
Relationship between the Change in E/T Ratio and the Cooking Performance of Eucalyptus and Acacia Woods during Kraft Pulping Process




Abstract
1. Introduction
2. Results and Discussion
2.1. Chemical Composition Analysis of Raw Materials
2.1.1. Analysis of Components
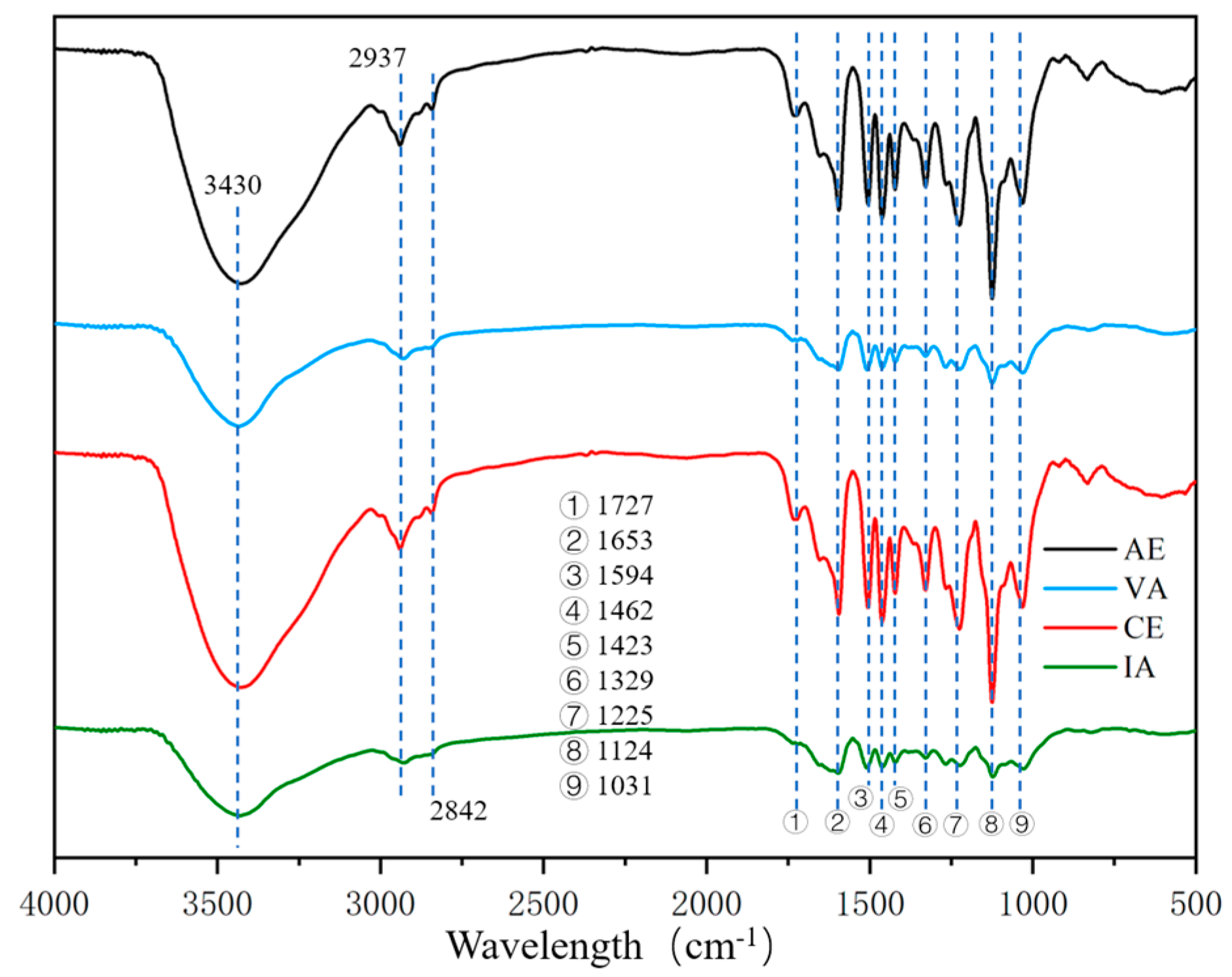
2.1.2. FTIR Spectroscopy
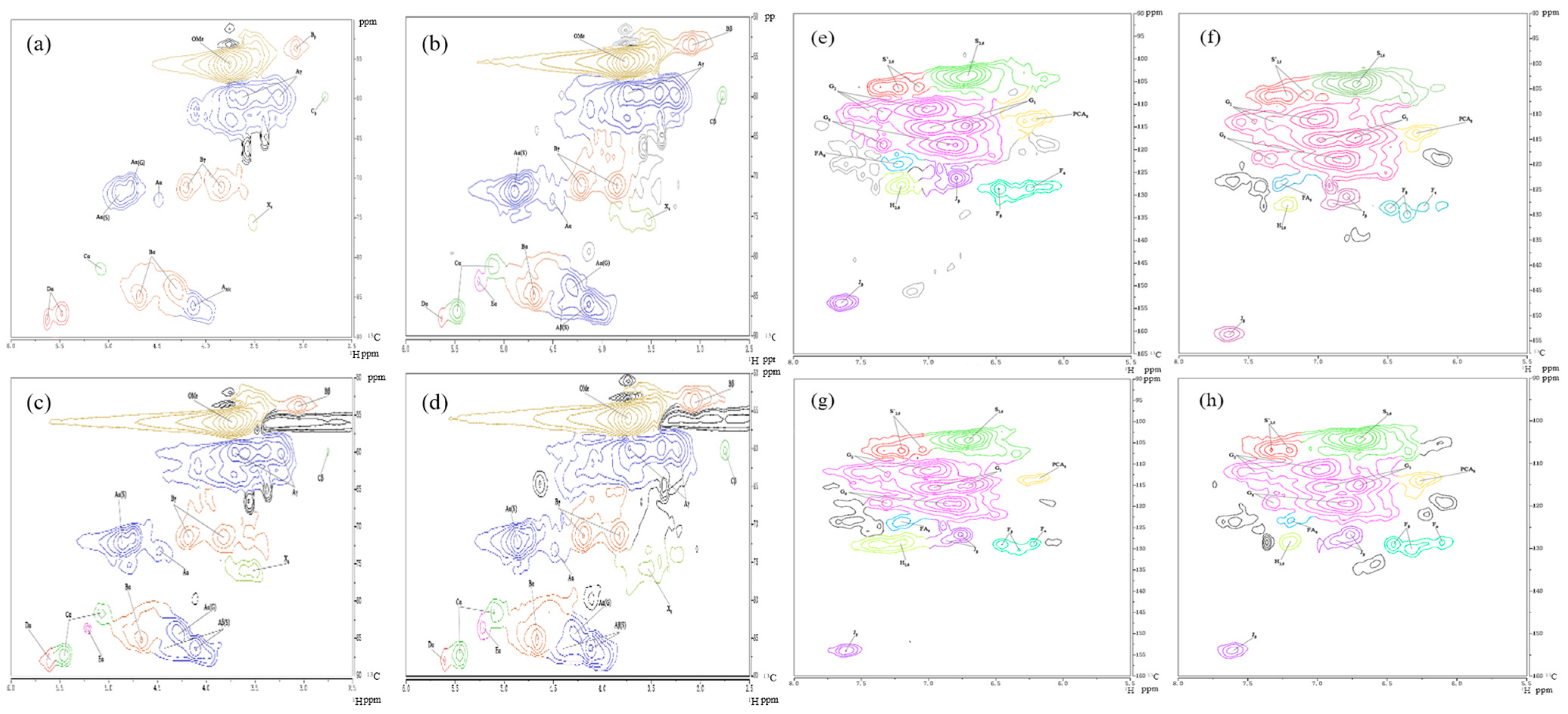
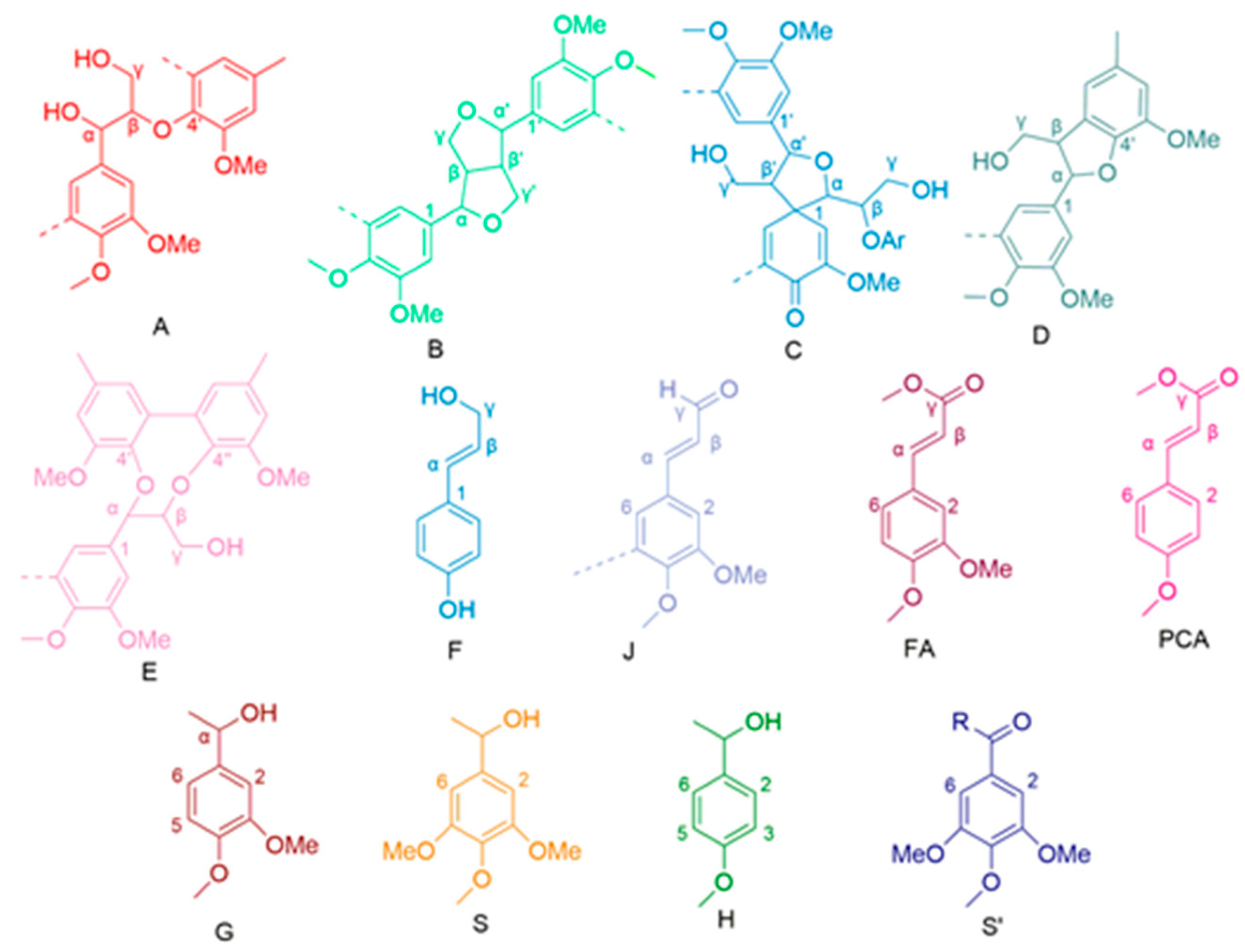
2.1.3. 1H-13C HSQC NMR Analysis
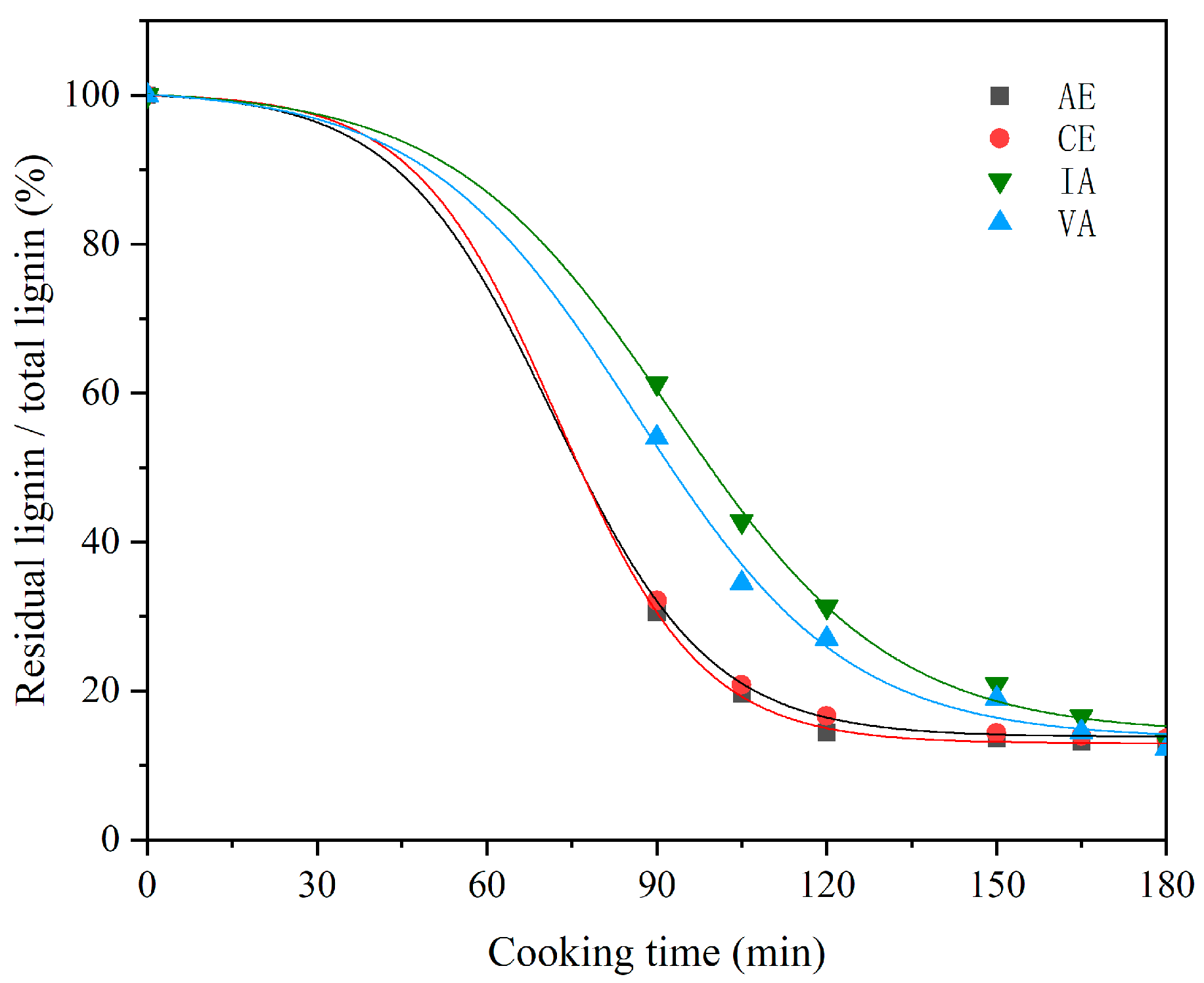
2.2. Analysis of Cooking Efficiency
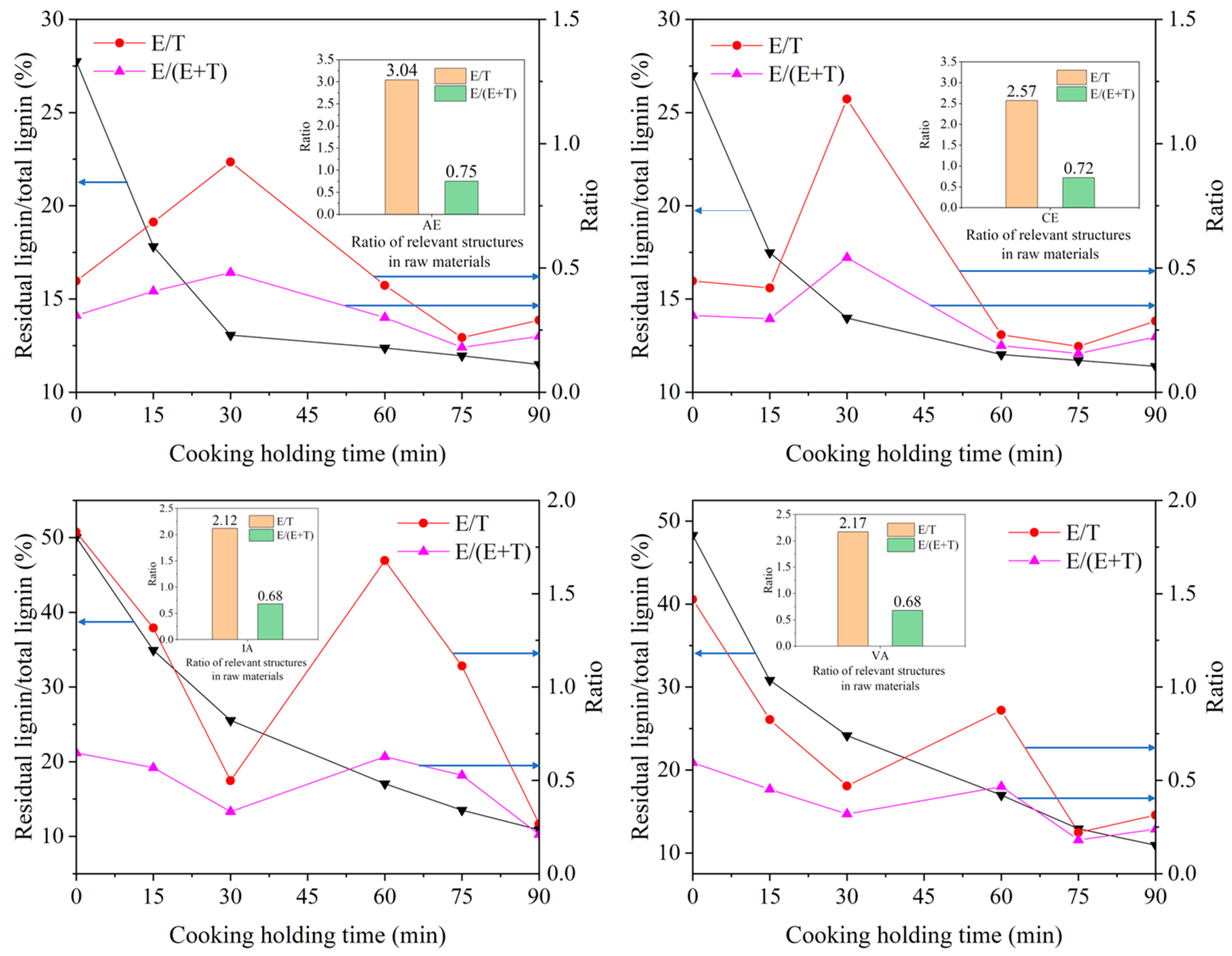
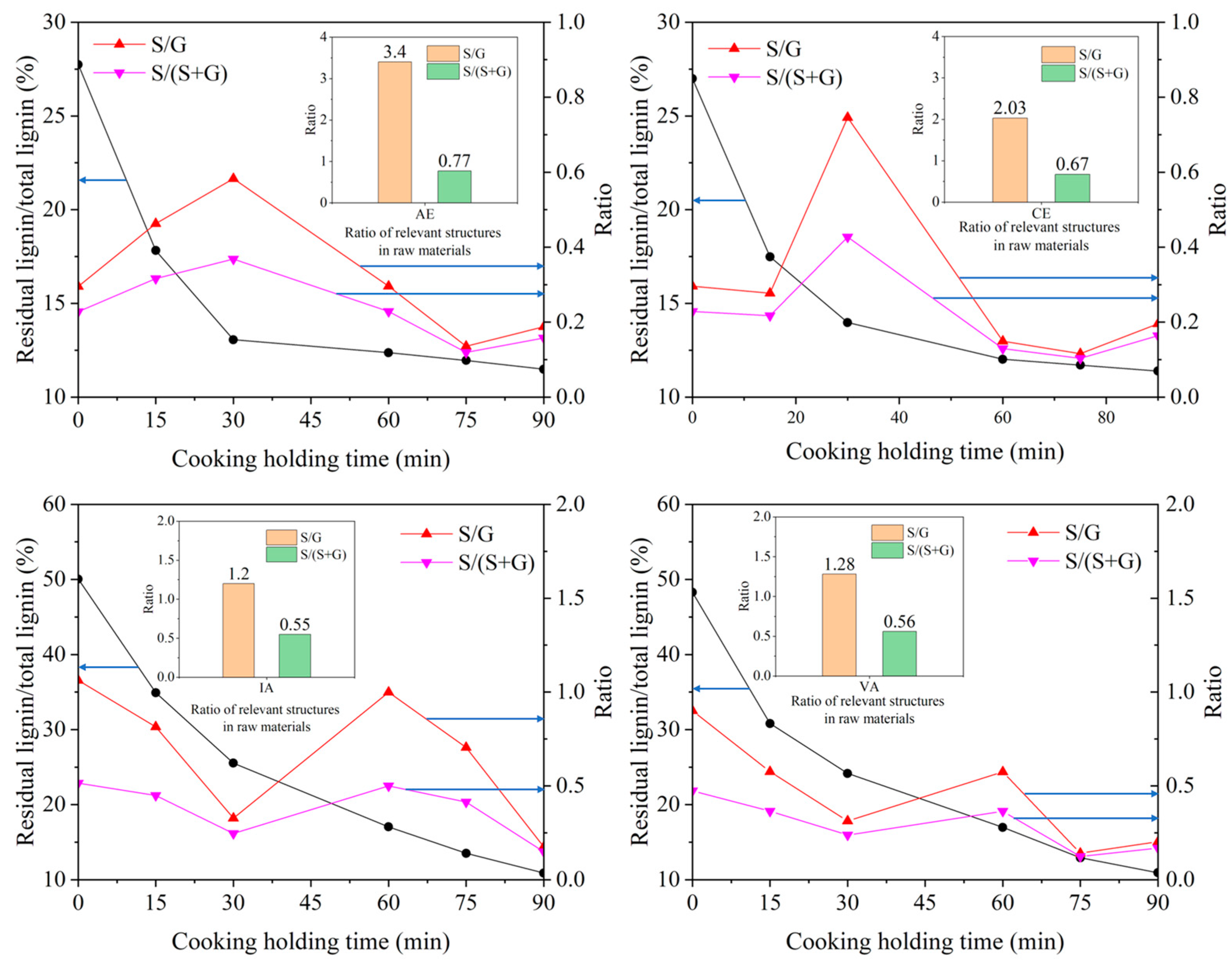
2.3. Effect of Lignin Side Chain Conformation on Delignification Properties of Hardwoods
2.3.1. Relationship between E/T Ratio and Cooking Performance of Different Samples
2.3.2. Relationship between S/G Ratio and Cooking Performance of Different Samples
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Analysis of Lignin and Saccharides of Raw Materials
3.2.2. Preparation of a Series of Lignocellulosic Biomass Using Different Cooking Conditions
3.2.3. Determination of Lignin Content
3.2.4. Preparation of Milled Wood Lignin Samples (MWLs) from Ground Wood
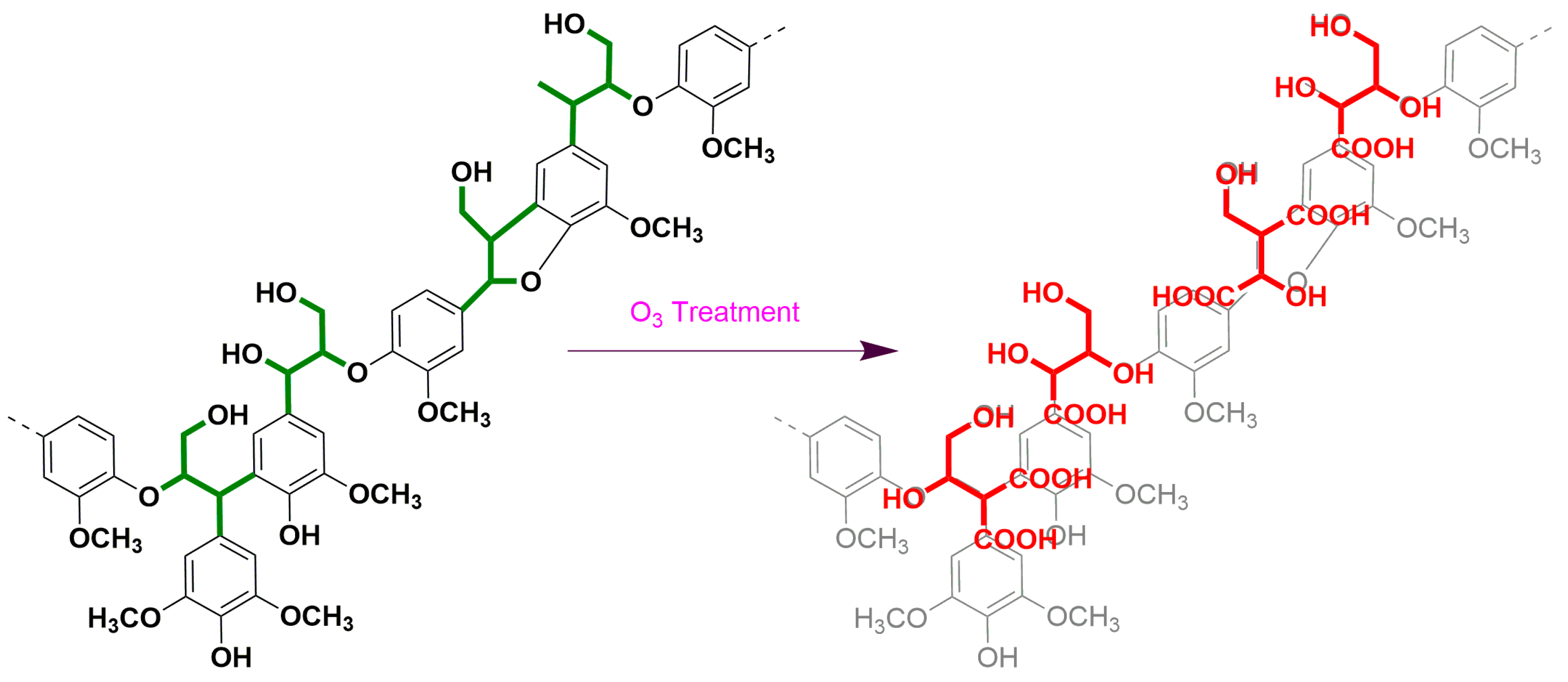
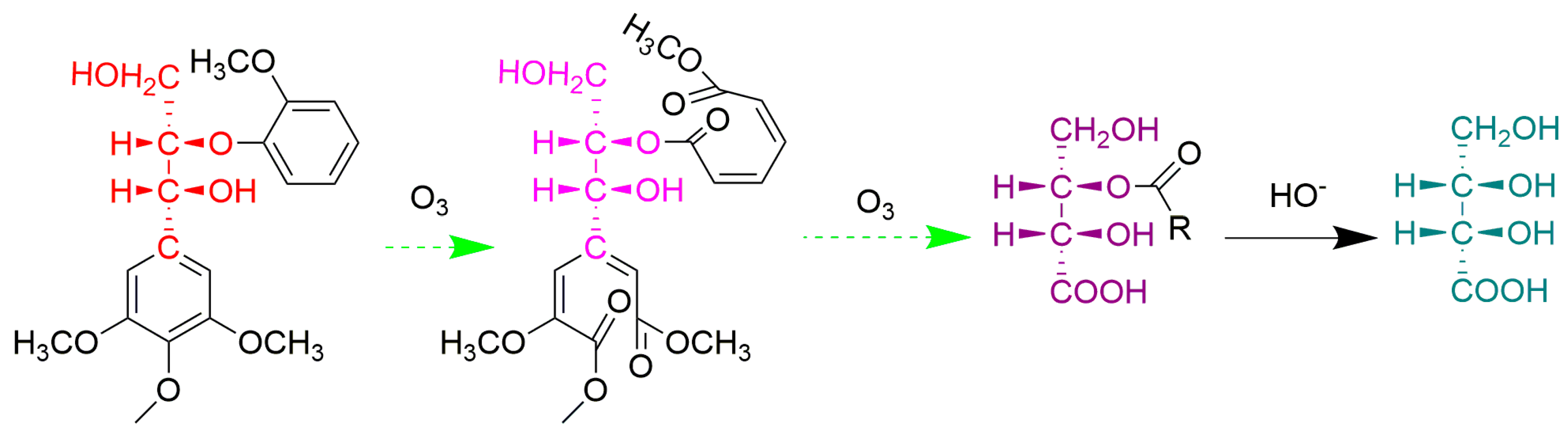
3.2.5. Ozone Oxidation
3.2.6. Nitrobenzene Oxidation
3.2.7. FTIR Spectra
3.2.8. 2D HSQC NMR
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.F.; Matsumoto, Y.; Tange, T.; Iiyama, K. Structural characteristics of lignin in primitive pteridophytes: Selaginella species. J. Wood Sci. 2007, 53, 412–418. [Google Scholar] [CrossRef]
- Karlsson, O.; Ikeda, T.; Kishimoto, T.; Magara, K.; Matsumoto, Y.; Hosoya, S. Ozonation of a lignin-carbohydrate complex model compound of the benzyl ether type. J. Wood Sci. 2000, 46, 263–265. [Google Scholar] [CrossRef]
- Akiyama, T.; Sugimoto, T.; Matsumoto, Y.; Meshitsuka, G. Erythro/threo ratio of β-O-4 structures as an important structural characteristic of lignin. I: Improvement of ozonation method for the quantitative analysis of lignin side-chain structure. J. Wood Sci. 2002, 48, 210–215. [Google Scholar] [CrossRef]
