3.1. Materials
All reactions were performed in oven-dried glassware under normal atmosphere, unless otherwise noted. All reagents and solvents were purchased from Fluorochem, TCI, or Merck and used without any further purification. Where specified, reactions were monitored via thin-layer chromatography (TLC) on POLYGRAM® Xtra Sil G/UV254 (0.2 mm layer thickness; Macherey-Nagel); spots were observed under a UV lamp at 254 or 365 nm. Gravimetric column chromatography was performed using silica gel (60 Å, particles size: 0.63–0.2 mm) as stationary phase. 1H, 13c, 19F, and 11B NMR spectra were acquired on a Bruker AVANCE I 400 instrument at 400.13, 100.61, 376.5, and 128.38 MHz, respectively, and are referenced using residual non-deuterated solvents (CHCl3 7.26 ppm in CDCl3, DMSO 2.50 ppm in d6-DMSO); deuterated solvents were purchased from Eurisotop; multiplicities are abbreviated as singlet (s), doublet (d), doublet of doublets (dd), and triplet (t); coupling constants are reported in Hz; IR spectra were acquired in attenuated total reflectance (ATR) mode using a FT-IR Thermo Scientific Nicolet iS10 Smart iTR instrument equipped with a diamond optical element, over a range 4000–650 cm−1 (at 4 cm−1 resolution). Intensities are denoted as: br = broadened signal, sh = shoulder, vs = very strong, s = strong, m = medium, w = weak and vw = very weak.
3.3. Synthesis of the Complexes
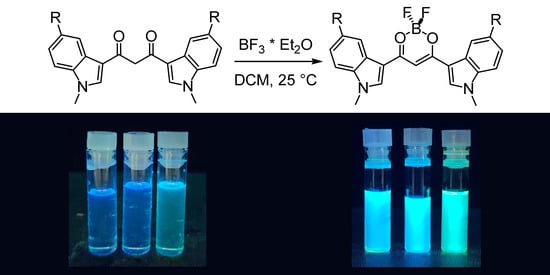
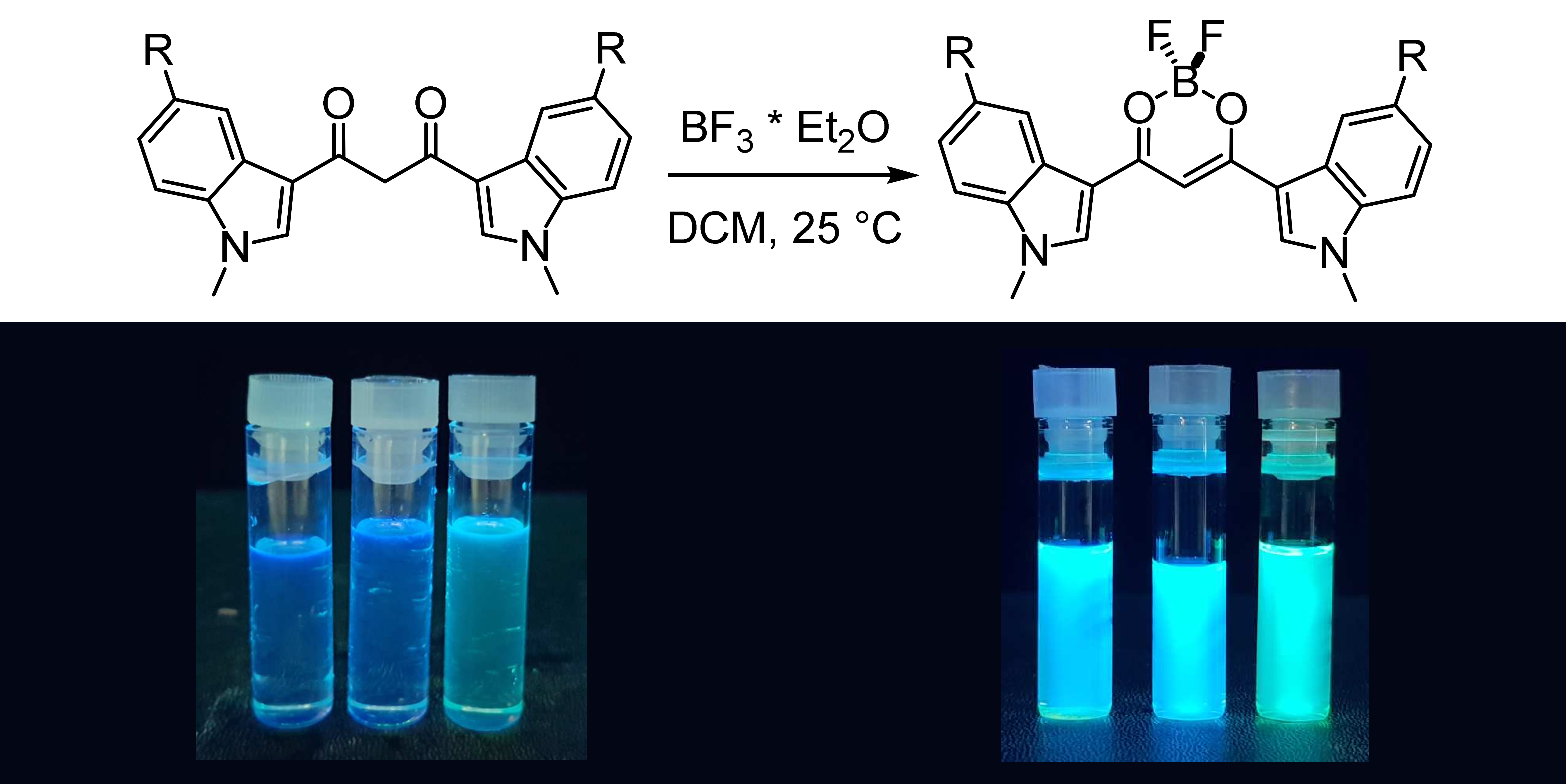
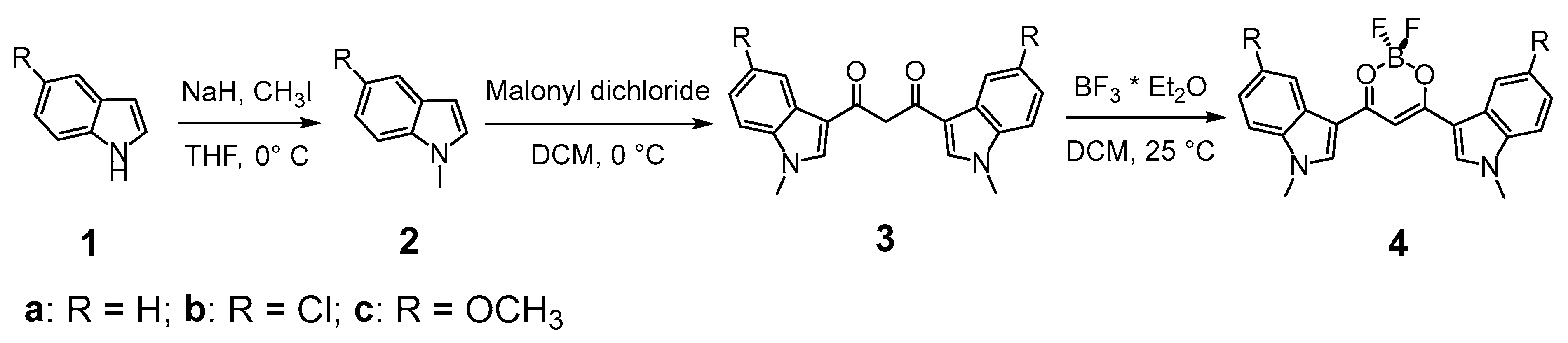
3.3.1. 1,3-Bis(1-methyl-3-indolyl)propane-1,3-dione Difluoroborate (4a)
To a solution of HBIP (100 mg, 0.30 mmol) in dichloromethane (100 mL), BF3·Et2O (1.1 mL, 9.0 mmol) is added, and the mixture is stirred at rt for 5 min: a colored precipitate forms. The solid is filtered on a Buchner filter funnel and washed with cold dichloromethane and then dried under vacuum. BIP difluoroborate is obtained as an orange solid (73 mg, 64% yield). 1H-NMR (d6-DMSO) δ = 8.77 (s, 2H), 8.19 (m, 2H), 7.67 (m, 2H), 7.38 (m, 4H), 7.18 (s, 1H, enolate C-H), 3.97 (s, 6H, N-CH3). 