
Diastereoselective Synthesis of the Borylated d-Galactose Monosaccharide 3-Boronic-3-Deoxy-d-Galactose and Biological Evaluation in Glycosidase Inhibition and in Cancer for Boron Neutron Capture Therapy (BNCT)

Abstract
1. Introduction
2. Results
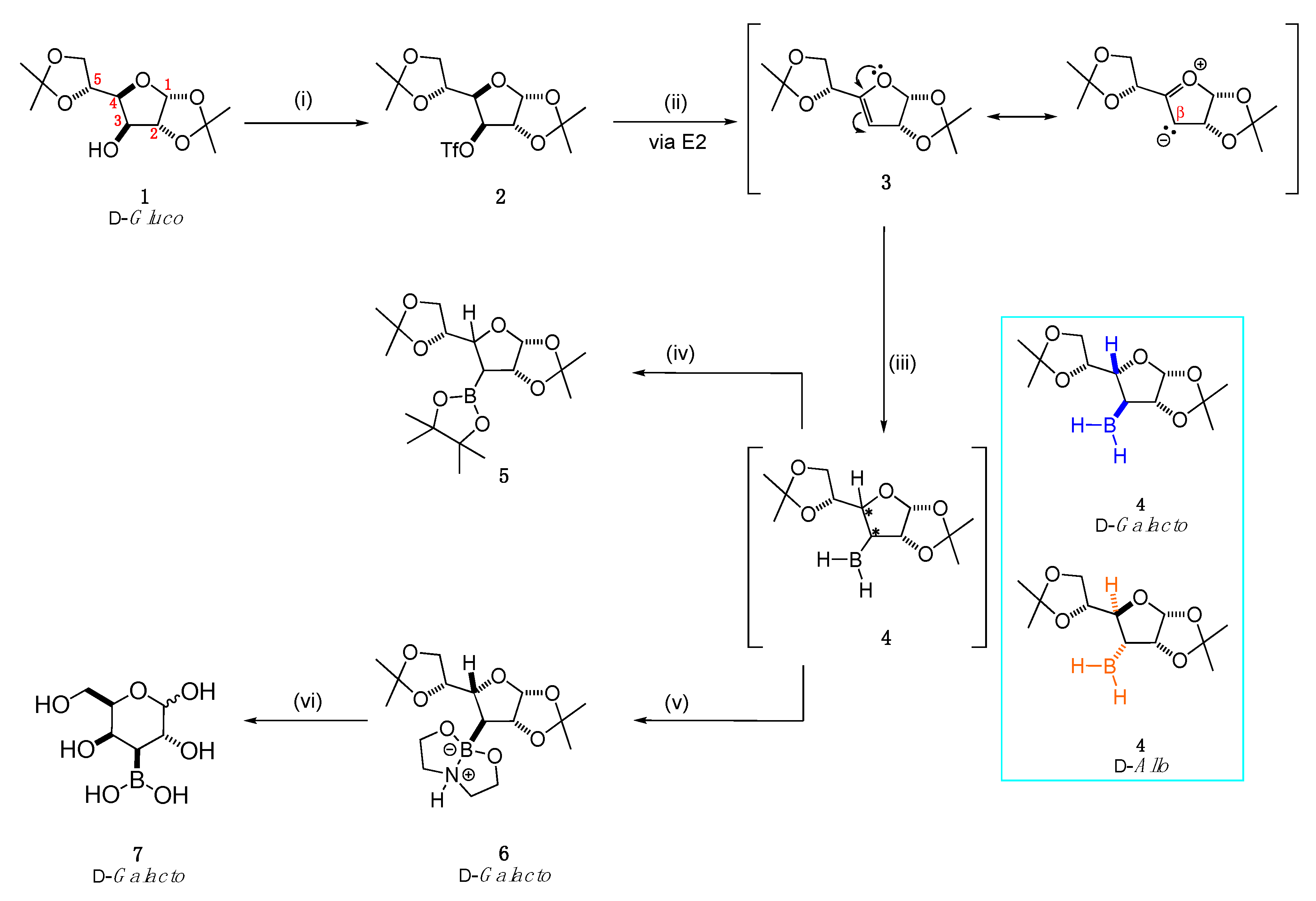
2.1. Synthesis
2.1.1. Hydroboration/Borane Trapping
2.1.2. Deprotection Step
3. Biological Evaluation in Glycosidase Inhibition and in Cancer for BNCT
3.1. Glycosidase Assay
3.1.1. Glycosidases
3.1.2. Controls
3.1.3. Intermediate 6 and Target Compound 7
3.2. Cancer Assay and Structure Activity Relationships
4. Materials and Methods
4.1. Glycosidase Inhibition Experimental (Table 1)
4.2. Cancer Screening Experimental (Table 2)
4.3. Chemistry
General Experimental
4.4. Syntheses
4.4.1. 3-Deoxy-1,2;5,6-di-O-isopropylidene-α-d-erythro-Hex-3-Enofuranose 3
4.4.2. 3-Deoxy-3-Boronodiethanolamine-1,2:5,6-Di-O-isopropylidene-α-d-galactofuranose 6
4.4.3. 3-Boronic-3-Deoxy-d-galactose 7
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Campkin, D.M.; Shimadate, Y.; Bartholomew, B.; Bernhardt, P.V.; Nash, R.J.; Sakoff, J.A.; Kato, A.; Simone, M. Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy. Molecules 2022, 27, 3447. [Google Scholar] [CrossRef]
- Garget, T.A.; Houston, T.A.; Kiefel, M.J.; Simone, M. Bicyclic Systems with Bridgehead (Ring Junction) Boron Atoms. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier Inc.: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Jenkinson, S.F.; Thompson, A.L.; Simone, M. Methyl 2-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-4-nitrobenzoate. Acta Cryst. 2012, E68, o2699–o2700. [Google Scholar] [CrossRef]
- Legge, W.J.; Shimadate, Y.; Sakoff, J.; Houston, T.A.; Kato, A.; Bernhardt, P.V.; Simone, M. Borylated methyl cinnamates: Green synthesis, characterization, crystallographic analysis and biological activities—In glycosidase inhibition and in cancer cells lines. Beilstein Arch. 2021, 2021, 4. [Google Scholar]
- Pappin, B.B.; Garget, T.A.; Healy, P.C.; Simone, M.; Kiefel, M.J.; Houston, T.A. Facile amidinations of 2-aminophenylboronic acid promoted by boronate ester formation. Org. Biomol. Chem. 2019, 17, 803–806. [Google Scholar] [CrossRef]
- Pappin, B.B.; Levonis, S.M.; Healy, P.C.; Kiefel, M.J.; Simone, M.; Houston, T.A. Crystallization Induced Amide Bond Formation Creates a Boron-Centred Spirocyclic System. Heterocycl. Commun. 2017, 23, 167–169. [Google Scholar] [CrossRef]