- Aulin-Erdtman, G.; Tomita, Y.; Forsen, S. Studies on the degradation of lignin and model compounds. I. The configuration of dehydrodiisoeugenol. Acta Chem. Scand. 1963, 17, 535–536. [Google Scholar] [CrossRef]
- Akiyama, T.; Magara, K.; Matsumoto, Y.; Meshitsuka, G.; Ishizu, A.; Lundquist, K. Proof of the presence of racemic forms of arylglycerol-β-aryl ether structure in lignin: Studies on the stereo structure of lignin by ozonation. J. Wood Sci. 2000, 46, 414–415. [Google Scholar] [CrossRef]
- Akiyama, T.; Magara, K.; Meshitsuka, G.; Lundquist, K.; Matsumoto, Y. Absolute configuration of β- and α-asymmetric carbons within β-O-4 structure in hardwood lignin. J. Wood Chem. Technol. 2015, 35, 8–16. [Google Scholar] [CrossRef]
- Nawawi, D.S.; Syafii, W.; Akiyama, T.; Matsumoto, Y. Characteristics of guaiacyl-syringyl lignin in reaction wood in the gymnosperm Gnetum gnemon L. Holzforschung 2016, 70, 593–602. [Google Scholar] [CrossRef]
- Shimizu, S.; Yokoyama, T.; Akiyama, T.; Matsumoto, Y. Reactivity of lignin with different composition of aromatic syringyl/guaiacyl structures and erythro/threo side chain structures in β-O-4 type during alkaline delignification: As a basis for the different degradability of hardwood and softwood lignin. J. Agric. Food Chem. 2012, 60, 6471–6476. [Google Scholar] [CrossRef]
- Akiyama, T.; Matsumoto, Y.; Okuyama, T.; Meshitsuka, G. Ratio of erythro and threo forms of β-O-4 structures in tension wood lignin. Phytochemistry 2003, 64, 1157–1162. [Google Scholar] [CrossRef]
- Akiyama, T.; Goto, H.; Nawawi, D.S.; Syafii, W.; Matsumoto, Y.; Meshitsuka, G. Erythro/threo ratio of β-O-4-structures as an important structural characteristic of lignin. Part 4: Variation in the erythro/threo ratio in softwood and hardwood lignins and its relation to syringyl/guaiacyl ratio. Holzforschung 2005, 59, 276–281. [Google Scholar] [CrossRef]
- Li, P.; Ren, J.; Jiang, Z.; Huang, L.; Wu, C.; Wu, W. Review on the preparation of fuels and chemicals based on lignin. RSC Adv. 2022, 12, 10289–10305. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Akiyama, T.; Matsumoto, Y.; Meshitsuka, G. The erythro/threo ratio of β-O-4 structures as an important structural characteristic of lignin-Part 2. Changes in erythro/threo (E/T) ratio of β-O-4 structures during delignification reactions. Holzforschung 2002, 56, 416–421. [Google Scholar] [CrossRef]