13c-NMR (d6-DMSO) δ = 174.8 (+), 138.8 (−), 137.9 (+), 125.1 (+), 123.5 (−), 122.9 (−), 121.4 (−), 111.6 (−), 109.4 (+), 91.0 (−), 33.8 (−). 11B-NMR (d6-DMSO) δ = 0.91 (br s). 19F-NMR (d6-DMSO) δ = −142.6 (br s), −142.7 (br s). IR (ATR) ῦ (cm−1) = 3567 (br, vs), 3134 (vw), 3053 (vw), 2899 (vw), 1610 (w), 1583 (s), 1560 (vs), 1538 (vs), 1527 (vs), 1510 (br, vs), 1489 (sh, s), 1464 (vs), 1452 (sh, s), 1411 (vw), 1384 (s), 1363 (s), 1313 (s), 1307 (s), 1257 (vw), 1235 (s), 1176 (sh, m), 1165 (s), 1148 (s), 1128 (vs), 1122 (vs), 1087 (vs), 1054 (m), 1015 (w), 967 (s), 944 (w), 916 (w), 884 (w), 841 (vs), 780 (w), 766 (w), 750 (vs), 744 (vs).
3.3.2. 1,3-Bis(5-chloro-1-methyl-3-indolyl)propane-1,3-dione Difluoroborate (4b)
To a solution of HBClIP (3b) (100 mg, 0.25 mmol) in dichloromethane (100 mL), BF3·Et2O (1 mL, 7.8 mmol) is added, and the mixture is stirred at rt for 5 min: a colored precipitate forms. The solid is filtered on a Buchner filter funnel and washed with cold dichloromethane. The residue is purified via column chromatography (DCM, Rf = 0.45) to obtain BClIP difluoroborate as an orange solid (73 mg, 68% yield). 1H-NMR (d6-DMSO) δ = 8.59 (s, 2H, H2), 8.12 (d, 4J = 2.05 Hz, 2H, H4), 7.71 (d, 3J = 9.0 Hz, 2H, H7), 7.4 (dd, 3J = 9.0 Hz, 4J = 2.2 Hz, 2H, H6), 7.20 (s, 1H, enolate C-H), 3.97 (s, 6H, N-CH3). 13c-NMR (d6-DMSO) δ = 174.8 (+), 140.1 (−), 136.5 (+), 127.8 (+), 126.1 (+), 123.5 (−), 120.5 (−), 113.5 (−), 91.1 (−), 40.4 (−). 11B-NMR (d6-DMSO) δ = 0.85 (br s). 19F-NMR (d6-DMSO) δ = −139.3 (br s), −139.4 (br s). IR (ATR) ῦ (cm−1) = 3123 (vw), 1616 (vw), 1558 (br, vs), 1527 (s), 1509 (m), 1466 (m), 1450 (m), 1428 (w), 1424 (vw), 1393 (vs), 1377 (m), 1368 (m), 1348 (vw), 1341 (vw), 1328 (s), 1296 (s), 1239 (sh, w), 1230 (s), 1177 (w), 1143 (m), 1137 (m), 1106 (s), 1078 (m), 1063 (m), 1034 (s), 1018 (sh, w), 993 (w), 988 (w), 928 (w), 919 (vw), 886 (w), 875 (w), 852 (w), 845 (vw), 826 (w), 802 (w), 793 (m), 783 (m), 777 (m), 736 (w), 683 (m), 638 (w).
