
Development and Characterization of Novel Selective, Non-Basic Dopamine D2 Receptor Antagonists for the Treatment of Schizophrenia

 ,
,  , , ,
, , ,  , ,
, ,  , and
, and 
Abstract
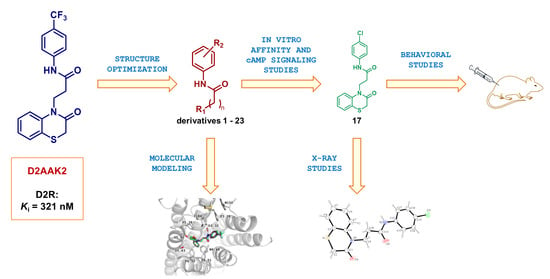
1. Introduction
2. Results and Discussion
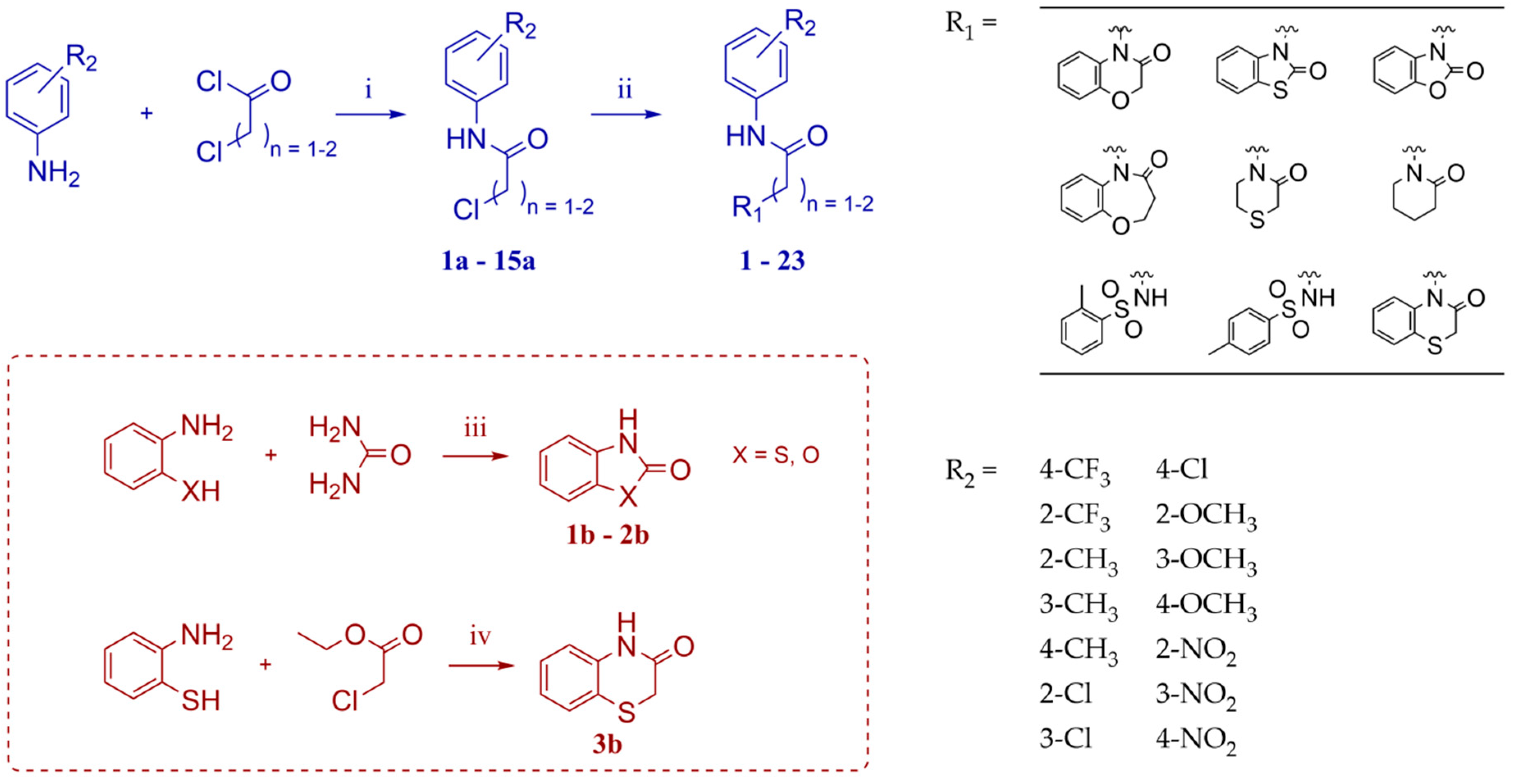
2.1. Chemistry
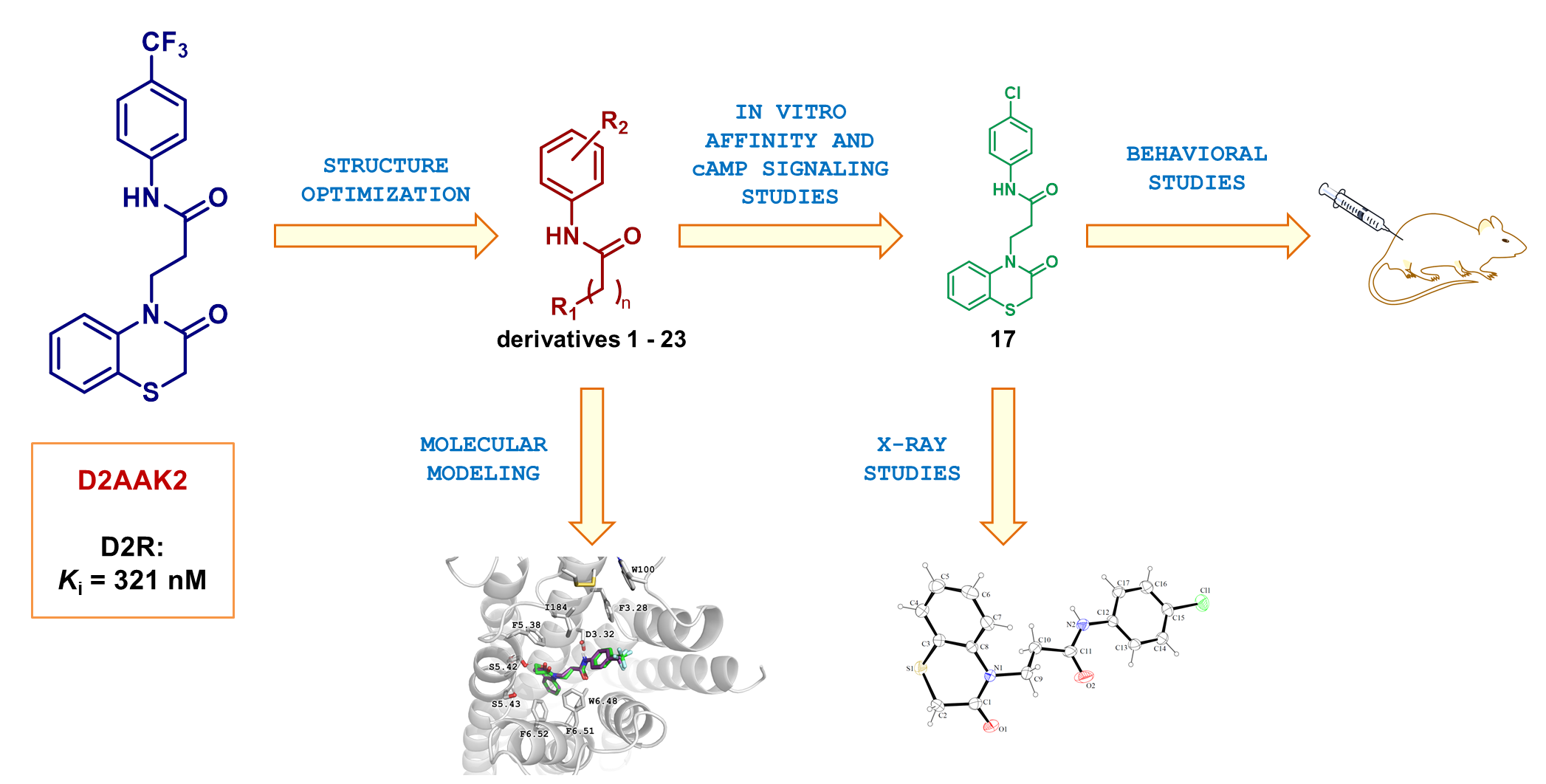
2.2. Affinity of Compounds at D2 Receptor and Structure-Activity Relationship
2.3. Determination of the Affinity Profile of Selected Compounds for Additional GPCRs of Interest
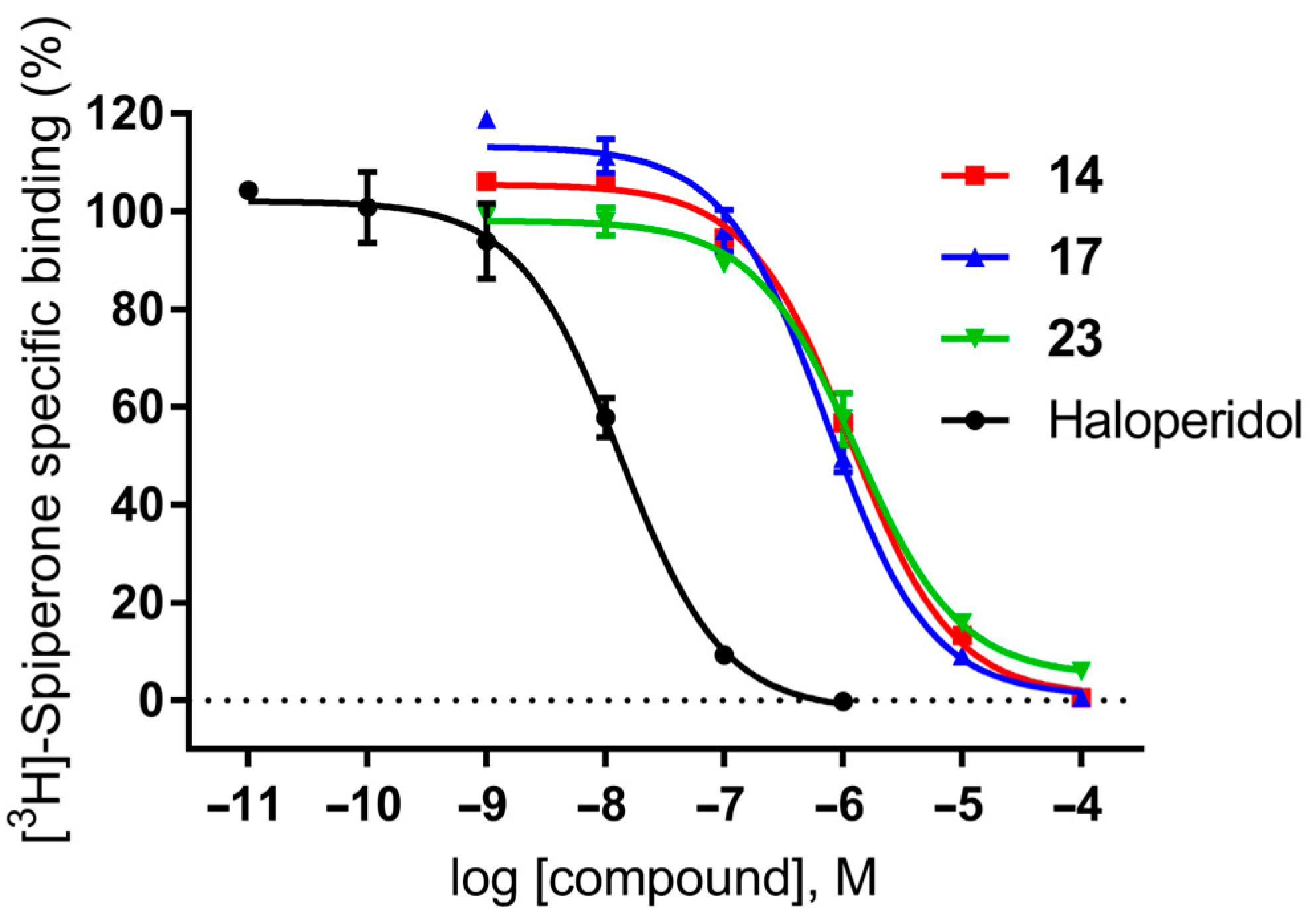
2.4. Efficacy of Selected Compounds at D2 Receptor
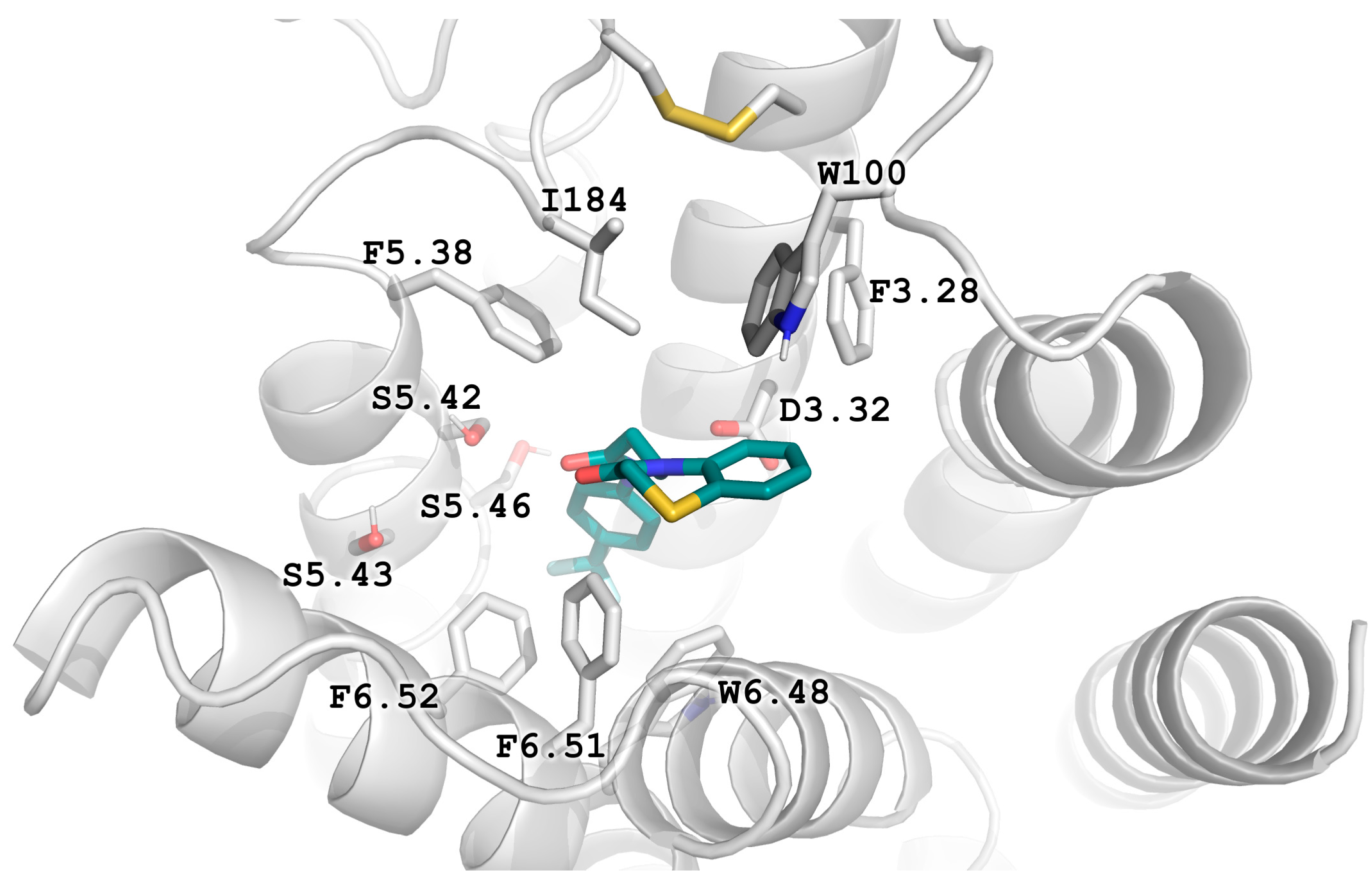
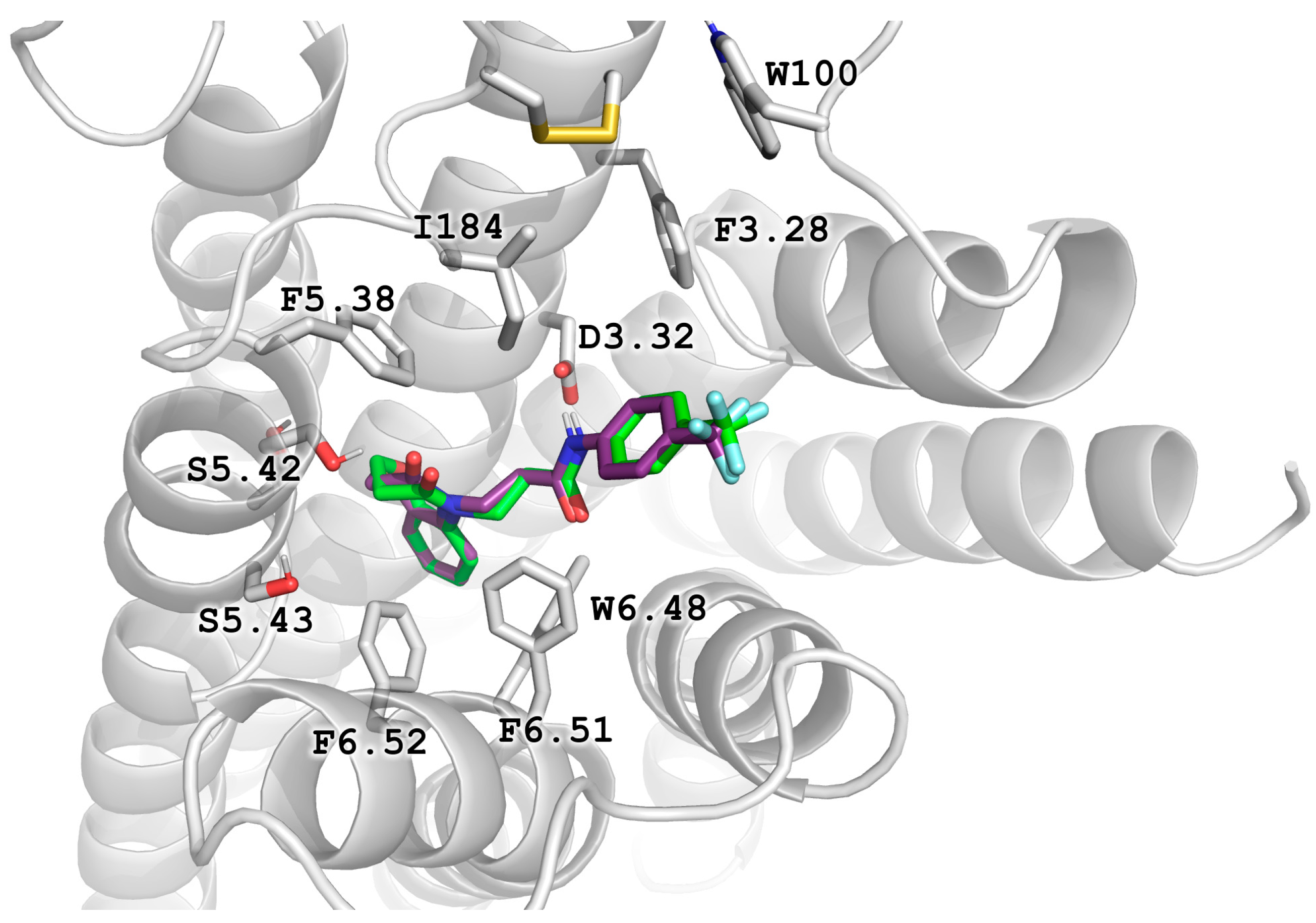