- Dexter, L.C.; Thomas, E.; Thomas, H.F.; Tor, P.S. Effect of the α- and γ-hydroxyls on the alkaline hydrolysis rate of nonphenolic β-0-4 lignin diastereomers. Holzforschung 2002, 56, 67–72. [Google Scholar]
- Miksche, G.E. Zum alkalischen abbau der p-alkoxy-arylglycerin-β-arylätherstrukturen des Lignins. Versuche mit erythro-veratryl-glycerin-β-guajacyläther. Acta Chem. Scand. 1972, 26, 3275–3281. [Google Scholar] [CrossRef]
- Posoknistakul, P.; Akiyama, T.; Yokoyama, T.; Matsumoto, Y. Stereo-preference in the degradation of the erythro and threo Isomers of β-O-4 Type lignin model compounds in oxidation processes: Part 1: In the reaction with active oxygen species under oxygen delignification conditions. J. Wood Chem. Technol. 2016, 36, 288–303. [Google Scholar] [CrossRef]
- Posoknistakul, P.; Akiyama, T.; Yokoyama, T.; Matsumoto, Y. Stereo-preference in the degradation of the erythro and threo isomers of β-O-4 type lignin model compounds in oxidation processes. Part 2: In the reaction with hydroxyl and oxyl anion radicals under hydrogen peroxide bleaching conditions. J. Wood Chem. Technol. 2017, 37, 87–98. [Google Scholar] [CrossRef]
- Posoknistakul, P.; Akiho, S.; Akiyama, T.; Yokoyama, T.; Matsumoto, Y. Stereo-preference in the degradation of the erythro and threo isomers of β-O-4-type lignin model compounds in oxidation processes III: In the reaction with chlorine- and manganese-based oxidants. J. Wood Sci. 2018, 64, 451–457. [Google Scholar] [CrossRef]
- Dimmel, D.R.; MacKay, J.J.; Courchene, C.E.; Kadla, J.; Scott, J.; O’Malley, D.; McKeand, S. Pulping and bleaching of partially CAD-deficient wood. J. Wood Chem. Technol. 2002, 22, 235–248. [Google Scholar] [CrossRef]
- Gupta, G.K.; Shukla, P. Insights into the resources generation from pulp and paper industry wastes: Challenges, perspectives and innovations. Bioresour. Technol. 2020, 297, 122496. [Google Scholar]
- Pettersen, R.C. The chemical composition of wood. In The Chemistry of Solid Wood; Advances in Chemistry Series; American Chemical Society: Washington, DC, USA, 1984; Volume 207, pp. 57–126. [Google Scholar]
