

An Automated Solid-Phase Extraction–UPLC–MS/MS Method for Simultaneous Determination of Sulfonamide Antimicrobials in Environmental Water
 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of Automated Solid Phase Extraction Procedures
2.1.1. Sorbent Type
2.1.2. Elution Solvent and Eluent Volume
2.1.3. Effect of Na2EDTA
2.1.4. Effect of Ionic Strength and pH Value
2.2. Matrix Effect
2.3. Evaluation of the Method Performance
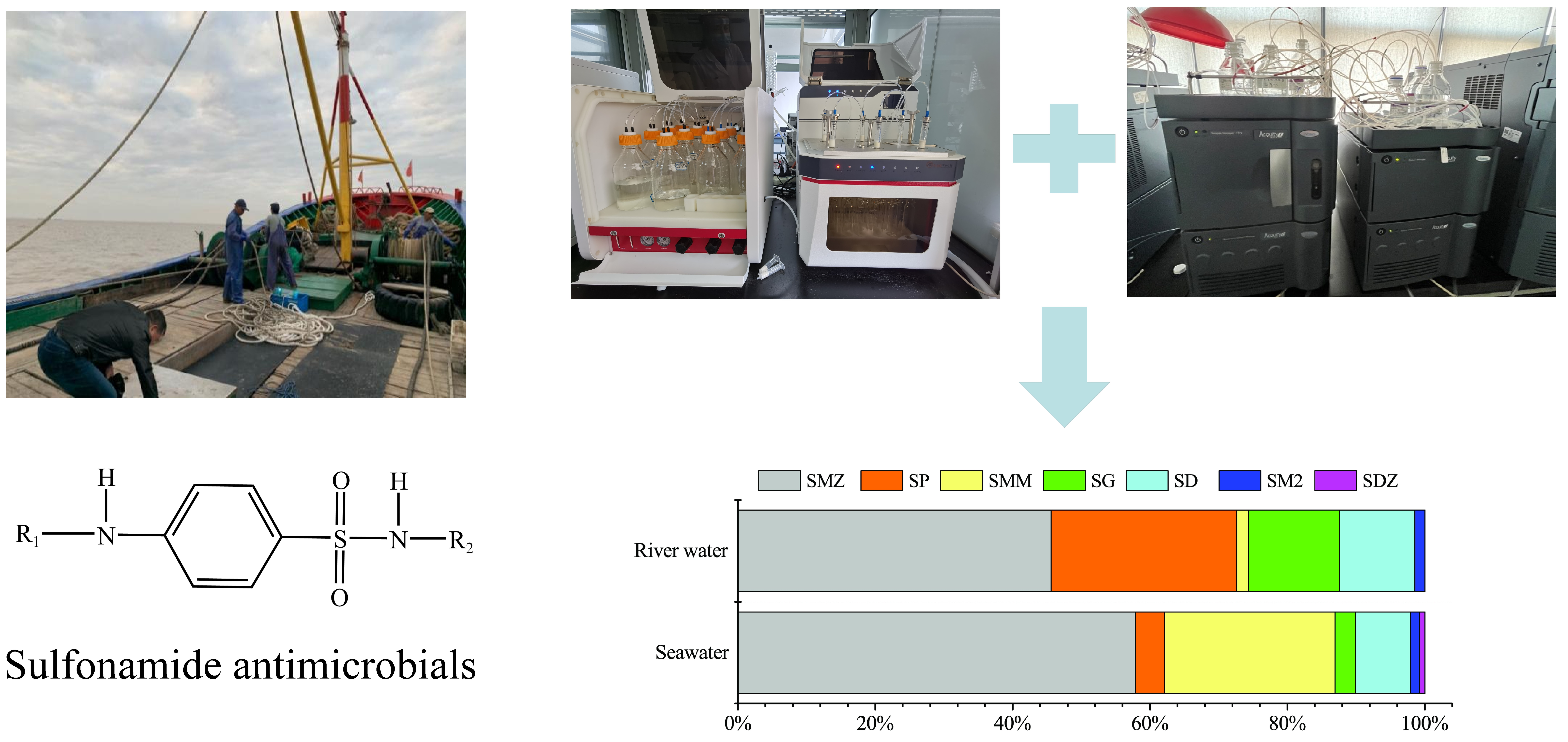
2.4. Real Water Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sampling and Preparation
3.3. Automated Solid-Phase Extraction
3.4. Instrumental Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kokoszka, K.; Wilk, J.; Felis, E.; Bajkacz, S. Application of UHPLC-MS/MS method to study occurrence and fate of sulfonamide antibiotics and their transformation products in surface water in highly urbanized areas. Chemosphere 2021, 283, 131189. [Google Scholar] [CrossRef]
- Semail, N.F.; Keyon, A.S.A.; Saad, B.; Kamaruzaman, S.; Zain, N.N.M.; Lim, V.; Miskam, M.; Abdullah, W.N.W.; Yahaya, N.; Chen, D.D.Y. Simultaneous preconcentration and determination of sulfonamide antibiotics in milk and yoghurt by dynamic pH junction focusing coupled with capillary electrophoresis. Talanta 2022, 236, 122833. [Google Scholar] [CrossRef]
- Kools, S.A.; Moltmann, J.F.; Knacker, T. Estimating the use of veterinary medicines in the European Union. Regul. Toxicol. Pharmacol. 2008, 50, 59–65. [Google Scholar] [CrossRef]
- Bruyndonckx, R.; Adriaenssens, N.; Versporten, A.; Hens, N.; Monnet, D.L.; Molenberghs, G.; Goossens, H.; Weist, K.; Coenen, S. Consumption of antibiotics in the community, European Union/European Economic Area, 1997–2017. J. Antimicrob. Chemother. 2021, 76, 7–13. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.S.Q.; Guo, Y.T.; Li, X.Y.; Wang, X.D.; Wang, J.X.; Qian, F.Y.; Gu, H.D.; Zhang, Z.N. Magnetic solid phase extraction of sulfonamides based oncarboxylated magnetic graphene oxide nanoparticles inenvironmental waters. J. Chromatogr. A 2018, 1575, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ben, W.W.; Ye, H.; Zhang, D.; Qiang, Z.M. Performance of ozonation and biological activated carbon in eliminating sulfonamides and sulfonamide-resistant bacteria: A pilot-scale study. Chem. Eng. J. 2018, 341, 327–334. [Google Scholar] [CrossRef]
- Cui, H.Y.; Chang, H.; Zheng, H.J.; Wan, Y. Determination and occurrence of sulfonamide transformation products in surface waters. Sci. Total Environ. 2021, 779, 146562. [Google Scholar] [CrossRef]
