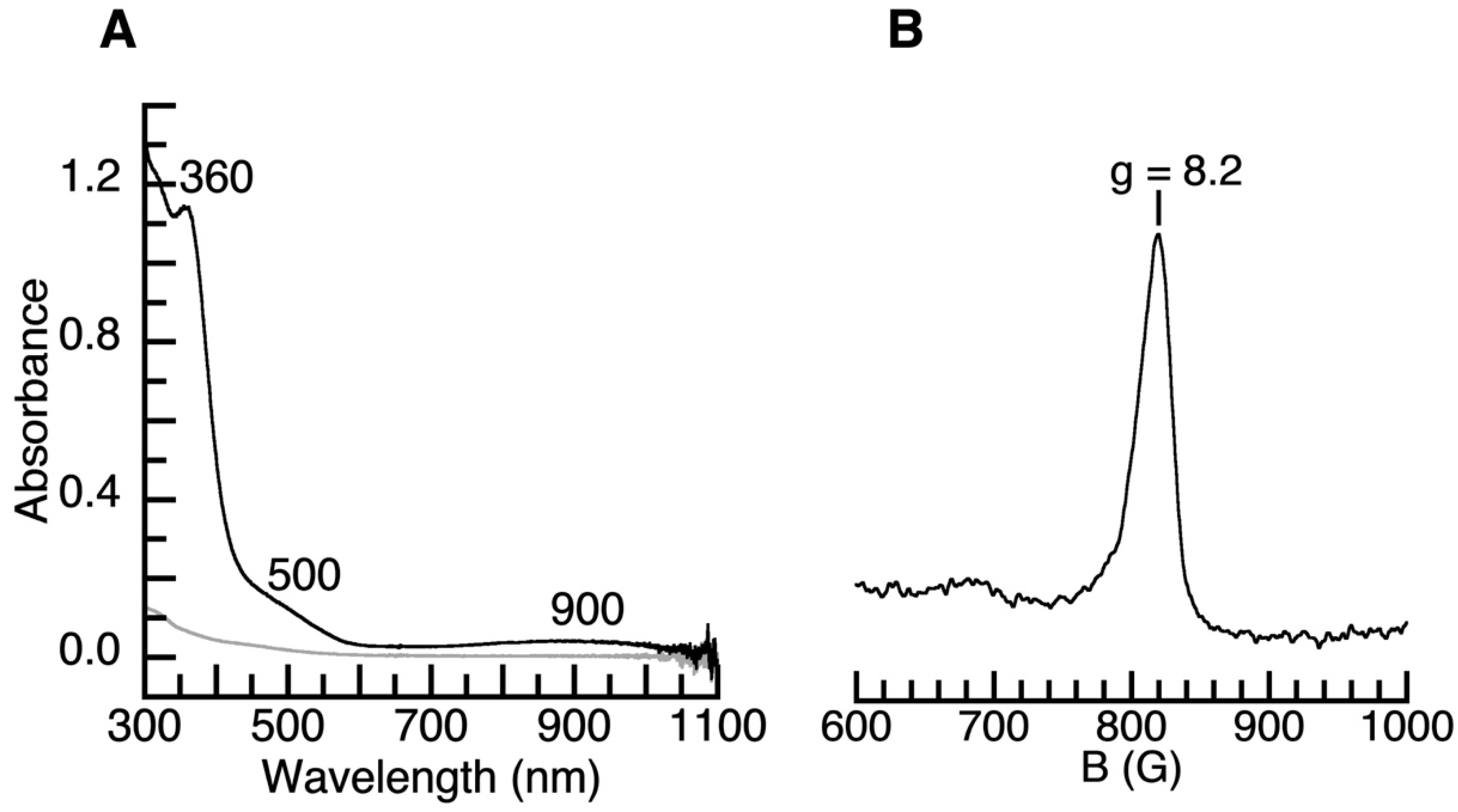
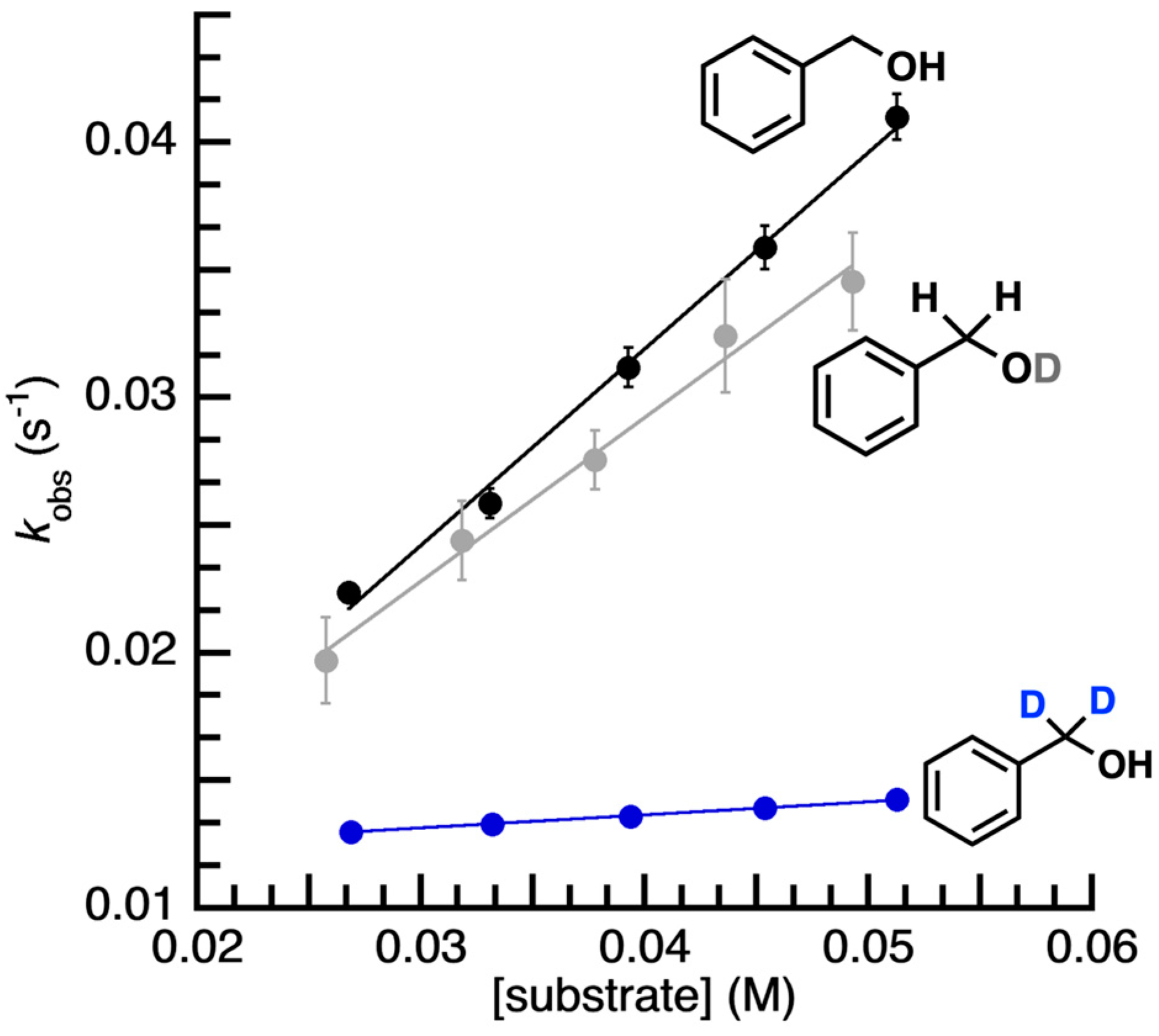
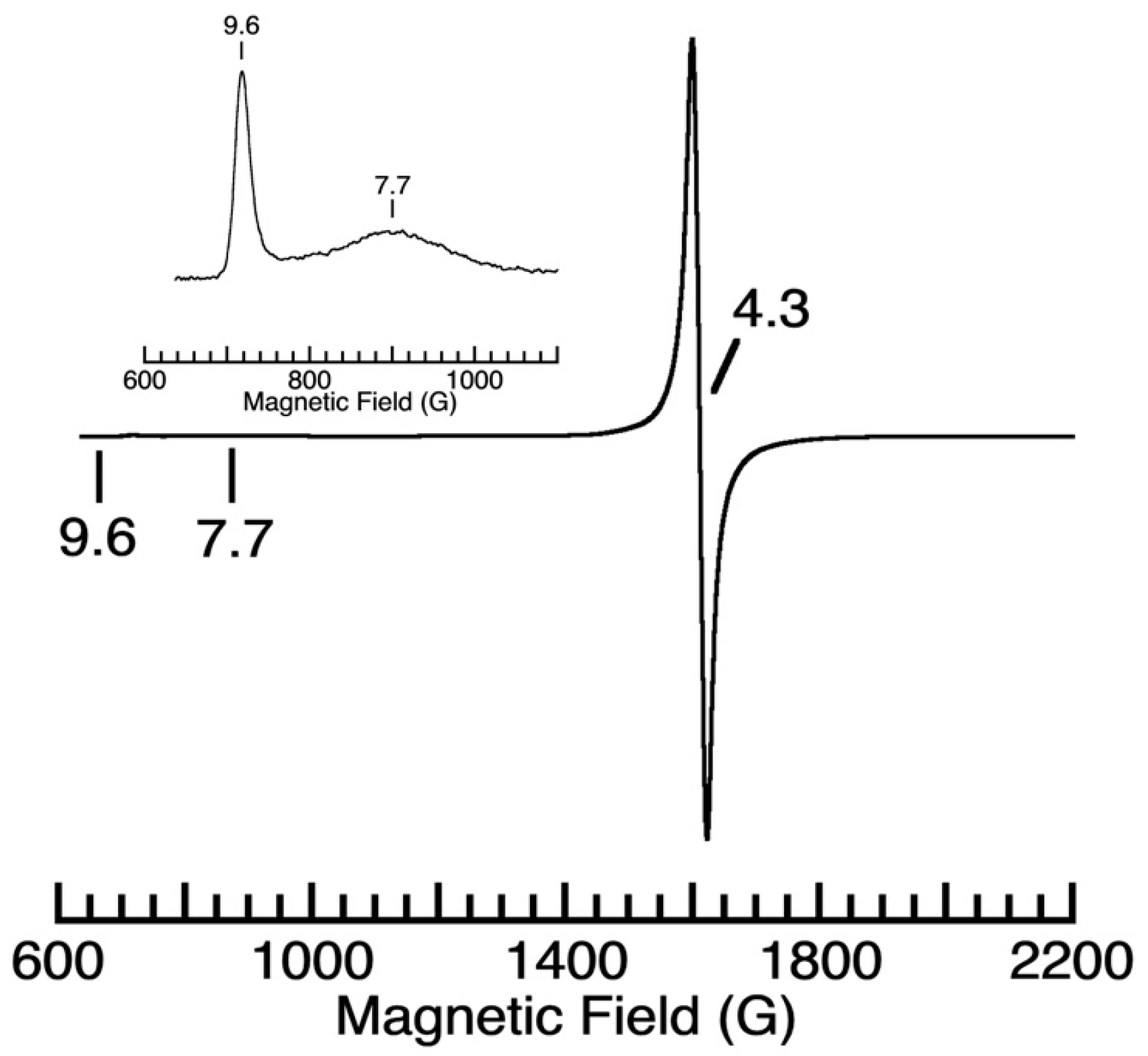
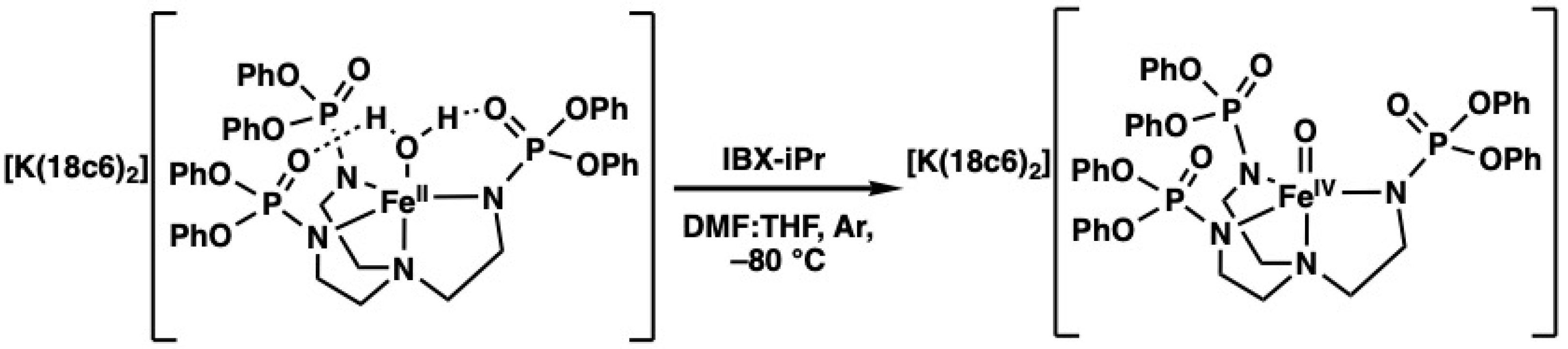
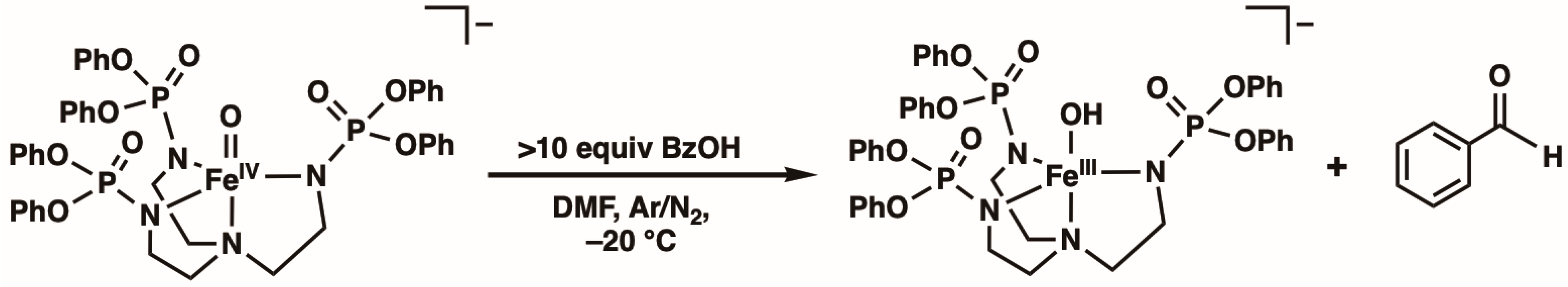
2. Results and Discussion
3. Summary
4. Experimental Section
4.1. General Procedures
4.2. Physical Methods
4.3. Preparative Methods
4.4. X-ray Crystallographic Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Srnec, M.; Wong, S.D.; England, J.; Que, L.; Solomon, E.I. π-Frontier Molecular Orbitals in S = 2 Ferryl Species and Elucidation of Their Contributions to Reactivity. Proc. Natl. Acad. Sci. USA 2012, 109, 14326–14331. [Google Scholar] [CrossRef]
- Paine, T.K.; Costas, M.; Kaizer, J.; Que, L. Oxoiron(IV) Complexes of the Tris(2-Pyridylmethyl)Amine Ligand Family: Effect of Pyridine α-Substituents. J. Biol. Inorg. Chem. 2006, 11, 272–276. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.R.; Que, L. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Krebs, C.; Fujimori, D.G.; Walsh, C.T.; Bollinger, J. Non-Heme Fe (IV)–Oxo Intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.U.; In, J.H.; Lim, M.H.; Brennessel, W.W.; Bukowski, M.R.; Stubna, A.; Münck, E.; Nam, W.; Que, L. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science 2003, 299, 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Banse, F.; Bartoli, J.F.; Mattioli, T.A.; Battioni, P.; Horner, O.; Bourcier, S.; Girerd, J.J. New Example of a Non-Heme Mononuclear Iron(IV) Oxo Complex. Spectroscopic Data and Oxidation Activity. Inorg. Chem. 2005, 44, 9592–9596. [Google Scholar] [CrossRef]
- Meyer, S.; Klawitter, I.; Demeshko, S.; Bill, E.; Meyer, F. A Tetracarbene-Oxoiron(IV) Complex. Angew. Chem.–Int. Ed. 2013, 52, 901–905. [Google Scholar] [CrossRef]
- Wang, D.; Ray, K.; Collins, M.J.; Farquhar, E.R.; Frisch, J.R.; Gómez, L.; Jackson, T.A.; Kerscher, M.; Waleska, A.; Comba, P.; et al. Nonheme Oxoiron(Iv) Complexes of Pentadentate N5 Ligands: Spectroscopy, Electrochemistry, and Oxidative Reactivity. Chem. Sci. 2013, 4, 282–291. [Google Scholar] [CrossRef]
- Klinker, E.J.; Kaizer, J.; Brennessel, W.W.; Woodrum, N.L.; Cramer, C.J.; Que, L. Structures of Nonheme Oxoiron(IV) Complexes from X-Ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem.–Int. Ed. 2005, 44, 3690–3694. [Google Scholar] [CrossRef]
- Lee, J.L.; Ross, D.L.; Barman, S.K.; Ziller, J.W.; Borovik, A.S. C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem. 2021, 60, 13759–13783. [Google Scholar] [CrossRef]