- Mittal, A.; Scott, G.M.; Amidon, T.E.; Kiemle, D.J.; Stipanovic, A.J. Quantitative analysis of sugars in wood hydrolyzates with 1H NMR during the autohydrolysis of hardwoods. Bioresour. Technol. 2009, 100, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, E.E. Screening of cellulolytic activity of some rhizospheric soil fungi isolated from different sources. Delta J. Sci. 2022, 44, 88–100. [Google Scholar]
- Li, Z.Z. Research on renewable biomass resource-lignin. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2012, 10, 21–29. [Google Scholar]
- Jiang, B.; Cao, T.Y.; Gu, F.; Wu, W.J.; Jin, Y.C. Comparison of the structural characteristics of cellulolytic enzyme lignin preparations isolated from wheat straw stem and leaf. ACS Sustain. Chem. Eng. 2017, 5, 342–349. [Google Scholar] [CrossRef]
- Maunu, S.L. NMR studies of wood and wood products. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 151–174. [Google Scholar] [CrossRef]
- Rencoret, J.; Marques, G.; Gutierrez, A.; Ibarra, D.; Li, J.; Gellerstedt, G.; Santos, J.I.; Jimenez-Barbero, J.; Martinez, A.T.; del Rio, J.C. Structural characterization of milled wood lignins from different eucalypt species. Holzforschung 2008, 62, 514–526. [Google Scholar] [CrossRef]
- Kim, H.; Ralph, J. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d(6)/pyridine-d(5). Org. Biomol. Chem. 2010, 8, 576–591. [Google Scholar] [CrossRef]
- Rio, J.C.; Rencoret, J.; Prinsen, P.; Martinez, A.T.; Ralph, J.; Gutierrez, A. Structural characterization of wheat straw lignin as revealed by analytical pyrolysis, 2D-NMR, and reductive cleavage methods. J. Agric. Food Chem. 2012, 60, 5922–5935. [Google Scholar] [CrossRef]
- Wen, J.L.; Sun, S.L.; Xue, B.L.; Sun, R.C. Recent advances in characterization of lignin polymer by solution-state nuclear magnetic resonance (NMR) methodology. Materials 2013, 6, 359–391. [Google Scholar] [CrossRef]
- Rencoret, J.; Prinsen, P.; Gutierrez, A.; Martinez, A.T.; del Rio, J.C. Isolation and structural characterization of the milled wood lignin, dioxane lignin, and cellulolytic lignin preparations from Brewer’s Spent Grain. J. Agric. Food Chem. 2015, 63, 603–613. [Google Scholar] [CrossRef]