- Ministry of Agriculture of the People’s Republic of China. Announcement No. 235 of the Ministry of Agriculture of the People’s Republic of China; Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2008.
- Fan, Y.M.; Zeng, G.L.; Ma, X.G. Effects of prepolymerization on surface molecularly imprinted polymer for rapid separation and analysis of sulfonamides in water. J. Colloid Interface Sci. 2020, 571, 21–29. [Google Scholar] [CrossRef]
- Lu, T.L.; Shahriman, M.S.; Sambasevam, K.P.; Zain, N.N.M.; Yahaya, N.; Lim, V.; Raoov, M. In-tip solid-phase microextraction: A method for determination of sulphonamide residues in environmental water samples. Int. J. Environ. Anal. Chem. 2022, 1–16. [Google Scholar] [CrossRef]
- García-Galán, M.J.; Garrido, T.; Fraile, J.; Ginebreda, A.; Díaz-Cruz, M.S.; Barceló, D. Application of fully automated online solid phase extraction-liquid chromatography-electrospray-tandem mass spectrometry for the determination of sulfonamides and their acetylated metabolites in groundwater. Anal. Bioanal. Chem. 2011, 399, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.K.; Terry, K.A.; Toito, J. Determination of sulfonamide antibiotics in wastewater: A comparison of solid phase microextraction and solid phase extraction methods. J. Chromatogr. A 2006, 1131, 1–10. [Google Scholar] [CrossRef]
- Jurado, A.; Margareto, A.; Pujades, E.; Vázquez-Suñé, E.; Diaz-Cruz, M.S. Fate and risk assessment of sulfonamides and metabolites in urban groundwater. Environ. Pollut. 2020, 267, 115480. [Google Scholar] [CrossRef]
- Xue, M.; Wu, H.; Liu, S.; Huang, X.; Jin, Q.; Ren, R. Simultaneous determination of 44 pharmaceutically active compounds in water samples using solid-phase extraction coupled with ultra-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 203–222. [Google Scholar] [CrossRef]
- Panditi, V.R.; Batchu, S.R.; Gardinali, P.R. Online solid-phase extraction-liquid chromatography-electrospray-tandem mass spectrometry determination of multiple classes of antibiotics in environmental and treated waters. Anal. Bioanal. Chem. 2013, 405, 5953–5964. [Google Scholar] [CrossRef]
- Wang, X.; Ye, N.S.; Hu, X.Y.; Liu, Q.Y.; Li, J.; Peng, L.; Ma, X.T. Open-tubular capillary electrochromatographic determination of ten sulfonamides in tap water and milk by a metal-organic framework-coated capillary column. Electrophoresis 2018, 39, 2236–2245. [Google Scholar] [CrossRef]
- Jamal, S.; Baderia, V.K.; Agrawal, Y.K.; Sanghi, S.K. Fluorescence detection and identification of eight sulphonamides using capillary electrophoresis on released excipients in lake water. Arab. J. Chem. 2019, 12, 1338–1344. [Google Scholar] [CrossRef]