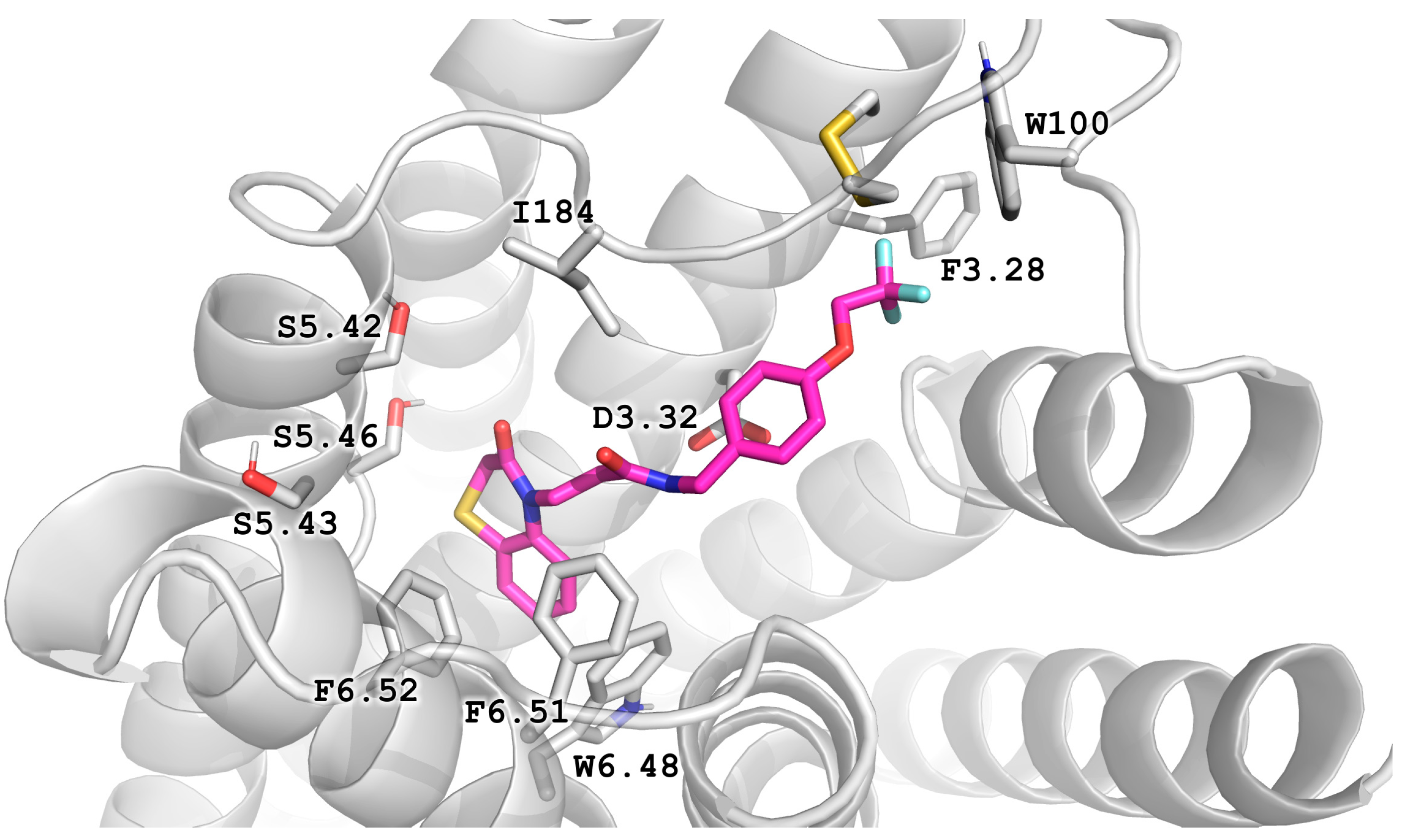
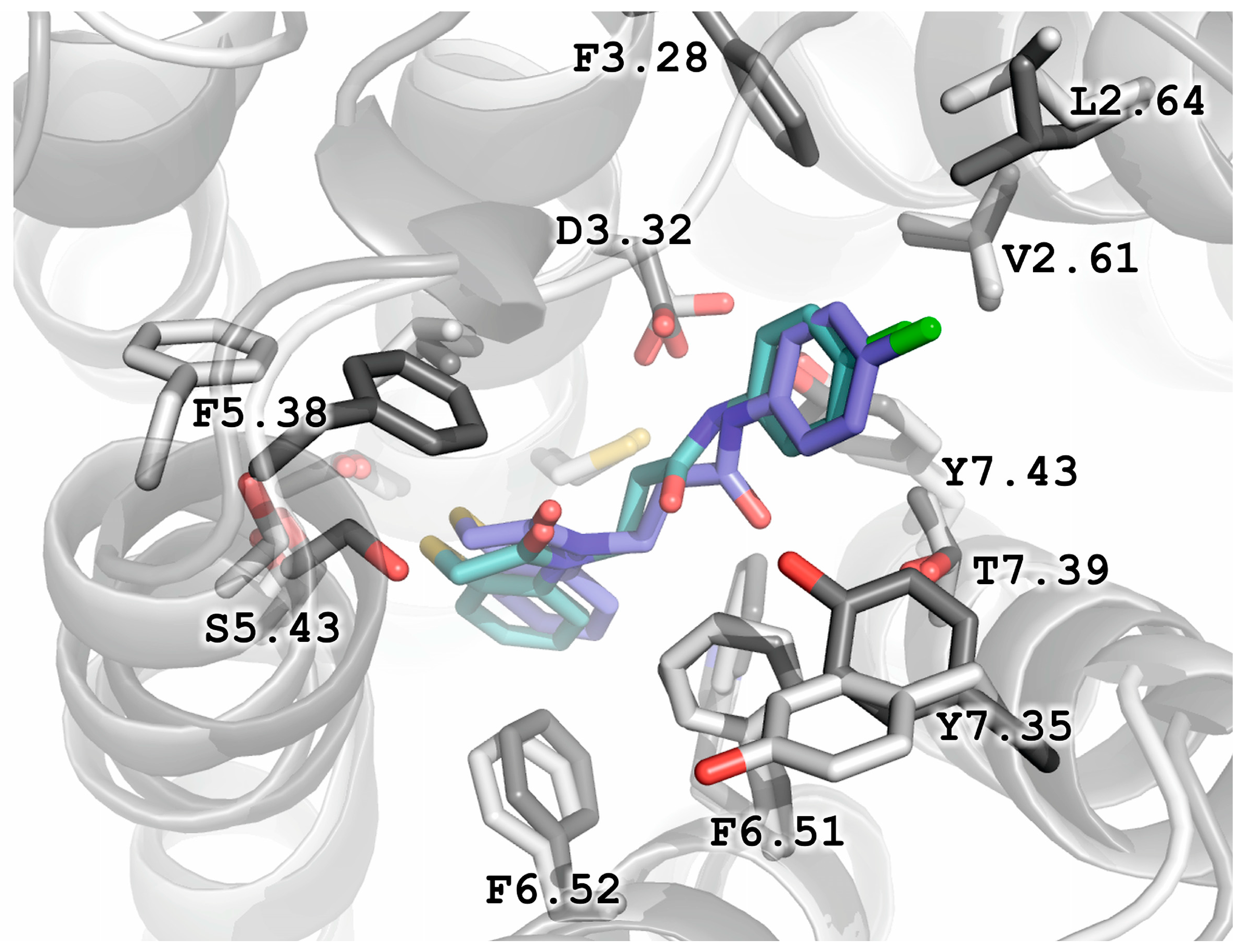
2.5. Molecular Modeling
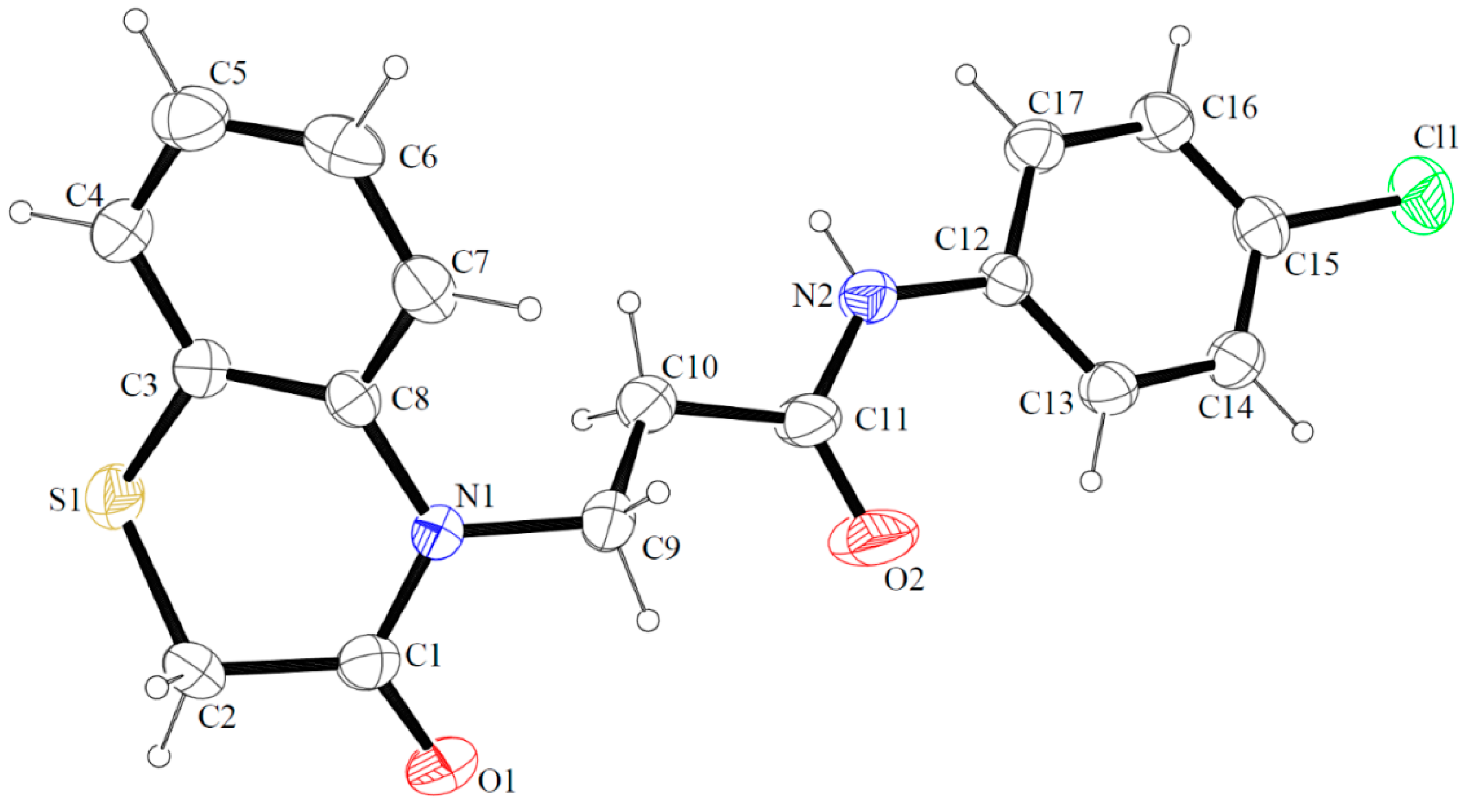
2.6. X-ray Studies
2.7. Behavioral Studies
2.7.1. Effect of Compound 17 on Locomotor Activity in Mice
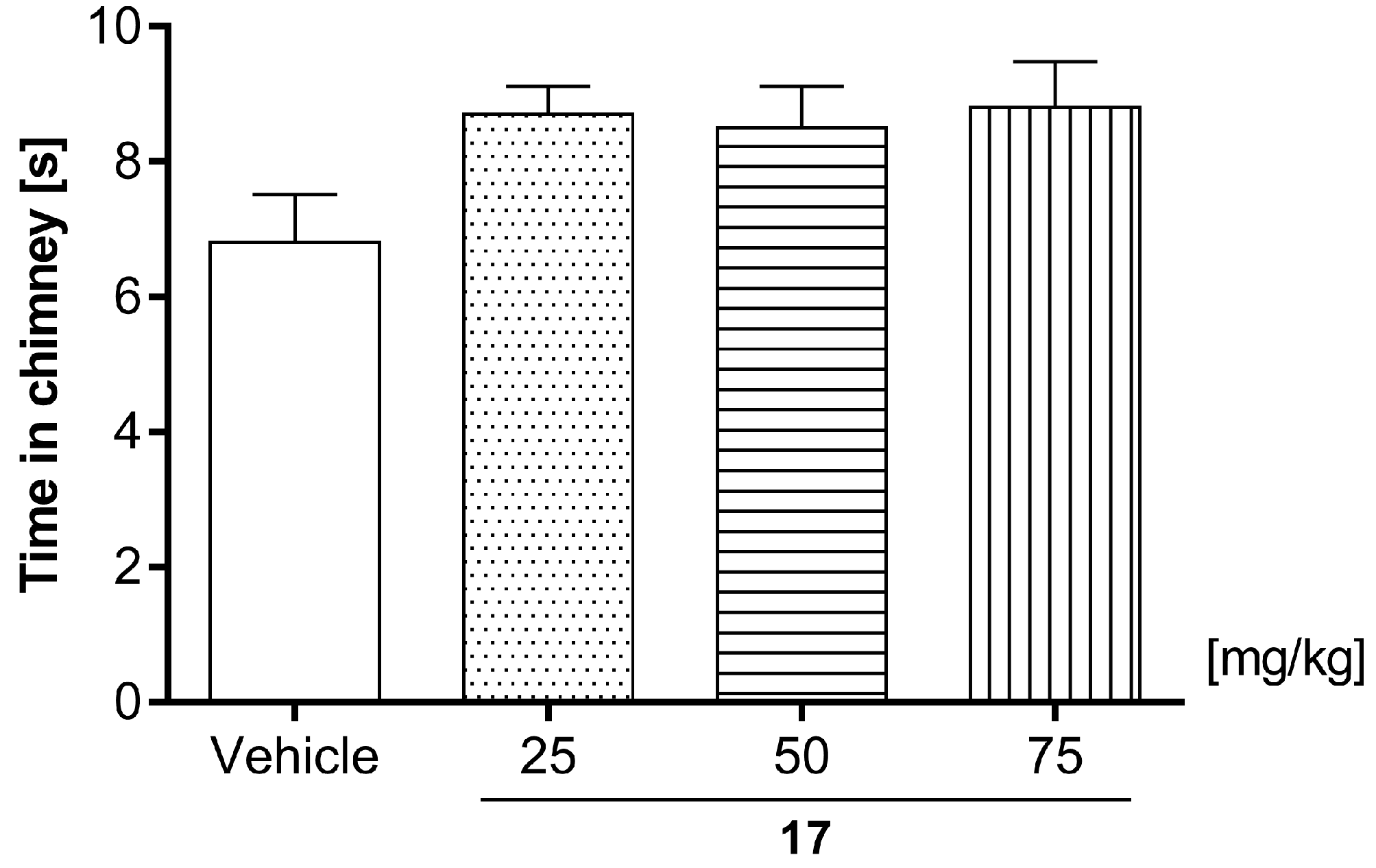
2.7.2. Motor Coordination Evaluated by Chimney Test
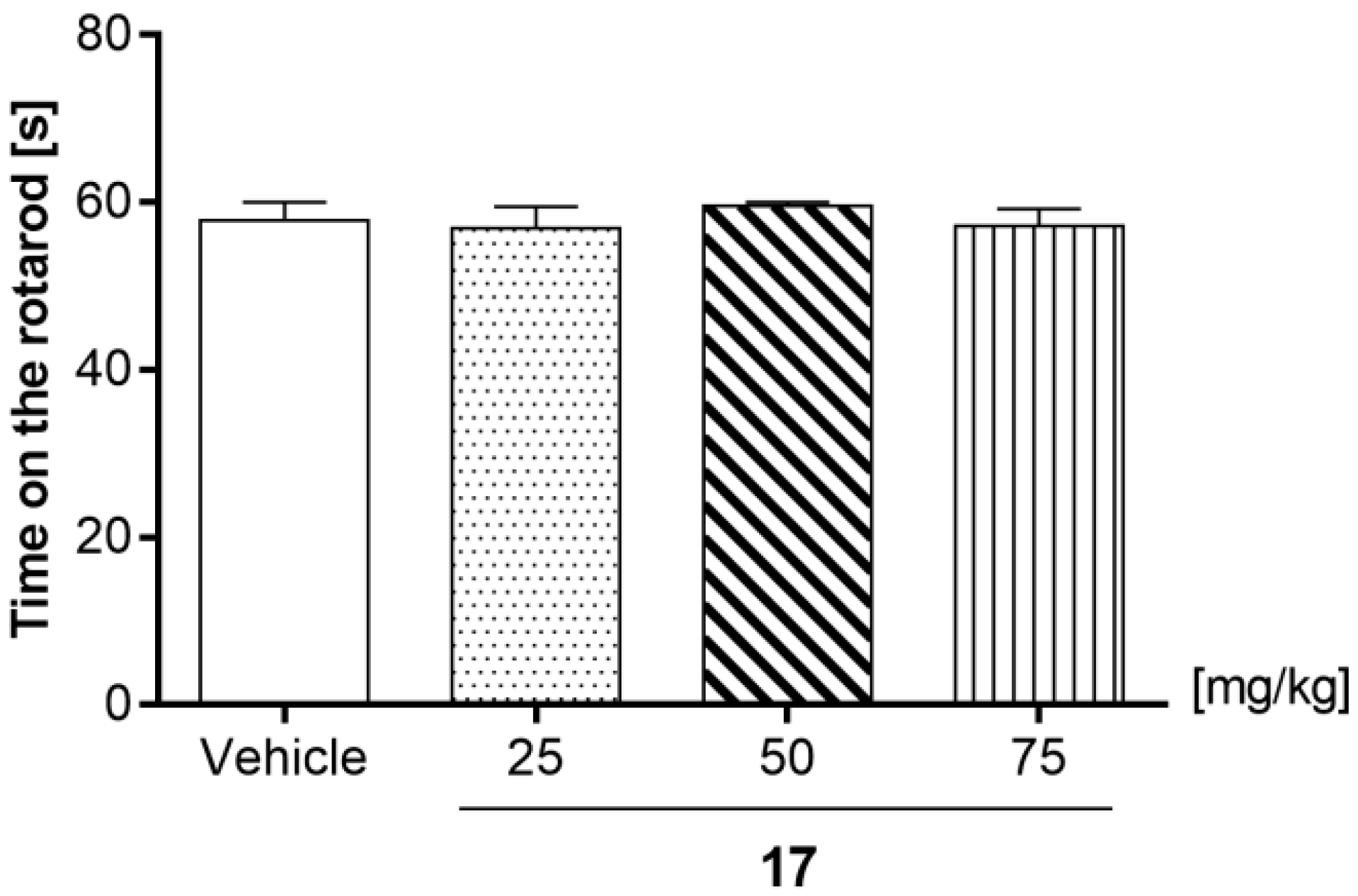
2.7.3. Effects of Compound 17 in the Rotarod Test
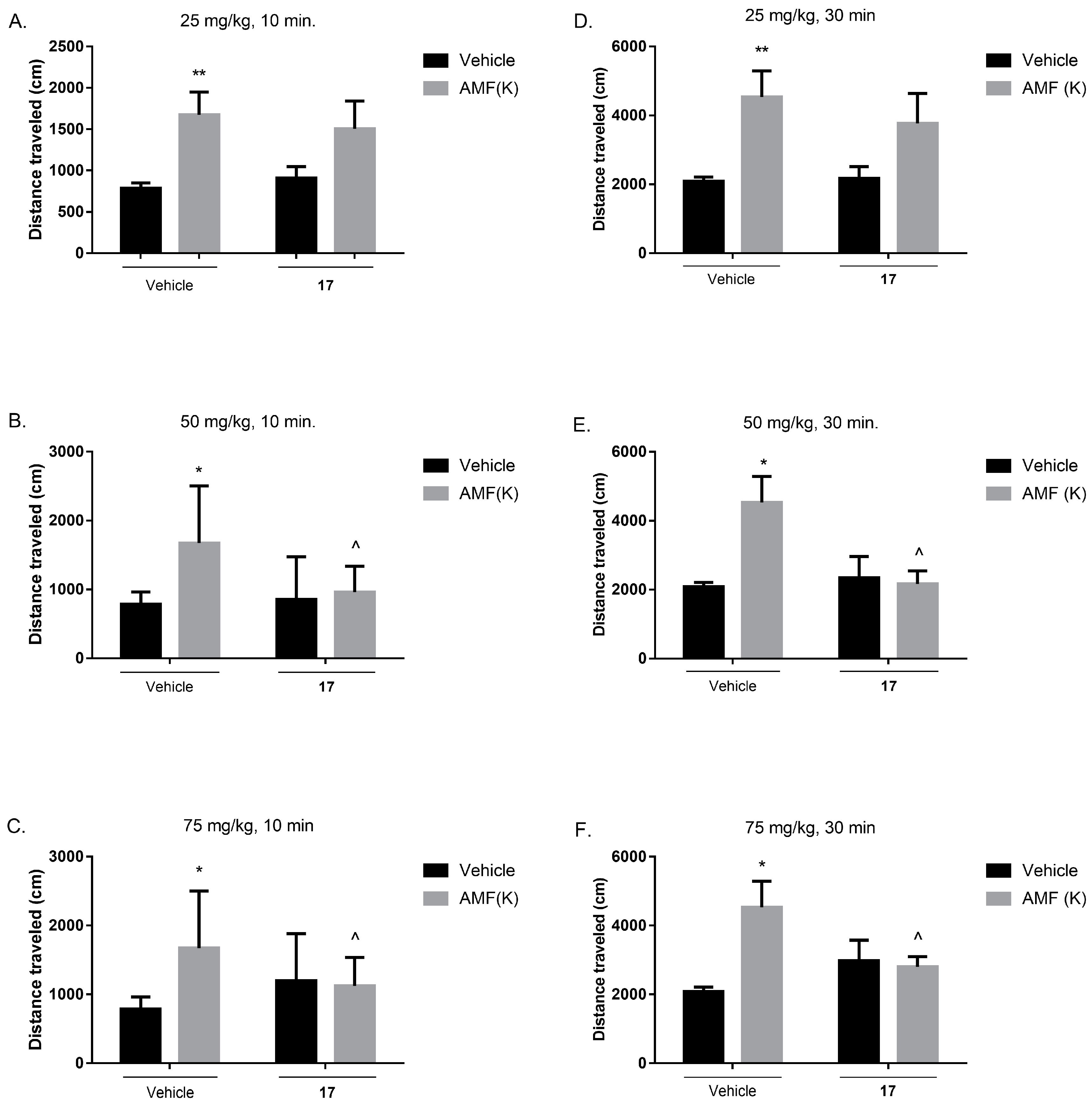
2.7.4. Spontaneous Locomotor Activity and Amphetamine-Induced Hyperactivity
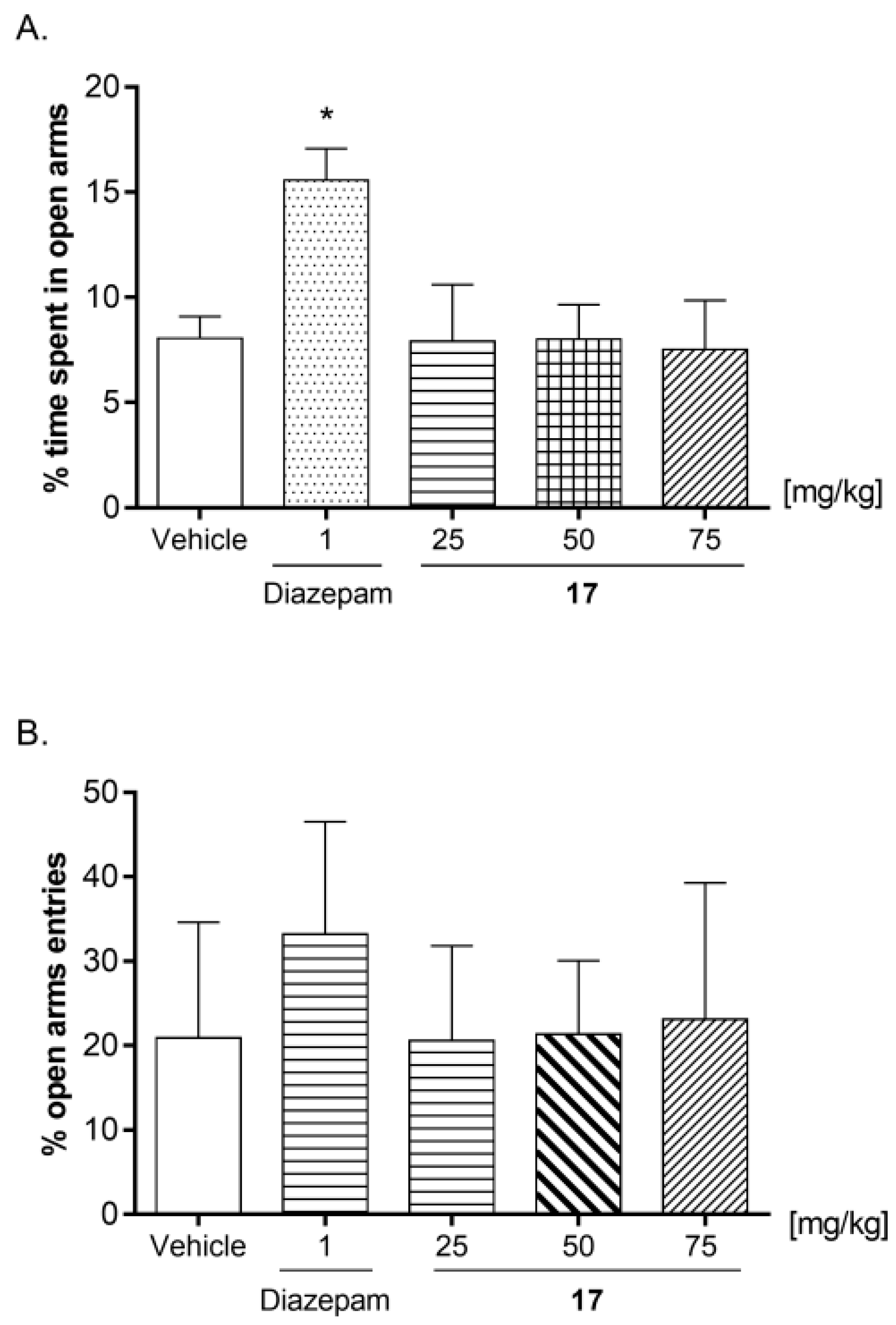
2.7.5. The Effect of Acute Administration of Compound 17 on Anxiety Responses in Mice
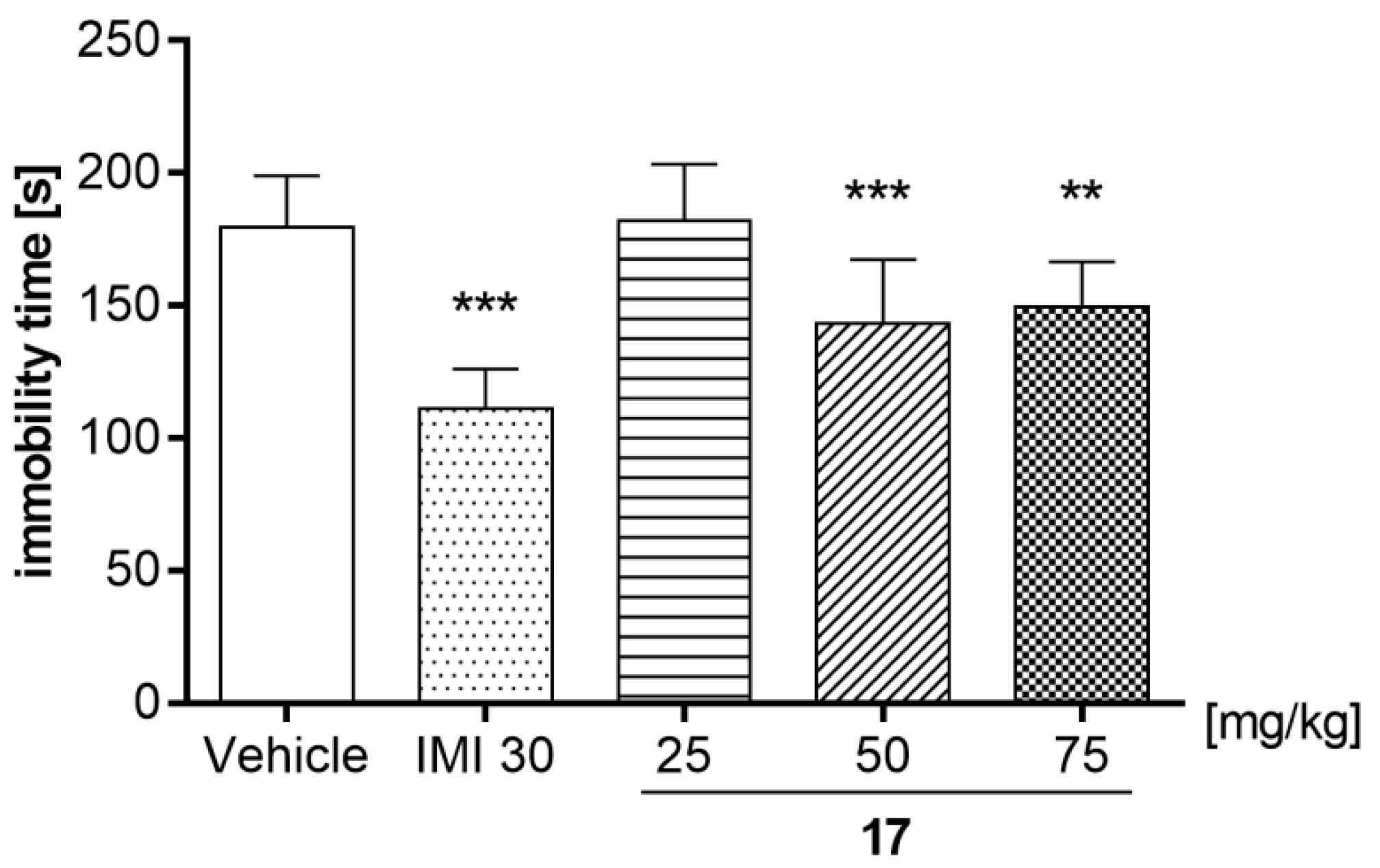