- Costa, C.A.E.; Coleman, W.; Dube, M.; Rodrigues, A.E.; Pinto, P.C.R. Assessment of key features of lignin from lignocellulosic crops: Stalks and roots of corn, cotton, sugarcane, and tobacco. Ind. Crops Prod. 2016, 92, 136–148. [Google Scholar] [CrossRef]
- Mattsson, C.; Andersson, S.I.; Belkheiri, T.; Amand, L.E.; Olausson, L.; Vamling, L.; Theliander, H. Using 2D NMR to characterize the structure of the low and high molecular weight fractions of bio-oil obtained from LignoBoost (TM) kraft lignin depolymerized in subcritical water. Biomass Bioenergy 2016, 95, 364–377. [Google Scholar] [CrossRef]
- Nieminen, K.; Kuitunen, S.; Paananen, M.; Sixta, H. Novel insight into lignin degradation during kraft cooking. Ind. Eng. Chem. Res. 2014, 53, 2614–2624. [Google Scholar] [CrossRef]
- Wilder, H. Delignification rate studies. Part II of a series on kraft pulping kinetics. Tappi 1965, 48, 293–297. [Google Scholar]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass; Report TP-510-42618; Laboratory Analytical Procedure, NREL: Golden, CO, USA, 2008. [Google Scholar]
- Goyal, G.C. Antraquinone Pulping: A Tappi Press Anthology of Published Papers; Tappi Press: Atlanta, GA, USA, 1997; pp. 1977–1996. [Google Scholar]
- Hakanen, A.; Teder, A. Modified kraft cooking with polysulfide: Yield, viscosity, and physical properties. Tappi J. 1997, 7, 189–196. [Google Scholar]
- Wang, Z.G.; Yokoyama, T.; Chang, H.M.; Matsumoto, Y. Dissolution of Beech and Spruce milled woods in LiCl/DMSO. J. Agric. Food Chem. 2009, 57, 6167–6170. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Matsumoto, Y.; Chang, H.M.; Meshitsuka, G. Quantitative evaluation of milling effects on lignin structure during the isolation process of milled wood lignin. J. Wood Sci. 2005, 51, 89–91. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.; Yang, Y.; Ren, H.; Zhai, H. A spectroscopic method for quantitating lignin in lignocellulosic biomass based on the completely dissolved solution of biomass in LiCl/DMSO. Green Chem. 2022, 24, 2212–2221. [Google Scholar] [CrossRef]