- Simone, M.; Houston, T.A. A brief overview of some latest advances in the applications of boronic acids. J. Glyc. Lipidom. 2014, 2014, e124–e129. [Google Scholar]
- Soengas, R.G.; Simone, M.; Hunter, S.; Nash, R.J.; Fleet, G.W.J. Hydroxymethyl-branched piperidines from hydroxymethyl-branched lactones: Synthesis and biological evaluation of 1,5-dideoxy-2-C-hydroxymethyl-1,5-imino-D-mannitol, 1,5-dideoxy-2-hydroxymethyl-1,5-imino-L-gulitol and 1,5-dideoxy-2-hydroxymethyl-1,5-imino-D-talitol. Eur. J. Org. Chem. 2012, 12, 2394–2402. [Google Scholar]
- Simone, M.; Soengas, R.G.; Jenkinson, S.F.; Evinson, E.L.; Nash, R.J.; Fleet, G.W.J. Synthesis of three branched iminosugars [(3R,4R,5S)-3-(hydroxymethyl)piperidine-3,4,5-triol, (3R,4R,5R)-3-(hydroxymethyl)piperidine-3,4,5-triol and (3S,4R,5R)-3-(hydroxymethyl)piperidine-3,4,5-triol] and a branched trihydroxynipecotic acid [(3R,4R,5R)-3,4,5-trihydroxypiperidine-3-carboxylic acid] from sugar lactones with a carbon substituent at C-2. Tetrahedron Asymmetry 2012, 23, 401–408. [Google Scholar]
- Lowering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Lovering, F. Escape from Flatland 2: Complexity and promiscuity. Med. Chem. Commun. 2013, 4, 515–519. [Google Scholar] [CrossRef]
- Itoh, T.; Tamura, K.; Ueda, H.; Tanaka, T.; Sato, K.; Kuroda, R.; Aoki, S. Design and synthesis of boron containing monosaccharides by the hydroboration of D-glucal for use in boron neutron capture therapy (BNCT). Bioorg. Med. Chem. 2018, 26, 5922–5933. [Google Scholar] [CrossRef]
- Imperio, D.; Panza, L. Sweet Boron: Boron-Containing Sugar Derivatives as Potential Agents for Boron Neutron Capture Therapy. Symmetry 2022, 14, 182. [Google Scholar] [CrossRef]
- Raymond, J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef]
- Yadav, J.S.; Prathap, I.; Tadi, B.P. Formal synthesis of fostriecin by a carbohydrate-based approach. Tetrahedron Lett. 2006, 47, 3773–3776. [Google Scholar] [CrossRef]
- Nemr, A.E.; Tsuchiya, T.; Kobayashi, Y. α-Hydrogen elimination in some carbohydrate triflates. Carbohydr. Res. 1996, 293, 31–59. [Google Scholar] [CrossRef]
- Flechtner, T.W. Carbohydrate triflates: Reactions with bases. Carbohydr. Res. 1979, 77, 262–266. [Google Scholar] [CrossRef]
- Horton, D.; Roski, J.P.; Norris, P. Cycloaddition of Cyclopentadiene to 3-Deoxy-1,2:5,6-di-O-isopropylidene-α-D-erythro-hex-3-enofuranose. Synthesis and Representative Chemistry of 1,6-Anhydro-2,3-dideoxy-α-D-glycero-hex-2-enopyran-4-ulose (“Isolevoglucosenone”). J. Org. Chem. 1996, 61, 3783–3793. [Google Scholar] [CrossRef]