- Zhu, Y.J.; He, P.F.; Hu, H.M.; Qi, M.Y.; Li, T.J.; Zhang, X.N.; Guo, Y.M.; Wu, W.Y.; Lan, Q.P.; Yang, C.C.; et al. Determination of quinolone antibiotics in environmental water using automatic solid-phase extraction and isotope dilution ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2022, 1208, 123390. [Google Scholar] [CrossRef] [PubMed]
- Mokh, S.; El Khatib, M.; Koubar, M.; Daher, Z.; Al Iskandarani, M. Innovative SPE-LC-MS/MS technique for the assessment of 63 pharmaceuticals and the detection of antibiotic-resistant-bacteria: A case study natural water sources in Lebanon. Sci. Total Environ. 2017, 609, 830–841. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Borges-Miquel, T.M.; Rodríguez-Delgado, M.Á. Dispersive liquid–liquid microextraction combined with ultra-high performance liquid chromatography for the simultaneous determination of 25 sulfonamide and quinolone antibiotics in water samples. J. Pharm. Biomed. Anal. 2013, 75, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, C.; Xu, Z.A.; Chakraborty, A. A coupled method of on-line solid phase extraction with the UHPLC–MS/MS for detection of sulfonamides antibiotics residues in aquaculture. Chemosphere 2020, 254, 126765. [Google Scholar] [CrossRef]
- Nakhjavan, B.; Bland, J.; Khosravifard, M. Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS. Molecules 2021, 26, 6627. [Google Scholar] [CrossRef]
- Zheng, M.; Tang, S.W.; Bao, Y.Y.; Daniels, K.D.; How, Z.T.; El-Din, M.G.; Wang, J.; Tang, L. Fully-automated SPE coupled to UHPLC-MS/MS method for multiresidue analysis of 26 trace antibiotics in environmental waters: SPE optimization and method validation. Environ. Sci. Pollut. Res. 2022, 29, 16973–16987. [Google Scholar] [CrossRef] [PubMed]
- Khurana, P.; Pulicharla, R.; Brar, S.K. Antibiotic-metal complexes in wastewaters: Fate and treatment trajectory. Environ. Int. 2021, 157, 106863. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shi, W.; You, M.; Zhang, R.; Kuang, Y.; Dang, C.; Sun, W.; Zhou, Y.; Wang, W.; Ni, J. Antibiotics in water and sediments of Danjiangkou Reservoir, China: Spatiotemporal distribution and indicator screening. Environ. Pollut. 2019, 246, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef]
- Zhou, L.J.; Ying, G.G.; Liu, S.; Zhao, J.L.; Chen, F.; Zhang, R.Q.; Peng, F.Q.; Zhang, Q.Q. Simultaneous determination of human and veterinary antibiotics in various environmental matrices by rapid resolution liquid chromatography–electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2012, 1244, 123–138. [Google Scholar] [CrossRef]