3.3.3. 1,3-Bis(5-methoxy-1-methyl-3-indolyl)propane-1,3-dione Difluoroborate (4c)
To a solution of HBMIP (3c) (100 mg, 0.26 mmol) in dichloromethane (100 mL), BF3·Et2O (1 mL, 7.80 mmol) is added, and the mixture was stirred at rt for 5 min: a colored precipitate forms. The solid is filtered on a Buchner filter funnel, washed with cold dichloromethane, and then dried under vacuum. BMIP difluoroborate is obtained as an orange solid (80 mg, 70%). 1H-NMR (d6-DMSO) δ = 8.64 (s, 2H, H2), 7.62 (d, 4J = 2.3 Hz, 2H, H4), 7.57 (d, 3J = 8.9 Hz, 2H, H7), 7.08 (s, 1H, enolate C-H), 7.02 (dd, 3J = 8.9 Hz, 4J = 2.4 Hz, 2H, H6), 3.93 (s, 6H), 3.85 (s, 6H). 13c-NMR (d6-DMSO) δ = 174.5 (+), 156.2 (+), 138.6 (−), 132.9 (+), 126 (+), 112.4 (−), 112.3 (−), 109 (+), 104.2 (−), 90.6 (−), 55.5 (−), 33.9 (−). 11B-NMR (d6-DMSO) δ = 0.88 (br s). 