- Lacy, D.C.; Gupta, R.; Stone, K.L.; Greaves, J.; Ziller, J.W.; Hendrich, M.P.; Borovik, A.S. Formation, Structure, and EPR Detection of a High Spin FeIV-Oxo Species Derived from Either an FeIII-Oxo or FeIII-OH Complex. J. Am. Chem. Soc. 2010, 132, 12188–12190. [Google Scholar] [CrossRef]
- England, J.; Martinho, M.; Farquhar, E.R.; Frisch, J.R.; Bominaar, E.L.; Münck, E.; Que, L. A Synthetic High-Spin Oxoiron(IV) Complex: Generation, Spectro-Scopic Characterization, and Reactivity. Angew. Chem.–Int. Ed. 2009, 48, 3622–3626. [Google Scholar] [CrossRef]
- Bigi, J.P.; Harman, W.H.; Lassalle-Kaiser, B.; Robles, D.M.; Stich, T.A.; Yano, J.; Britt, R.D.; Chang, C.J. A High-Spin Iron(IV)-Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J. Am. Chem. Soc. 2012, 134, 1536–1542. [Google Scholar] [CrossRef]
- Barman, S.K.; Yang, M.Y.; Parsell, T.H.; Green, M.T.; Borovik, A.S. Semiempirical Method for Examining Asynchronicity in Metal-Oxido-Mediated C-H Bond Activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2108648118. [Google Scholar] [CrossRef]
- Cook, S.A.; Borovik, A.S. Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res. 2015, 48, 2407–2414. [Google Scholar] [CrossRef] [PubMed]