- Yang, L.H.; Shi, Y.; Li, J.J.; Luan, T.G. In situ derivatization and hollow-fiber liquid-phase microextraction to determine sulfonamides in water using UHPLC with fluorescence detection. J. Sep. Sci. 2018, 41, 1651–1662. [Google Scholar] [CrossRef]
- Li, L.; Liu, D.; Zhang, Q.; Song, K.; Zhou, X.H.; Tang, Z.; Zhou, X. Occurrence and ecological risk assessment of selected antibiotics in the freshwater lakes along the middle and lower reaches of Yangtze River Basin. J. Environ. Manag. 2019, 249, 109396. [Google Scholar] [CrossRef]
- Shimizu, A.; Takada, H.; Koike, T.; Takeshita, A.; Reungsang, A. Ubiquitous occurrence of sulfonamides in tropical Asian waters. Sci. Total Environ. 2013, 452–453, 108–115. [Google Scholar] [CrossRef]
- Duan, W.Y.; Cui, H.W.; Jia, X.Y.; Huang, X. Occurrence and ecotoxicity of sulfonamides in the aquatic environment: A review. Sci. Total Environ. 2022, 820, 153178. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SPE Cartridge | Dimensions | Sorbent Properties |
|---|---|---|
| CNW Poly-Sery HLB | 50–80 μm a, 500 mg b, 6 mL c | hydrophilic–lipophilic balanced reversed-phase sorbent |
| CNW Poly-Sery MCX | 92 μm, 500 mg, 6 mL | mixed-mode cation exchange sorbent |
| CNW Poly-Sery MAX | 35–45 μm, 500 mg, 6 mL | mixed-mode anion exchange sorbent |
| CNW Poly-Sery XAD2 | 694 μm, 500 mg, 6 mL | non-ionic reticulated styrene–divinylbenzene polymer sorbent |
| CNWBOND LC-C18 | 40–63 μm, 500 mg, 6 mL | C18-bonded non-polar silica |
| Abbreviation | Full Name | Molecular Weight | Log KOW a | pKa b | Use | Retention Time (min) | Precursor Ion (m/z) | Product Ion (m/z) | Cone Voltage (V) | Collision Energy (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| SG | sulfaguanidine | 214.24 | NA | NA | human | 1.03 | 215 | 108, 156 * | 34 | 20, 15 |
| SG-D4 | 218.27 | 1.03 | 219 | 160 * | 34 | 15 | ||||
| SP | sulfapyridine | 249.29 | 0.35 | 2.90, 8.54 | human and veterinary | 3.12 | 250 | 156 *, 184 | 28 | 15, 16 |
| SP-13C6 | 255.24 | 3.12 | 256 | 162 * | 28 | 16 | ||||
| SD | sulfadiazine | 250.28 | −0.09 | 2.00, 6.48 | human and veterinary | 2.30 | 251 | 92, 156 * | 23 | 27, 15 |
| SD-13C6 | 256.24 | 2.30 | 257 | 162 * | 23 | 15 | ||||
| ST | sulfathiazole | 255.32 | 0.05 | 2.20, 7.24 | human and veterinary | 2.81 | 256 | 92, 156 * | 26 | 25, 15 |
| ST-D4 | 259.34 | 2.76 | 260 | 160 * | 26 | 15 | ||||
| SM1 | sulfamerazine | 264.30 | 0.14 | 2.06, 6.90 | human and veterinary | 3.51 | 265 | 92, 156 * | 24 | 25, 15 |