19F-NMR (d6-DMSO) δ = −139.8 (br s), −139.9 (br s). IR (ATR) ῦ (cm−1) = 3131 (vw), 2946 (vw), 1619 (w), 1575 (sh, m), 1554 (br, vs), 1523 (vs), 1471 (vs), 1459 (sh, s), 1440 (m), 1425 (w), 1391 (vs), 1367 (s), 1345 (vw), 1330 (m), 1298 (m), 1282 (m), 1273 (m), 1228 (sh, m), 1221 (s), 1212 (sh, m), 1143 (w), 1126 (m), 1096 (s), 1060 (m), 1024 (m), 984 (m), 894 (w), 860 (vw), 850 (w), 811 (w), 800 (w), 777 (m), 736 (vw), 702 (sh, m), 698 (s).
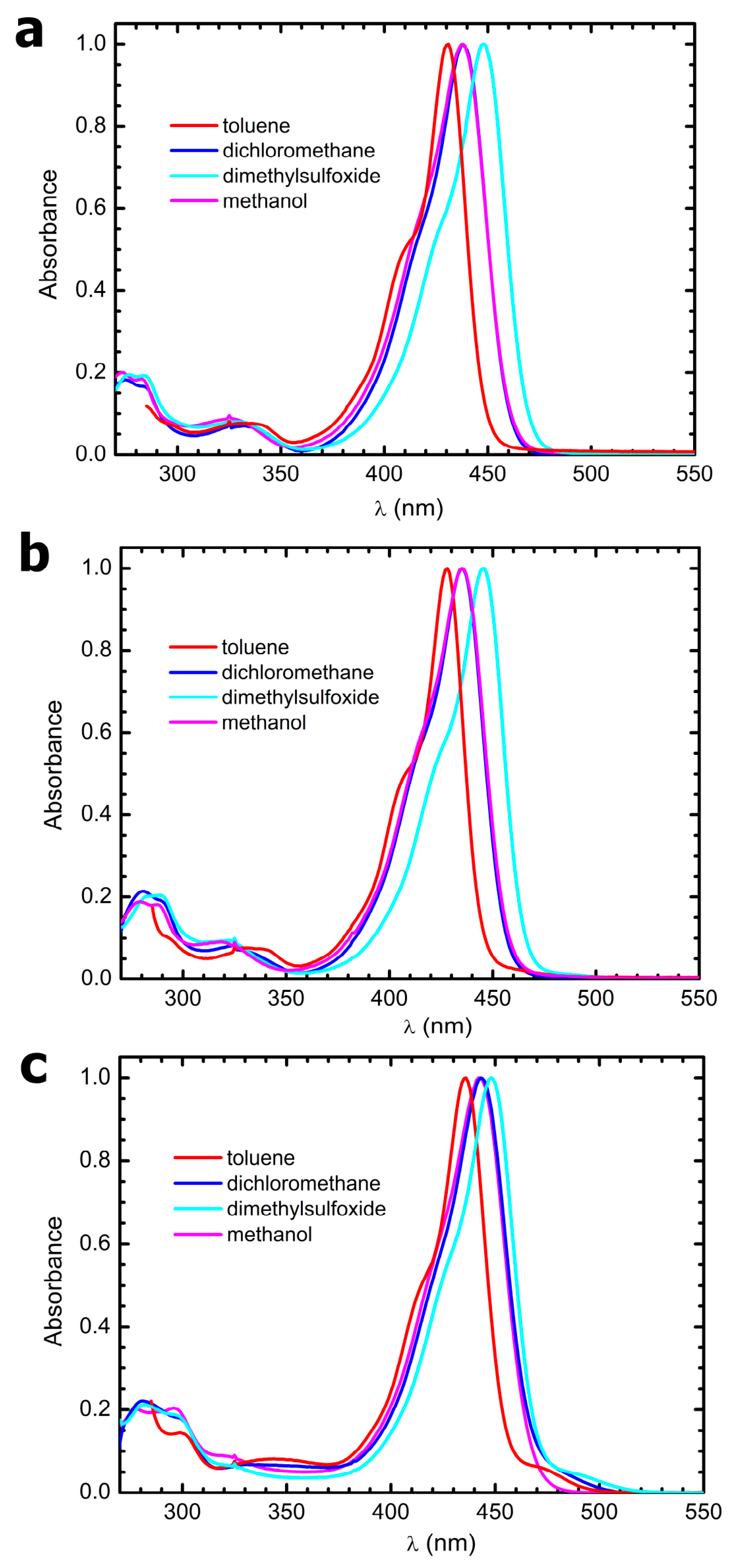
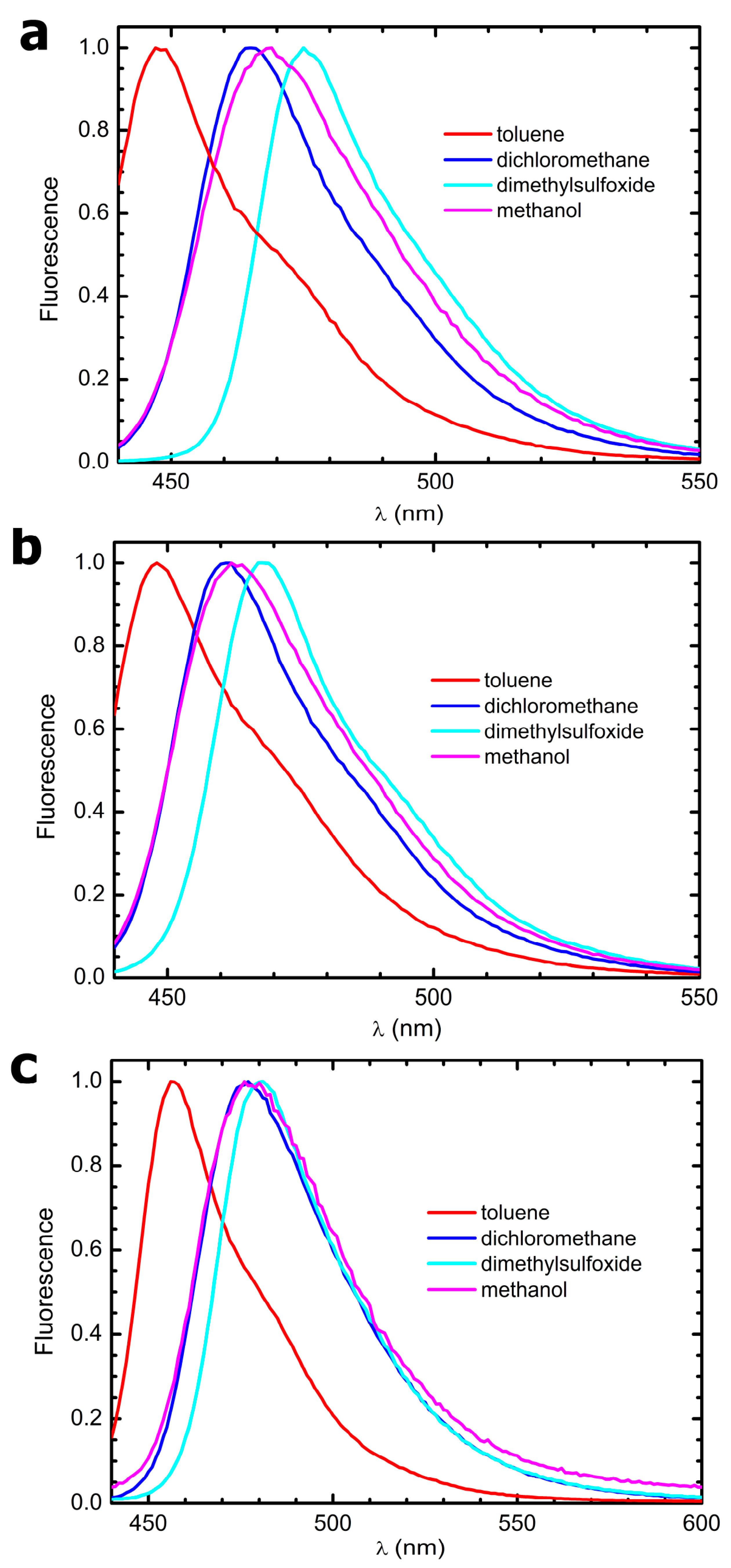
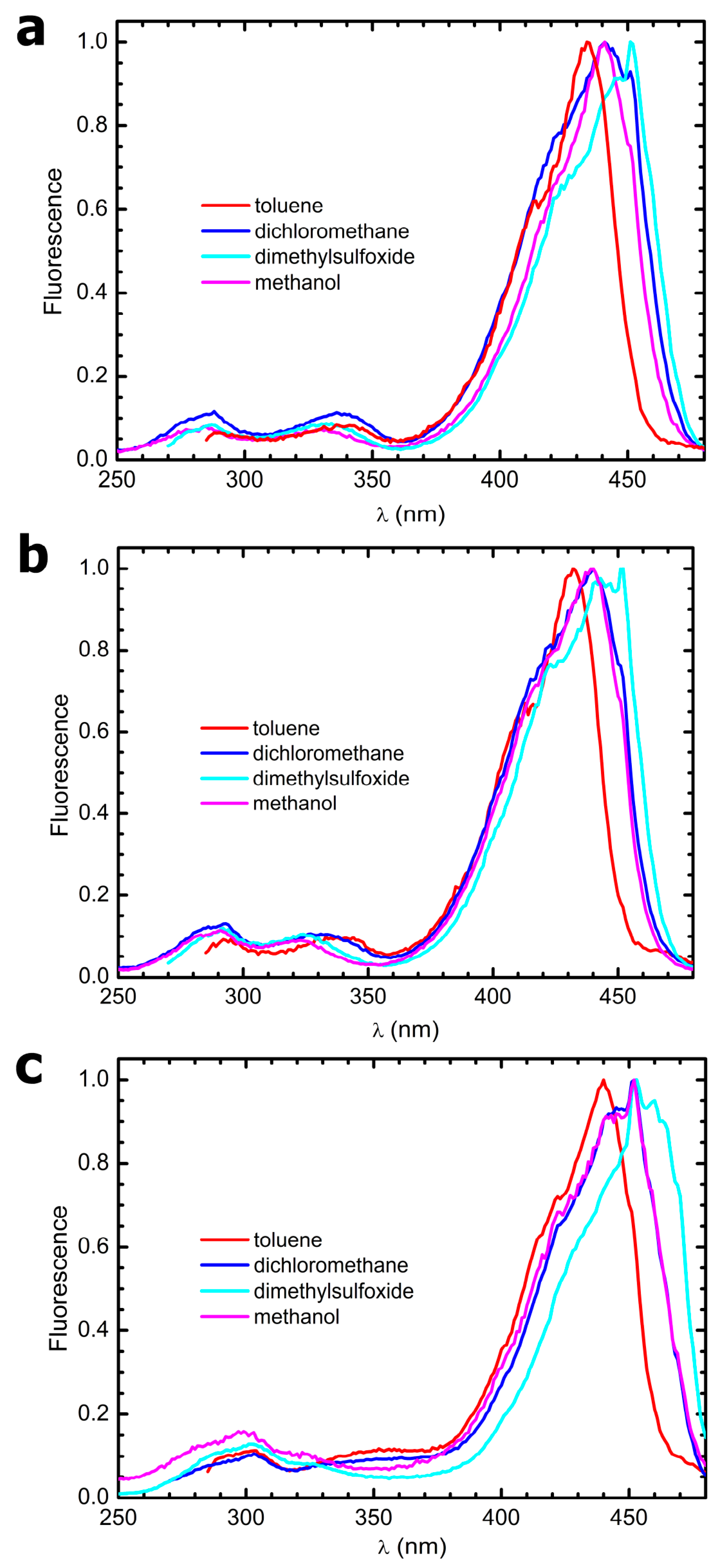
3.4. Steady-State Electronic-State Transition Spectroscopy Measurements
The UV-Vis absorption spectra were recorded with a Perkin Elmer Lambda2 spectrophotometer. The fluorescence emission and excitation spectra were recorded with a PTI fluorescence master system spectrofluorimeter. The instrument was interfaced with the acquisition software Felix 2000 (version 1), which performed online correction of the data with respect to the excitation lamp spectral radiance and detector spectral quantum efficiency.