- Björkman, A. Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Sven. Papp. 1956, 59, 477–485. [Google Scholar]
- Verhaar, L.T.; De, W.H. The gas chromatographic determination of potyhydroxy monocarbonic acids obtained by oxygeneration of hexoses in aqueous alkaline solutions. J. Chromatogr. 1969, 41, 168–179. [Google Scholar] [CrossRef]
- Chen, C.L. Nitrobenzene and cupric oxide oxidations. In Methods in Lignin Chemistry; Springer: Berlin/Heidelberg, Germany, 1992; pp. 301–321. [Google Scholar]
- Kim, H.; Ralph, J.; Akiyama, T. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d(6). Bioenergy Res. 2008, 1, 56–66. [Google Scholar] [CrossRef]
- Rencoret, J.; Marques, G.; Gutierrez, A.; Nieto, L.; Jimenez-Barbero, J.; Martinez, A.T.; del Rio, J.C. Isolation and structural characterization of the milled-wood lignin from Paulownia fortunei wood. Ind. Crops Prod. 2009, 30, 137–143. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Lignin | Carbohydrate | NBO | Ozonation | ||||
|---|---|---|---|---|---|---|---|---|
| KL | ASL | TL | Glucan | Xylan | Arabinan | S/V/H | E/T | |
| AE | 18.04 ± 0.90 | 6.81 ± 0.61 | 24.85 ± 0.7 | 41.37 ± 0.51 | 17.48 ± 0.14 | 0.64 ± 0.04 | 75/24/1 | 2.54 |
| CE | 22.08 ± 0.21 | 1.74 ± 0.03 | 23.82 ± 0.24 | 43.7 ± 0.47 | 13.66 ± 0.25 | 0.59 ± 0.06 | 67/32/0 | 2.63 |
| IA | 22.54 ± 0.08 | 1.67 ± 0.04 | 24.21 ± 0.12 | 49.37 ± 0.23 | 13.69 ± 0.46 | - | 54/45/1 | 2.13 |
| VA | 20.68 ± 0.02 | 3.43 ± 0.07 | 24.11 ± 0.1 | 44.14 ± 0.11 | 15.89 ± 0.17 | - | 52/47/1 | 2.17 |
| Wavelength (cm−1) | Assignment |
|---|---|
| 3430 | Stretching vibration of –OH (phenolic and alcoholic hydroxyl) |
| 2937 | C–H stretching vibration in the aromatic methoxyl |
| 2842 | C–H stretching in methyl and methylene of the side chains |
| 1727 | Stretching vibration of non-conjugate C=O |
| 1653 | Stretching vibration of conjugate C=O |
| 1594 | Stretching vibration of benzene ring |
| 1506 | Stretching vibration of benzene ring |
| 1462 | Bending vibration of C–H (CH2, CH3) |
| 1423 | Stretching vibration of benzene ring |
| 1329 | Stretching vibration of C–O in S-unit |
| 1267 | Stretching vibration of C–O in G-unit |
| 1225 | Stretching vibration of C–O in S-unit |
| 1124 | In-plane bending vibration of C–H in benzene ring of S-unit |