| SM1-13C6 | 270.26 | 3.51 | 271 | 162 * | 24 | 15 | ||||
| SML | sulfamethizole | 270.33 | 0.54 | 1.86, 5.29 | veterinary | 5.42 | 271 | 92, 156 * | 19 | 30, 15 |
| SML-13C6 | 276.29 | 5.42 | 277 | 162 * | 19 | 15 | ||||
| SMZ | sulfamethoxazole | 253.28 | 0.89 | 1.85, 5.60 | human and veterinary | 9.28 | 254 | 92, 156 * | 27 | 26, 16 |
| SMZ-13C6 | 259.23 | 9.27 | 260 | 162 * | 27 | 16 | ||||
| SIZ | sulfisoxazole | 267.30 | 1.01 | 1.66, 4.71 | human and veterinary | 10.19 | 268 | 92, 156 * | 22 | 28, 13 |
| SIZ-13C6 | 273.26 | 10.19 | 274 | 162 * | 22 | 13 | ||||
| SIM | sulfisomidine | 278.33 | NA | NA | human | 1.96 | 279 | 156 *, 186 | 30 | 18, 25 |
| SIM-D4 | 282.31 | 1.96 | 283 | 160 * | 30 | 18 | ||||
| SM2 | sulfamethazine | 278.33 | 0.89 | 2.65, 7.65 | human and veterinary | 5.19 | 279.1 | 92, 186 * | 30 | 28, 16 |
| SM2-D4 | 282.35 | 5.11 | 283.1 | 186 * | 30 | 16 | ||||
| SMM | sulfamonomethoxine | 280.30 | 0.70 | 1.98, 5.96 | human and veterinary | 7.73 | 281 | 92 *, 156 | 28 | 31, 22 |
| SMM-D4 | 284.33 | 7.63 | 285 | 96 * | 28 | 22 | ||||
| SMP | sulfamethoxypyridazine | 280.30 | 0.32 | 2.09, 6.95 | human and veterinary | 5.80 | 281 | 156 *, 215 | 34 | 20, 15 |
| SMP-D3 | 283.32 | 5.69 | 284 | 156 * | 34 | 20 | ||||
| SM | sulfameter | 280.30 | 0.41 | 1.87, 6.50 | human and veterinary | 5.24 | 281 | 156 *, 215 | 32 | 20, 18 |
| SM-D4 | 284.33 | 5.16 | 285 | 160 * | 32 | 20 | ||||
| SDZ | sulfachloropyridazine | 284.72 | 0.31 | 1.87, 5.45 | human and veterinary | 7.80 | 285.1 | 92, 156 * | 22 | 28, 15 |
| SDZ-13C6 | 290.68 | 7.80 | 291.1 | 162 * | 22 | 15 | ||||
| SQ | sulfaquinoxaline | 300.34 | 1.68 | 1.86, 5.56 | veterinary | 11.50 | 301.1 | 92.2, 156.1 * | 26 | 30, 16 |
| SQ-13C6 | 306.29 | 11.50 | 307.1 | 162.1 * | 26 | 16 | ||||
| SDM | sulfadoxine | 310.33 | 0.70 | 1.52, 6.01 | human | 9.50 | 311 | 92, 156 * | 20 | 32, 15 |
| SDM-D3 | 313.35 | 9.42 | 314 | 156 * | 25 | 20 | ||||
| SPM | sulfadimethoxine | 310.33 | 1.63 | 1.87, 5.86 | veterinary | 11.40 | 311.1 | 92, 156 * | 20 | 32, 21 |
| SPM-D6 | 316.37 | 11.33 | 317.1 | 162.1 * | 25 | 20 |
| Analyte | ILIS | Linear Range (μg/L) | Regression Equation | r2 | LOD a (ng/L) | LOQ b (ng/L) | EFs | Precision, RSD (%, n = 5) | |
|---|---|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | ||||||||
| SG | SG-D4 | 0.05−100 | y = 0.87x + 0.28 | 0.9999 | 0.05 | 0.15 | 1008 | 1.44 | 5.52 |
| SP | SP-13C6 | 0.05−100 | y = 0.93x + 0.21 | 0.9998 | 0.01 | 0.03 | 1007 | 1.96 | 3.54 |
| SD | SD-13C6 | 0.05−100 | y = 0.79x + 0.24 | 0.9995 | 0.01 | 0.03 | 990 | 2.64 | 4.06 |