The solvents used to prepare the solutions for all electronic-state transition spectroscopy experiments were purchased from Merck and were of HPLC grade. They were used without further purification. Due to the poor solubility of the compounds in some of the solvents, a concentrated stock in acetone was prepared weighing some powder and dissolving it in 3 mL of this solvent. Afterwards, 50 µL aliquots of this stock were put in glass vials and let to evaporate for 48 h at 50 °C. Finally, the as-obtained films were resuspended in 3 mL of the desired solvent, and the samples were sonicated for 1 h to favor disaggregation. From this procedure, clear solutions were obtained in all solvents.
Molar extinction coefficients were estimated by performing linear regressions on absorbance vs. concentration plots, obtained through progressive dilution of the above stock solutions to one-tenth of the initial concentration in several steps (N > 5). An exemplary plot is reported in
Figure S10.
Fluorescence quantum yields, ϕ
Fluo, were determined by comparing the integrated fluorescence intensity with that of dimethyl-POPOP, a dye used in particle physics as a reactant of scintillator chambers. Due to its high-fluorescence quantum yield, which is tabulated to be ϕ
Fluo = 0.95 when dissolved in cyclohexane and excited in its UVA absorption band [
26], dimethyl-POPOP has been also widely used as a fluorescence standard since the 1970s. Fluorescence was elicited exciting the samples at their absorption maxima; the emission spectra were normalized with respect to their peak absorbances. Fluorescence quantum yield values were corrected for the solvent’s refractive indexes.
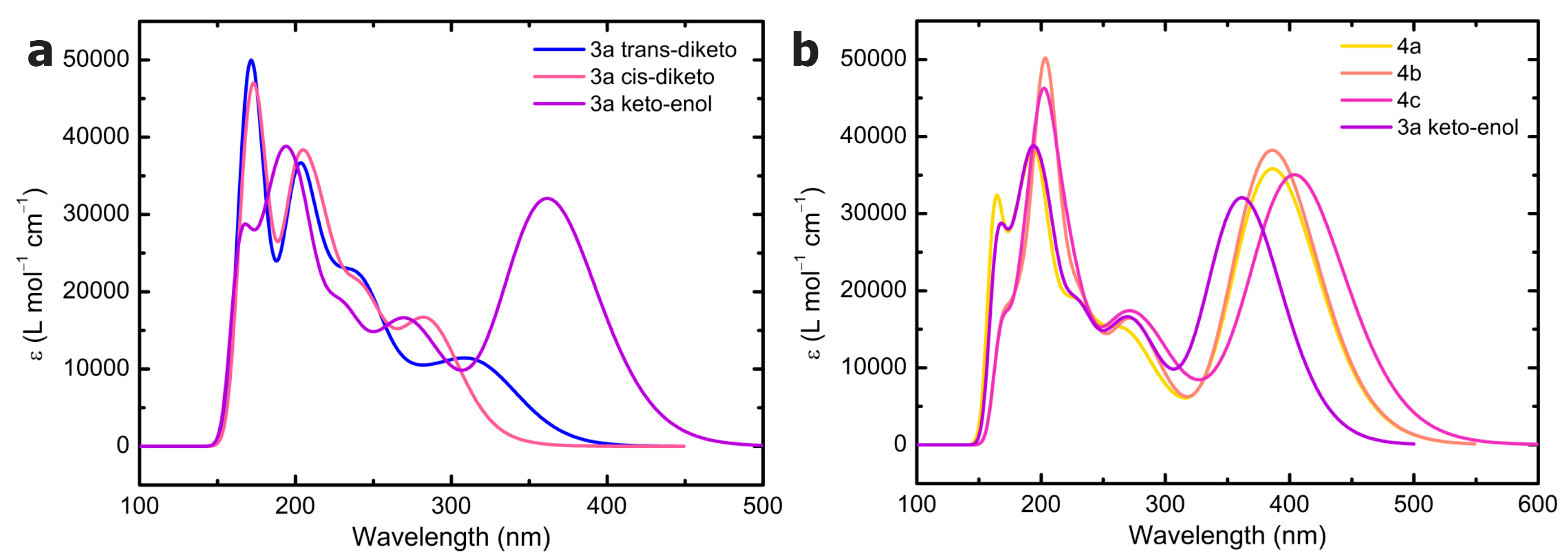
3.7. Density Functional Calculations
All DFT calculations were performed using the Gaussian 16 program [
33]. Four density functionals were considered: M06-2X [
34], ωB97X-D [
35], B3LYP-D3, and CAM-B3LYP-D3, i.e., B3LYP [
36,
37] and CAM-B3LYP [
38], including the D3 version of Grimme’s dispersion (D3) with Becke–Johnson damping (BJ) [
39]. M06-2X and ωB97X-D have been shown to be among those having the highest accuracy in a recent assessment performed by Liang et al. [
30] for the determination of electronic excitations in the UV-Vis region in organic and main-group molecules. They were used in combination with the triple zeta def2-TZVPD basis sets [
40,
41,
42], as suggested in Ref. [
30], although identical results were obtained for B3LYP-D3 in combination with def2-TZVP basis sets. A (99,590) pruned grid was used (i.e., 99 radial points and 590 angular points per radial point), corresponding to the grid = ultrafine option.