- Price, J.C.; Barr, E.W.; Tirupati, B.; Bollinger, J.M.; Krebs, C. Erratum: The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2004, 43, 1134. [Google Scholar] [CrossRef]
- O’Brien, J.R.; Schuller, D.J.; Yang, V.S.; Dillard, B.D.; Lanzilotta, W.N. Substrate-Induced Conformational Changes in Escherichia coli Taurine/α-Ketoglutarate Dioxygenase and Insight into the Oligomeric Structure. Biochemistry 2003, 42, 5547–5554. [Google Scholar] [CrossRef] [PubMed]
- Grzyska, P.K.; Ryle, M.J.; Monterosso, G.R.; Liu, J.; Ballou, D.P.; Hausinger, R.P. Steady-State and Transient Kinetic Analyses of Taurine/α- Ketoglutarate Dioxygenase: Effects of Oxygen Concentration, Alternative Sulfonates, and Active-Site Variants on the FeIV-Oxo Intermediate. Biochemistry 2005, 44, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Satpathy, J.K.; Bagha, U.K.; Mubarak, M.Q.E.; Sastri, C.V.; De Visser, S.P. Inspiration from Nature: Influence of Engineered Ligand Scaffolds and Auxiliary Factors on the Reactivity of Biomimetic Oxidants. ACS Catal. 2021, 11, 9761–9797. [Google Scholar] [CrossRef]
- Oswald, V.F.; Lee, J.L.; Biswas, S.; Weitz, A.C.; Mittra, K.; Fan, R.; Li, J.; Zhao, J.; Hu, M.Y.; Alp, E.E.; et al. Effects of Noncovalent Interactions on High-Spin Fe(IV)-Oxido Complexes. J. Am. Chem. Soc. 2020, 142, 11804–11817. [Google Scholar] [CrossRef]