- Brown, H.C.; Zweifel, G. A stereospecific cis hydration of the double bond in cyclic derivatives. J. Am. Chem. Soc. 1959, 81, 247. [Google Scholar] [CrossRef]
- Cheema, M.K. Design and Synthesis of FRET-Based Boronic Acid Receptors to Detect Carbohydrate Clustering and Development of Diacylglycerol-Based Lipid Probesto Investigate Lipid-Protein Binding Interactions. Master’s Thesis, University of Tennessee—Knoxville, TRACE: Tennessee Research and Creative Exchange, Knoxville, NC, USA, 2009. [Google Scholar]
- Haefele, A.; Zedde, C.; Retailleau, P.; Ulrich, G.; Ziessel, R. Boron Asymmetry in a BODIPY Derivative. Org. Lett. 2010, 12, 1672–1675. [Google Scholar] [CrossRef]
- Ghorbani, M.; Simone, M. Developing new inexpensive room temperature ionic liquids with high thermal stability and a greener synthetic profile. ACS Omega 2020, 5, 12637–12648. [Google Scholar] [CrossRef]
- Watterson, M.P.; Pickering, L.; Smith, M.D.; Hudson, S.J.; Marsh, P.R.; Mordaunt, J.E.; Watkin, D.J.; Newman, C.J.; Fleet, G.W.J. 3-Azidotetrahydrofuran-2-carboxylates: Monomers for five-ring templated β-amino acid foldamers? Tetrahedron Asymmetry 1999, 10, 1855–1859. [Google Scholar] [CrossRef]
- Rochepeau-Jobron, L.; Jacquinet, J.-C. Diastereoselective hydroboration of substituted exo-glucals revisited. A convenient route for the preparation of L-iduronic acid derivatives. Carbohydr. Res. 1997, 303, 395–406. [Google Scholar] [CrossRef]
- Paulsen, H.; Behre, H. Umwandlung von 1,2:5,6-Di-O-isopropyliden-3-desoxy-α-D-glucose-3-en in 1,2:5,6-Di-O-isopropyliden-a-d-galactofuranose durch selektive hydroborierung. Carbohydr. Res. 1996, 2, 80–81. [Google Scholar] [CrossRef]
- Bonin, H.; Delacroix, T.; Gras, E. Dioxazaborocanes: Old adducts, new tricks. Org. Biomol. Chem. 2011, 9, 4714–4724. [Google Scholar] [CrossRef]
- Ni, W.; Fang, H.; Springsteen, G.; Wang, B. The Design of Boronic Acid Spectroscopic Reporter Compounds by Taking Advantage of the pKa-Lowering Effect of Diol Binding: Nitrophenol-Based Color Reporters for Diols. J. Org. Chem. 2004, 69, 1999–2007. [Google Scholar] [CrossRef]
- Cao, H.; McGill, T.; Heagy, M.D. Substituent Effects on Monoboronic Acid Sensors for Saccharides Based on N-Phenyl-1,8-naphthalenedicarboximides. J. Org. Chem. 2004, 69, 2959–2966. [Google Scholar] [CrossRef]
- Musashi, M.; Matsuo, M.; Oi, T.; Nomura, M. An anion-exchange chromatographic study on boron isotopic fractionation at 2 MPa at 293 K. J. Chromat. A 2006, 1131, 97–102. [Google Scholar] [CrossRef]