| ST | ST-D4 | 0.05−100 | y = 0.84x + 0.21 | 0.9995 | 0.012 | 0.04 | 1033 | 2.39 | 3.83 |
| SM1 | SM1-13C6 | 0.05−100 | y = 0.76x + 0.18 | 0.9994 | 0.01 | 0.03 | 1021 | 3.35 | 2.58 |
| SML | SML-13C6 | 0.05−100 | y = 0.79x + 0.16 | 0.9999 | 0.02 | 0.06 | 1021 | 3.05 | 3.73 |
| SMZ | SMZ-13C6 | 0.05−100 | y = 0.82x + 0.18 | 0.9997 | 0.01 | 0.03 | 988 | 2.58 | 3.26 |
| SIZ | SIZ-13C6 | 0.05−100 | y = 0.81x + 0.21 | 0.9994 | 0.01 | 0.03 | 1018 | 2.76 | 3.64 |
| SIM | SIM-D4 | 0.05−100 | y = 0.96x + 0.09 | 0.9996 | 0.04 | 0.12 | 1004 | 3.76 | 8.9 |
| SM2 | SM2-D4 | 0.05−100 | y = 0.79x + 0.17 | 0.9997 | 0.01 | 0.03 | 982 | 2.57 | 7.94 |
| SMM | SMM-D4 | 0.05−100 | y = 0.74x + 0.17 | 0.9998 | 0.02 | 0.06 | 1019 | 2.64 | 3.56 |
| SMP | SMP-D3 | 0.05−100 | y = 0.70x + 0.19 | 0.9995 | 0.01 | 0.03 | 1019 | 3.54 | 2.08 |
| SM | SM-D4 | 0.05−100 | y = 0.76x + 0.27 | 0.9993 | 0.015 | 0.05 | 995 | 3.84 | 3.00 |
| SDZ | SDZ-13C6 | 0.05−100 | y = 0.82x + 0.25 | 0.9993 | 0.02 | 0.06 | 1006 | 2.34 | 3.61 |
| SQ | SQ-13C6 | 0.05−100 | y = 0.82x + 0.60 | 0.9994 | 0.02 | 0.06 | 987 | 2.59 | 3.62 |
| SDM | SDM-D3 | 0.05−100 | y = 1.18x + 0.68 | 0.9992 | 0.01 | 0.03 | 1007 | 1.86 | 2.67 |
| SPM | SPM-D6 | 0.05−100 | y = 1.54x + 0.40 | 0.9998 | 0.01 | 0.03 | 1019 | 1.87 | 2.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, M.; He, P.; Hu, H.; Zhang, T.; Li, T.; Zhang, X.; Qin, Y.; Zhu, Y.; Guo, Y. An Automated Solid-Phase Extraction–UPLC–MS/MS Method for Simultaneous Determination of Sulfonamide Antimicrobials in Environmental Water. Molecules 2023, 28, 4694. https://doi.org/10.3390/molecules28124694
Qi M, He P, Hu H, Zhang T, Li T, Zhang X, Qin Y, Zhu Y, Guo Y. An Automated Solid-Phase Extraction–UPLC–MS/MS Method for Simultaneous Determination of Sulfonamide Antimicrobials in Environmental Water. Molecules. 2023; 28(12):4694. https://doi.org/10.3390/molecules28124694
Chicago/Turabian StyleQi, Mengyu, Pengfei He, Hongmei Hu, Tongtong Zhang, Tiejun Li, Xiaoning Zhang, Yilin Qin, Yingjie Zhu, and Yuanming Guo. 2023. "An Automated Solid-Phase Extraction–UPLC–MS/MS Method for Simultaneous Determination of Sulfonamide Antimicrobials in Environmental Water" Molecules 28, no. 12: 4694. https://doi.org/10.3390/molecules28124694
APA StyleQi, M., He, P., Hu, H., Zhang, T., Li, T., Zhang, X., Qin, Y., Zhu, Y., & Guo, Y. (2023). An Automated Solid-Phase Extraction–UPLC–MS/MS Method for Simultaneous Determination of Sulfonamide Antimicrobials in Environmental Water. Molecules, 28(12), 4694. https://doi.org/10.3390/molecules28124694