- Sun, C. Secondary Coordination Sphere Effects on Properties and Reactivities of Metal Complexes; University of California: Irvine, CA, USA, 2021. [Google Scholar]
- Lee, J.L.; Oswald, V.F.; Biswas, S.; Hill, E.A.; Ziller, J.W.; Hendrich, M.P.; Borovik, A.S. Stepwise Assembly of Heterobimetallic Complexes: Synthesis, Structure, and Physical Properties. Dalt. Trans. 2021, 50, 8111–8119. [Google Scholar] [CrossRef] [PubMed]
- Matson, E.M.; Bertke, J.A.; Fout, A.R. Isolation of Iron(II) Aqua and Hydroxyl Complexes Featuring a Tripodal H-Bond Donor and Acceptor Ligand. Inorg. Chem. 2014, 53, 4450–4458. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.; Ziller, J.W.; Borovik, A.S. Sulfonamido Tripods: Tuning Redox Potentials via Ligand Modifications. Polyhedron 2015, 85, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Rhoda, R.N.; Fraioli, A.V.; Taylor, W.L.; Kleinberg, J. Iron(II) Formate. In Inorganic Syntheses, Vol. 4; John Wiley & Sons: Hoboken, NJ, USA, 1953; pp. 159–161. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Koposov, A.Y.; Litvinov, D.N.; Ferguson, M.J.; McDonald, R.; Luu, T.; Tykwinski, R.R. Esters of 2-Iodoxybenzoic Acid: Hypervalent Iodine Oxidizing Reagents with a Pseudobenziodoxole Structure. J. Org. Chem. 2005, 70, 6484–6491. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Litvinov, D.N.; Koposov, A.Y.; Luu, T.; Ferguson, M.J.; McDonald, R.; Tykwinski, R. Preparation and structure of 2-iodoxybenzoate esters: Soluble and stable periodinane oxidizing reagents. Chem. Commun. 2004, 2004, 106–107. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Jaimes, J.L.; Follmer, A.H.; Ziller, J.W.; Borovik, A.S. Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex. Molecules 2023, 28, 4755. https://doi.org/10.3390/molecules28124755
Sun C, Jaimes JL, Follmer AH, Ziller JW, Borovik AS. Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex. Molecules. 2023; 28(12):4755. https://doi.org/10.3390/molecules28124755
Chicago/Turabian StyleSun, Chen, Jennifer L. Jaimes, Alec H. Follmer, Joseph W. Ziller, and Andrew S. Borovik. 2023. "Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex" Molecules 28, no. 12: 4755. https://doi.org/10.3390/molecules28124755
APA StyleSun, C., Jaimes, J. L., Follmer, A. H., Ziller, J. W., & Borovik, A. S. (2023). Selective C–H Bond Cleavage with a High-Spin FeIV–Oxido Complex. Molecules, 28(12), 4755. https://doi.org/10.3390/molecules28124755