- Uenishi, J.; Matsui, K.; Wada, A. Trienylboronic acid, a versatile coupling tool for retinoid synthesis; stereospecific synthesis of 13-aryl substituted (11Z)-retinal. Tetrahedron Lett. 2003, 44, 3093–3096. [Google Scholar] [CrossRef]
- McGregor, N.; Pardin, C.; Skene, W.G. Using Quenching Kinetics and Thermodynamics of Amino-Fluorophores as Empirical Tools for Predicting Boronic Acid Sensors Suitable for Use in Physiological Conditions. Aust. J. Chem. 2011, 64, 1438–1446. [Google Scholar] [CrossRef]
- Hargrove, A.E.; Ellington, A.D.; Anslyn, E.V.; Sessler, J.L. Chemical Functionalization of Oligodeoxynucleotides with Multiple Boronic Acids for the Polyvalent Binding of Saccharides. Bioconj. Chem. 2011, 22, 388–396. [Google Scholar] [CrossRef]
- Zhong, Q.; Ngim, K.K.; Sun, M.; Li, J.; Deese, A.; Chetwyn, N.P. Strategies for the analysis of highly reactive pinacolboronate esters. J. Chromat. A 2012, 1229, 216–222. [Google Scholar] [CrossRef]
- Wolkenstein, K.; Gross, J.H.; Falk, H. Boron-containing organic pigments from a Jurassic red alga. Proc. Natl. Acad. Sci. USA 2010, 107, 19374–19378. [Google Scholar] [CrossRef] [PubMed]
- Claessens, C.G.; Torres, T. Subphthalocyanine enantiomers: First resolution of a C3 aromatic compound by HPLC. Tetrahedron Lett. 2000, 41, 6361–6365. [Google Scholar] [CrossRef]
- Nanjappan, P.; Ramallngam, K.; Nowotnik, D.P. Synthesis of the Four Stereoisomers of 1-Azabicyclo[2.2,2]oct-3-yl-α -hydroxy-α-(4-phenylboronic acid)-α-phenylacetate (QNB-Boronic acid), including a preparative HPLC method to separate diastereoisomeric mixtures with high optical purity. Tetrahedron Asymmetry 1992, 3, 1271–1282. [Google Scholar] [CrossRef]
- Moriyasu, M.; Endo, M.; Ichimaru, M.; Mizutani, T.; Kato, A. High Performance Liquid Chromatographic Behavior of Difluoroborane Derivatives of β-Diketones. Anal. Sci. 1990, 6, 45–48. [Google Scholar] [CrossRef]
- Sun, J.; Perfetti, M.T.; Santos, W.L. A Method for the Deprotection of Alkylpinacolyl Boronate Esters. J. Org. Chem. 2011, 76, 3571–3575. [Google Scholar] [CrossRef] [PubMed]
- Prichard, K.; Campkin, D.; O’Brien, N.; Kato, A.; Fleet, G.W.J.; Simone, M.I. Biological Activities of 3,4,5-Trihydroxypiperidines and their O- and N-Alkylated Derivatives. Chem. Biol. Drug Des. 2018, 92, 1171–1197. [Google Scholar] [CrossRef]