Geometry optimization was carried out by means of the Berny optimization algorithm with analytical gradient and default convergence thresholds. Unscaled, harmonic vibrational frequencies were computed analytically. Zero-point vibrational energies (ZPVE) were calculated at 1 atm and 298 K from conventional ideal gas, rigid rotator, particle in a box, and quantum mechanical harmonic oscillator partition functions. Charge and spin densities were obtained using Charge Model 5 (CM5) [
43] and Hirshfeld population analysis [
44], respectively.
Time-dependent (TD) density functional theory in the frame of the linear response theory [
45] is the most common approach for the study of molecular electronic excited states [
46,
47]. All the calculations were performed using the Tamm–Dancoff approximation (TDA, [
30,
48]). Liang et al. [
30] showed that TDA offers a good balance between computational efficiency and accuracy. For comparison, B3LYP-D3 and ωB97X-D calculations have also been performed without TDA (TD-B3LYP-D3). The electronic absorption (emission) spectra were computed by single-point calculations on the ground (first excited)-state-optimized structures by considering the first 100 excitations. All the calculations were performed on the gas phase single molecule, other than for the evaluation of the relative stability of
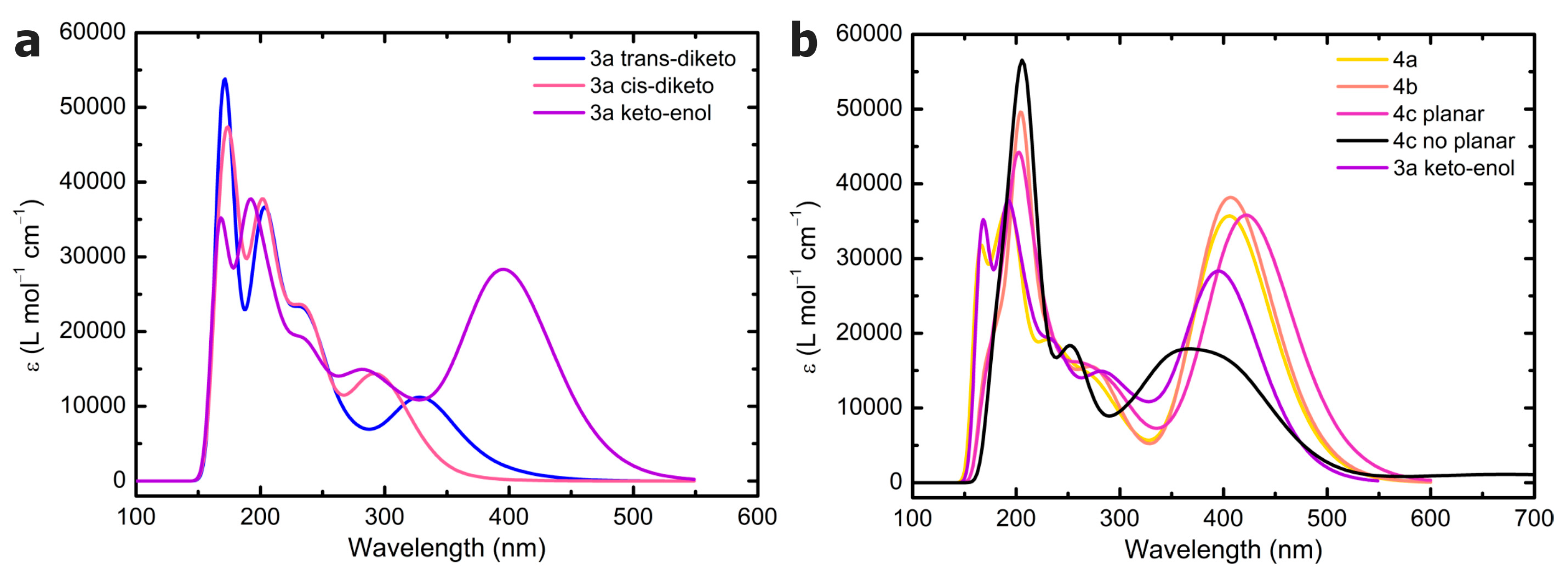
3a conformers, where a polarizable continuum toluene model was also adopted (ε = 2.3741 [
49]). A Gaussian broadening was applied to the TDDFT excitations using a standard deviation of 0.3 eV. No scaling factors were adopted. Visualization of the computed spectra and of the total density maps was performed using GaussView 6.0.