| 1031 | Stretching vibration of C–O (alcoholic hydroxyl and alkyl ether) |
| δH (ppm) | δC (ppm) | Label | Assignment |
|---|---|---|---|
| 2.78 | 59.78 | Cβ | Cβ-Hβ in spirodienones (C) |
| 3.06 | 53.48 | Bβ | Cβ-Hβ in β-β′ (resinol) substructures (B) |
| 3.25 | 59.78 | Aγ | Cγ-Hγ in β-O-4′ substructure (A) |
| 3.50 | 75.54 | X4 | C4-H4 in β-D-xylopyranoside (X) |
| 3.63 | 59.78 | Aγ | Cγ-Hγ in β-O-4′ substructure (A) |
| 3.75 | 55.73 | OMe | C-H in methoxyls (OMe) |
| 3.85 | 71.04 | Bγ | Cγ-Hγ in β-β′ (resinol) substructures (B) |
| 4.13 | 85.90 | Aβ(S) | Cβ-Hβ in β-O-4′ substructures (A) linked to S(Erythro) |
| 4.19 | 71.04 | Bγ | Cγ-Hγ in β-β′ (resinol) substructures (B) |
| 4.29 | 84.10 | Aβ(G) | Cβ-Hβ in β-O-4′ unit (A) linked to a G-unit |
| 4.32 | 83.65 | Bα | Cα-Hα in β-β′ (resinol) substructures (B) |
| 4.48 | 72.84 | Aα | Cα-Hα in β-O-4′ unit (A) |
| 4.69 | 85.00 | Bα | Cα-Hα in β-β′ resinol substructures (B) |
| 4.79 | 71.04 | Aα(G) | Cα-Hα in β-O-4′ unit (A) linked to a G-unit (A) |
| 4.88 | 71.94 | Aα(S) | Cα-Hα in β-O-4′ unit (A) linked to an S-unit (A) |
| 5.10 | 81.40 | Cα | Cα-Hα in spirodienones (C) |
| 5.48/5.64 | 86.80 | Dα | Cα-Hα in phenylcoumaran substructure (D) |
| 6.21 | 113.37 | pCA8 | C8-H8 in p-coumarate (pCA) |
| 6.25 | 128.68 | Fɑ | Cα-Hα in p-hydroxycinnamyl alcohol (F) |
| 6.48 | 128.68 | Fβ | Cβ-Hβ in p-hydroxycinnamyl alcohol (F) |
| 6.72 | 103.91 | S2,6 | C2,6-H2,6 in syringyl units (S) |
| 6.73 | 114.72 | G5 | C5-H5 in guaiacyl units (G) |
| 6.79 | 126.43 | Jβ | Cβ-Hβ in cinnamyl aldehyde end-groups (J) |
| 6.80/6.85 | 118.77 | G6 | C6-H6 in guaiacyl units (G) |
| 6.98 | 115.17 | Aα | C5-H5 in etherified guaiacyl units (G) |
| 7.01/7.34 | 111.12/112.47 | G2 | C2-H2 in guaiacyl units (G) |
| 7.20 | 127.78 | H2,6 | C2,6-H2,6 in H units (H) |
| 7.22 | 106.61 | S′2,6 | C2,6-H2,6, C(α)=O in syringyl units (S′) |
| 7.23 | 123.27 | FA6 | C6-H6 in ferulate (FA) |
| 7.33 | 118.77 | G6 | C6-H6 in guaiacyl units (G) |
| 7.65 | 153.89 | Jα | Cα-Hα in cinnamyl aldehyde end-groups (J) |
| Lignin Aromatic Units (%) | AE | CE | IA | VA |
|---|---|---|---|---|
| G | 23.85% | 33.01% | 44.79% | 40.91% |