- Glenister, A.; Simone, M.; Hambley, T.W. A Warburg effect targeting vector designed to increase the uptake of compounds by cancer cells demonstrates glucose and hypoxia dependent uptake. PLoS ONE 2019, 14, e0217712. [Google Scholar] [CrossRef]
- Glenister, A.; Chen, C.K.J.; Renfrew, A.K.; Simone, M.; Hambley, T.W. Warburg Effect Targeting Cobalt(III) Cytotoxin Chaperone Complexes. J. Med. Chem. 2021, 64, 2678–2690. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakai, K.; Matsumura, A. Boron Neutron Capture Therapy for Glioblastoma. Cancer Lett. 2008, 262, 143–152. [Google Scholar] [CrossRef]
- Malouff, T.D.; Seneviratne, D.S.; Ebner, D.K.; Stross, W.C.; Waddle, M.R.; Trifiletti, D.M.; Krishnan, S. Boron Neutron Capture Therapy: A Review of Clinical Applications. Front. Oncol. 2021, 11, 601820. [Google Scholar] [CrossRef]
- Dymova, M.A.; Taskaev, S.Y.; Richter, V.A.; Kuligina, E.V. Boron neutron capture therapy: Current status and future perspectives. Cancer Comm. 2020, 40, 406–421. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, J.; Jiang, P.; Tian, S.; Wang, H.; Fan, R.; Liu, J.; Yang, Y.; Liu, Z.; Wang, J. The basis and advances in clinical application of boron neutron capture therapy. Radiat. Oncol. 2021, 16, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Pooh, K.; Kobayashi, T.; Kageji, T.; Uyama, S.; Matsumura, A.; Kumada, H. Clinical Review of the Japanese Experience with Boron Neutron Capture Therapy and a Proposed Strategy using Epithermal Neutron Beams. J. Neuro-Oncol. 2003, 62, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Zavjalov, E.; Zaboronok, A.; Kanygin, V.; Kasatova, A.; Kichigin, A.; Mukhamadiyarov, R.; Razumov, I.; Sycheva, T.; Mathis, B.J.; Maezono, S.E.B.; et al. Accelerator-based Boron Neutron Capture Therapy for Malignant Glioma: A Pilot Neutron Irradiation Study using Boron Phenylalanine, Sodium Borocaptate and Liposomal Borocaptate with a Heterotopic U87 Glioblastoma Model in SCID Mice. Int. J. Radiat. Biol. 2020, 96, 868–878. [Google Scholar] [CrossRef]
- Lee, J.W.; Ban, M.J.; Park, J.H.; Lee, S.M. Effect of F-18 Fluorodeoxyglucose Uptake by Bone Marrow on the Prognosis of Head and Neck Squamous Cell Carcinoma. J. Clin. Med. 2019, 9, 1169. [Google Scholar] [CrossRef]
- Pauwels, E.K.J.; Ribeiro, M.J.; Stoot, J.H.M.B.; McCready, V.R.; Bourguignon, M.; Mazière, B. FDG accumulation and tumour biology. Nucl. Med. Biol. 1998, 25, 317–322. [Google Scholar] [CrossRef]