2.7.6. Compound 17 Dose-Effect Relationship in the FST
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for Synthesis of the Intermediates 1a–15a
4.1.2. General Procedure for Synthesis of the Intermediates 1b–2b
4.1.3. Synthesis of the Intermediate 3b
4.1.4. General Procedure for Synthesis of the Final Compounds 1–3, 5, 7–23
4.1.5. Synthesis of the Final Compound 4
4.1.6. Synthesis of the Final Compound 6
4.1.7. Purification and Spectral Data for the Synthesized Final Compounds 1–23
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]oxazin-4-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (1). Compound purified by crystallization from ACN: white solid. Yield: 38%. 1H NMR (600 MHz, DMSO) δ 10.41 (s, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.07 (dq, J = 8.5, 4.3 Hz, 1H), 7.02 (d, J = 4.4 Hz, 2H), 4.64 (s, 2H), 4.22 (t, J = 7.4 Hz, 2H), 2.70 (t, J = 7.4 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 169.92, 164.49, 145.45, 143.01, 128.78, 126.49 (q, J = 3.9 Hz), 124.85 (q, J = 271.3 Hz), 124.05, 123.69 (q, J = 32.6 Hz), 123.25, 119.50, 117.12, 115.83, 67.54, 37.47, 34.69. HRMS (ESI) m/z [M + H]+ calculated for C18H15F3N2O3: 365.1108, found: 365.1109.
- 3-(2-Oxobenzo[d]thiazol-3(2H)-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (2). Compound was purified by washing with ACN: white solid. Yield: 34%. 