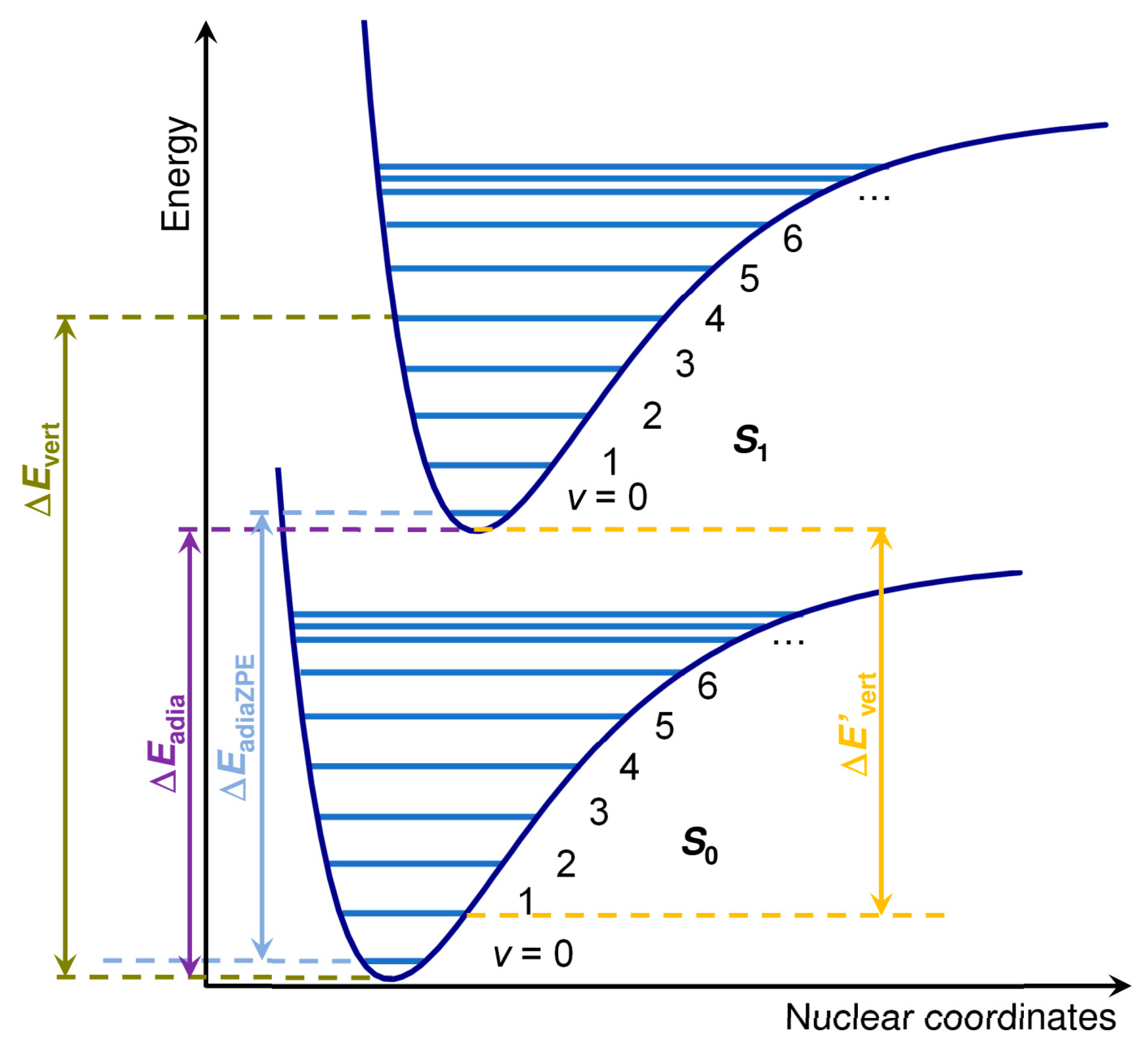
The vertical excitation energies (Δ
Evert) and purely electronic adiabatic excitation energies (Δ
Eadia) for
S0 →
S1 were obtained as the energy difference between the ground state energy and the energy of the first excited state computed through single-point and geometry optimization, respectively. No difference was observed between geometry optimization, including the first 5 and 100 excitations in an initial assessment using ωB97X-D. For this reason, only the first five excitations were considered in the geometry optimization. ZPVE-corrected vertical adiabatic excitation energies were also obtained (Δ