- Clavo, A.C.; Brown, R.S.; Wahl, R.L. Fluorodeoxyglucose Uptake in Human Cancer Cell Lines Is Increased by Hypoxia. J. Nucl. Med. 1995, 36, 1625–1632. [Google Scholar]
- Sung, Y.; Tetrault, M.-A.; Takahashi, K.; Ouyang, J.; Pratx, G.; Fakhri, G.E.; Normandin, M.D. Dependence of fluorodeoxyglucose (FDG) uptake on cell cycle and dry mass: A single-cell study using a multi-modal radiography platform. Sci. Rep. 2020, 10, 4280. [Google Scholar] [CrossRef]
- Ong, L.-C.; Jin, Y.; Song, I.-C.; Yu, S.; Zhang, K. 2-[18F]-2-deoxy-D-glucose (FDG) uptake in human tumor cells is related to the expression of Glut-1 and Hexokinase II. Acta Radiol. 2008, 49, 1145–1153. [Google Scholar] [CrossRef]
- Ballinger, J.; Gnanasegaran, G. Chapter 4: 18F-FDG and Non-FDG PET Radiopharmaceuticals. In PET/CT Imaging. Basics and Practice; Agrawal, A.S.K., Esmail, A., Usmani, S., Eds.; BNMS British Nuclear Medicine Society; Springer: Nottingham, UK; Cham, Switzerland, 2022; pp. 27–32. [Google Scholar]
- Ponto, L.L.B.; Walsh, S.; Huang, J.; Mundt, C.; Thede-Reynolds, K.; Watkins, G.L.; Sunderland, J.; Acevedo, M.; Donovan, M. Pharmacoimaging of Blood-Brain Barrier Permeable (FDG) and Impermeable (FLT) Substrates After Intranasal (IN) Administration. AAPS J. 2018, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H. Response of Normal Tissues to Boron Neutron Capture Therapy (BNCT) with 10B-Borocaptate Sodium (BSH) and 10B-Paraboronophenylalanine (BPA). Cells 2021, 10, 2883. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.L.; Siqueira, J.A.; Batista-Silva, W.; Cardoso, F.B.; Nunes-Nesi, A.; Araújo, W.L. Boron: More Than an Essential Element for Land Plants? Front. Plant Sci. 2021, 11, 610307. [Google Scholar] [CrossRef] [PubMed]
- Warington, K. The effect of boric acid and borax on the broad bean and certain other plants. Ann. Bot. 1923, 37, 629–672. [Google Scholar] [CrossRef]
- Uluisika, I.; Karakaya, H.C.; Koc, A. The Importance of Boron in Biological Systems. J. Trace Elem. Med. Biol. 2018, 45, 156–162. [Google Scholar] [CrossRef]
- Health Effects. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp3-c3.pdf. (accessed on 17 April 2023).
- ATSDR. Benzene. In Benzene Overview. Available online: https://www.atsdr.cdc.gov/sites/toxzine/benzene_toxzine.html. (accessed on 17 April 2023).
- CDC. Benzene. Available online: https://www.cdc.gov/niosh/idlh/71432.html. (accessed on 17 April 2023).
- Sigma-Aldrich, S.D.S. Phenylboronic Acid. Available online: https://www.sigmaaldrich.com/AU/en/sds/aldrich/p20009 (accessed on 17 January 2023).