| S | 74.95% | 66.99% | 53.53% | 52.18% |
| H | 1.20% | 0.00% | 1.67% | 6.91% |
| S/G ratio | 3.40 | 2.03 | 1.20 | 1.28 |
| Lignin interunit linkages (%) | ||||
| β-O-4′ (A) | 69.72% | 65.61% | 61.96% | 60.20% |
| Resinol (B) | 26.19% | 13.86% | 22.18% | 27.17% |
| Spirodienones (C) | 4.61% | 6.30% | 3.25% | 3.95% |
| Phenylcoumaran (D) | 3.51% | 7.42% | 8.48% | 8.68% |
| Dibenzodioxocin substructutres (E) | - | 2.82% | - | - |
| p-hydroxycinamates (%) | ||||
| p-coumarates (PCA) and p-hydroxycinnamyl alcohol (F) | 3.93% | 3.58% | 4.32% | 5.81% |
| Ferulate (FA) | 1.04% | 3.43% | 1.91% | 1.82% |
| p-coumarates and p-ydroxycinnamyl alcohol/ferulate ratio | 3.79 | 1.04 | 2.26 | 3.19 |
| Lignin end-groups | ||||
| Cinnamyl aldehyde end-groups (J) | 3.31% | 3.31% | 6.97% | 7.71% |
| Samples | Regression Equation | Rate Constant (100 × 1/min) | R2 |
|---|---|---|---|
| AE | y = 17.61exp(−x/14.29) + 12.97 | 7.01 | 0.99 |
| CE | y = 18.17exp(−x/15.49) + 13.97 | 6.46 | 0.99 |
| VA | y = 39.80exp(−x/25.40) + 13.83 | 3.94 | 0.99 |
| IA | y = 48.55exp(−x/31.63) + 12.70 | 3.61 | 0.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Xu, X.; Miao, C.; Li, P.; Tong, G. Relationship between the Change in E/T Ratio and the Cooking Performance of Eucalyptus and Acacia Woods during Kraft Pulping Process. Molecules 2023, 28, 4637. https://doi.org/10.3390/molecules28124637
Yu J, Xu X, Miao C, Li P, Tong G. Relationship between the Change in E/T Ratio and the Cooking Performance of Eucalyptus and Acacia Woods during Kraft Pulping Process. Molecules. 2023; 28(12):4637. https://doi.org/10.3390/molecules28124637
Chicago/Turabian StyleYu, Jiangdong, Xuewen Xu, Chen Miao, Penghui Li, and Guolin Tong. 2023. "Relationship between the Change in E/T Ratio and the Cooking Performance of Eucalyptus and Acacia Woods during Kraft Pulping Process" Molecules 28, no. 12: 4637. https://doi.org/10.3390/molecules28124637
APA StyleYu, J., Xu, X., Miao, C., Li, P., & Tong, G. (2023). Relationship between the Change in E/T Ratio and the Cooking Performance of Eucalyptus and Acacia Woods during Kraft Pulping Process. Molecules, 28(12), 4637. https://doi.org/10.3390/molecules28124637