EadiaZPVE). A graphical definition of these energies is reported in
Figure 8. The corresponding values for the
S1 →
S0 transition are indicated with a prime (see
Figure 8).
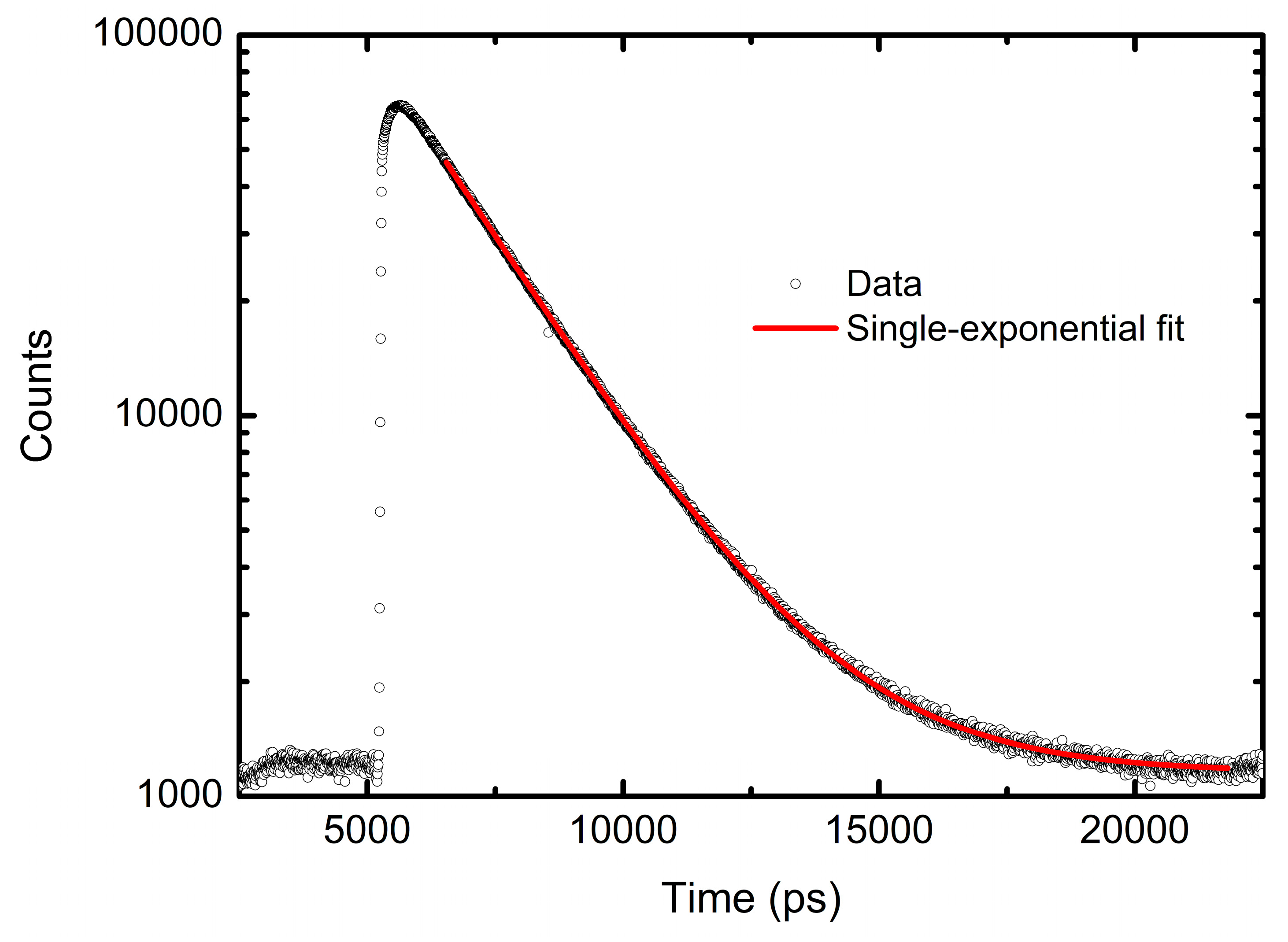
The radiative decay rates
of the S
1 species can be estimated as the Einstein coefficient for the spontaneous emission, as defined in Refs. [
50,
51]:
where
, c, and ħ represent the vacuum permittivity, the speed of light, and the reduced Planck constant, respectively, while
is the ground to excited state transition electric dipole moment.




 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
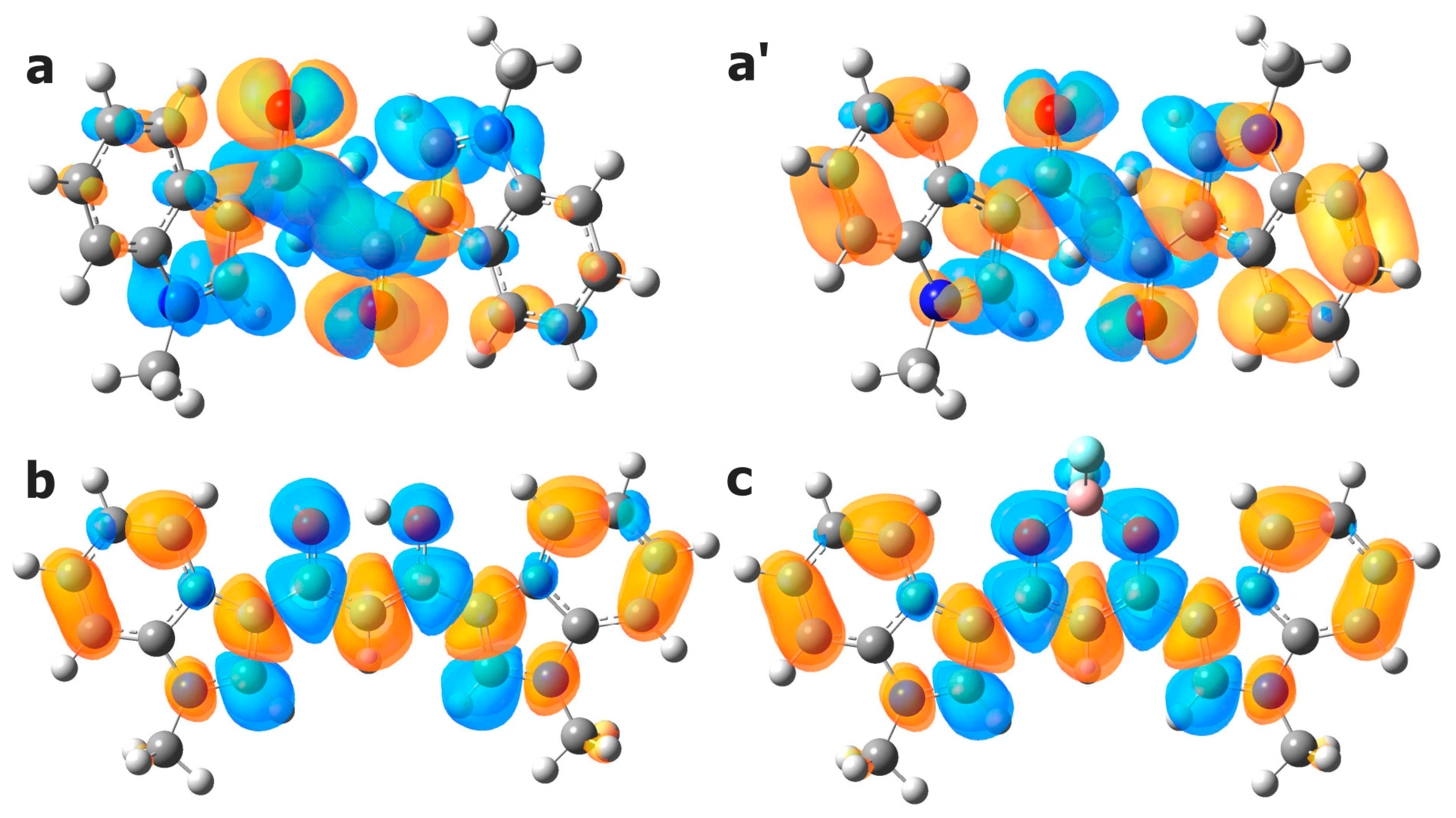{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