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.G.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: Version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef]
- Zhu, A.; Lee, D.; Shim, H. Metabolic PET Imaging in Cancer Detection and Therapy Response. Semin. Oncol. 2011, 38, 55–69. [Google Scholar] [CrossRef]
- Kessler, M.; Acuto, O.; Storelli, C.; Murer, H.; Müller, M.; Semenza, G. A modified procedure for the rapid preparation of efficiently transporting vesicles from small intestinal brush border membranes. Their use in investigating some properties of D-glucose and choline transport systems. Biochim. Biophys. Acta 1978, 506, 136–154. [Google Scholar] [CrossRef]
- McNaught, A.D. Nomenclature of Carbohydrates. Pure Appl. Chem. 1996, 68, 1919–2008. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Hall, L.D.; Miller, D.C. Fluorinated sulphonic esters of sugars: Their synthesis and reactions with pyridine. Carbohydr. Res. 1976, 47, 299–305. [Google Scholar] [CrossRef]
- Chiu, T.; Watanabe, K.; Fox, J. Nucleosides LXXXIII. Synthetic Studies on Nucleoside Antibiotics. 11. Synthesis of Methyl 4-amino-3,4-dideoxy-β-D-ribo-hexopyranoside and -hexopyranosiduronic acid (Derivatives related to the Carbohydrate moiety of Gougerotin)*. Carbohydr. Res. 1974, 32, 211. [Google Scholar] [CrossRef]
- Srivastava, H.; Srivastava, V. Synthesis of 6-deoxy-1,2:3,5-di-O-methylidene-α-D-xylo-hex-5-enofuranose. Carbohydr. Res. 1978, 60, 210. [Google Scholar] [CrossRef]
- Tewson, T.J.; Welch, M.J. New approaches to the synthesis of 3-deoxy-3-fluoro-D-glucose. J. Org. Chem. 1978, 43, 1090–1092. [Google Scholar] [CrossRef]
- Zinner, H.; Wulf, G.; Heinatz, R. Derivate der Zucker-mercaptale, XXXV. Darstellung und Mercaptalbildung der 2-Desoxy-D-xylose. Chem. Ber. 1964, 97, 3536–3540. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | α-Glucosidase | β-Glucosidase | α-Galactosidase | β-Galactosidase | α-Mannosidase | β-Mannosidase | α-L-Rhamnosidase | α-L-Fucosidase | β-Glucuronidase | Trehalase | Amyloglucosidase | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Yeast | Rat Intestinal Maltase | Almond | Bovine Liver | Coffee Beans | Bovine Liver | Jack Bean | Snail | Penicillium Decumbens | Bovine Kidney | E. coli | Bovine Liver | Porcine Kidney | A. niger | ||
 | BSH | aNI b(0%) | aNI b(6.9%) | aNI b(0%) | aNI b(0%) | aNI b(15%) | aNI b(12.3%) | aNI b(0%) | aNI b(7.9%) | aNI b(19.2%) | aNI b(0.2%) | aNI b(0%) | aNI b(6%) | aNI b(19.6%) | aNI b(4.2%) | aNI b(0%) |
| 10B-BSH | aNI b(0%) | aNI b(5.6%) | aNI b(0%) | aNI b(0%) | aNI b(11.9%) | aNI b(3.9%) | aNI b(22.3%) | aNI b(7.7%) | aNI b(15%) | aNI b(0%) | aNI b(6.5%) | aNI b(3.2%) | aNI b(12.5%) | aNI b(2.3%) | aNI b(0%) | |
 | BPA | cNI d(0%) | cNI d(0%) | cNI d(0%) | cNI d(0%) | cNI d(0%) | cNI d(4.3%) | cNI d(10.9%) | cNI d(1.1%) | cNI d(2.1%) | cNI d(0%) | cNI d(0%) | cNI d(0.5%) | cNI d(7.4%) | cNI d(0%) | cNI d(0%) |
| 10B-BPA | cNI d(0%) | cNI d(0%) | cNI d(0%) | cNI d(0%) | cNI d(14.2%) | cNI d(1.2%) | cNI d(0%) | cNI d(0%) | cNI d(0.3%) | cNI d(0%) | cNI d(0%) | cNI d(2.4%) | cNI d(0%) | cNI d(0%) | cNI d(0%) | |
 6 | cNI d(5.9%) | cNI d(0%) | cNI d(3.1%) | cNI d(0.3%) | cNI d(12.2%) | cNI d(0.5%) | cNI d(16.3%) | cNI d(0%) | cNI d(0%) | cNI d(0.3%) | cNI d(6.5%) | cNI d(3.5%) | cNI d(12.4%) | cNI d(0%) | cNI d(0%) | |
 7 | aNI b(33.8%) | aNI b(12.1%) | aNI b(18.3%) | aNI b(1.3%) | aNI b(12.6%) | aNI b(0%) | aNI b(40.7%) | aNI b(0%) | aNI b(0%) | aNI b(0%) | aNI b(1.3%) | aNI b(3.7%) | aNI b(32.4%) | aNI b(0.5%) | aNI b(1.5%) | |
| Compound | HT29 | U87 | MCF-7 | A2780 | H460 | A431 | Du145 | BE2-C | SJ-G2 | MIA-Pa-Ca2 | MCF10A |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Colon carcinoma | Glioblastoma | Breast carcinoma | Ovarian carcinoma | Lung carcinoma | Skin carcinoma | Prostate carcinoma | Neuro-blastoma | Glio-blastoma | Pancreatic carcinoma | Breast (Normal) | |
| BSH | 3 ± 2 | <0 | 15 ± 3 | 2 ± 5 | 8 ± 2 | <0 | 0 ± 8 | 10 ± 6 | 3 ± 8 | 2 ± 6 | 8 ± 3 |
| 10B-BSH | 5 ± 1 | 0 ± 2 | 5 ± 3 | 5 ± 4 | 4 ± 2 | <0 | 7 ± 7 | 8 ± 7 | 1 ± 9 | 2 ± 4 | 13 ± 4 |
| BPA | 14 ± 0 | <0 | <0 | 4 ± 1 | 7 ± 8 | 4 ± 6 | 19 ± 10 | 13 ± 10 | 5 ± 8 | 3 ± 3 | 4 ± 1 |
| 10B-BPA | 15 ± 4 | <0 | 1 ± 3 | 8 ± 4 | 8 ± 5 | 4 ± 4 | 15 ± 9 | 10 ± 6 | 5 ± 10 | 11 ± 3 | <0 |
| 6 | 10 ± 5 | 8 ± 4 | 14 ± 5 | 10 ± 5 | 9 ± 2 | 0 ± 14 | 0 ± 8 | 2 ± 5 | <0 | 3 ± 1 | 5 ± 1 |
| 7 | −2 ± 2 | −6 ± 3 | 2 ± 2 | 8 ± 3 | 4 ± 1 | 3 ± 0 | 7 ± 4 | 1 ± 2 | 7 ± 4 | 1 ±2 | 7 ± 3 |
| 7 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simone, M.I. Diastereoselective Synthesis of the Borylated d-Galactose Monosaccharide 3-Boronic-3-Deoxy-d-Galactose and Biological Evaluation in Glycosidase Inhibition and in Cancer for Boron Neutron Capture Therapy (BNCT). Molecules 2023, 28, 4321. https://doi.org/10.3390/molecules28114321
Simone MI. Diastereoselective Synthesis of the Borylated d-Galactose Monosaccharide 3-Boronic-3-Deoxy-d-Galactose and Biological Evaluation in Glycosidase Inhibition and in Cancer for Boron Neutron Capture Therapy (BNCT). Molecules. 2023; 28(11):4321. https://doi.org/10.3390/molecules28114321
Chicago/Turabian StyleSimone, Michela I. 2023. "Diastereoselective Synthesis of the Borylated d-Galactose Monosaccharide 3-Boronic-3-Deoxy-d-Galactose and Biological Evaluation in Glycosidase Inhibition and in Cancer for Boron Neutron Capture Therapy (BNCT)" Molecules 28, no. 11: 4321. https://doi.org/10.3390/molecules28114321
APA StyleSimone, M. I. (2023). Diastereoselective Synthesis of the Borylated d-Galactose Monosaccharide 3-Boronic-3-Deoxy-d-Galactose and Biological Evaluation in Glycosidase Inhibition and in Cancer for Boron Neutron Capture Therapy (BNCT). Molecules, 28(11), 4321. https://doi.org/10.3390/molecules28114321