1H NMR (600 MHz, DMSO) δ 10.45 (s, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.66 (dd, J = 8.4, 3.1 Hz, 3H), 7.45 (d, J = 8.1 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 4.27 (t, J = 7.0 Hz, 2H), 2.80 (t, J = 7.0 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 169.67, 169.18, 142.91, 137.18, 127.04, 126.51 (q, J = 3.7 Hz), 124.82 (q, J = 270.8 Hz), 123.73 (q, J = 32.0 Hz), 123.60, 123.35, 121.93, 119.51, 111.99, 39.27, 35.03. HRMS (ESI) m/z [M + H]+ calculated for C17H13F3N2O2S: 367.0723, found: 367.0724.
- 3-(2-Oxobenzo[d]oxazol-3(2H)-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (3). Compound purified by crystallization from ACN: yellowish solid. Yield: 43%. 1H NMR (600 MHz, DMSO) δ 10.42 (s, 1H), 7.73 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 7.36–7.31 (m, 2H), 7.24–7.18 (m, 1H), 7.14–7.09 (m, 1H), 4.14 (t, J = 6.7 Hz, 2H), 2.86 (t, J = 6.7 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 169.71, 154.06, 142.85, 142.39, 131.39, 126.47 (q, J = 4.0 Hz), 124.78 (q, J = 272.0 Hz), 124.23, 123.71 (q, J = 32.0 Hz), 122.58, 119.46, 110.02, 109.93, 38.66, 34.89. HRMS (ESI) m/z [M + H]+ calculated for C17H13F3N2O3: 351.0951, found: 351.0950.
- 3-(4-Oxo-3,4-dihydrobenzo[b][1,4]oxazepin-5(2H)-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (4). Compound purified by DCVC (hexane/ethyl acetate 1:1): yellowish solid. Yield: 27%. 1H NMR (600 MHz, DMSO) δ 10.29 (s, 1H), 7.71 (d, J = 8.5 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.50 (dt, J = 7.8, 1.6 Hz, 1H), 7.24 (dtt, J = 20.0, 7.5, 1.5 Hz, 2H), 7.12 (dt, J = 7.7, 1.5 Hz, 1H), 4.45 (td, J = 6.7, 1.3 Hz, 2H), 4.10–3.98 (m, 2H), 2.61–2.57 (m, 2H), 2.52 (t, J = 4.5 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 170.69, 169.96, 150.18, 143.00, 136.99, 127.54, 126.43 (q, J = 3.9 Hz), 125.76, 124.21, 123.69 (q, J = 271.2 Hz), 123.68 (q, J = 31.9 Hz), 123.20, 119.41, 74.56, 43.82, 35.49, 35.00. HRMS (ESI) m/z [M + H]+ calculated for C19H17F3N2O3: 379.1264, found: 379.1266.
- 3-(3-Oxothiomorpholino)-N-(4-(trifluoromethyl)phenyl)propanamide (5). Compound was purified by washing with ACN: white solid. Yield: 20%. 1H NMR (600 MHz, DMSO) δ 10.39 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 3.67–3.58 (m, 4H), 3.23 (s, 2H), 2.85 (t, J = 5.7 Hz, 2H), 2.63 (t, J = 6.9 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 170.61, 167.07, 143.07, 126.52 (q, J = 3.7 Hz), 124.85 (q, J = 271.2 Hz), 123.67 (q, J = 31.9 Hz), 119.47, 49.40, 44.39, 35.59, 29.67, 26.39. HRMS (ESI) m/z [M + H]+ calculated for C14H15F3N2O2S: 333.0879, found: 333.0879.
- 3-(2-Oxopiperidin-1-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (6). Compound was purified by washing with ACN: white solid. Yield: 24%. 1H NMR (600 MHz, DMSO) δ 10.37 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 3.54 (t, J = 7.1 Hz, 2H), 3.29 (t, J = 5.6 Hz, 2H), 2.59 (t, J = 7.0 Hz, 2H), 2.20 (t, J = 6.3 Hz, 2H), 1.72–1.61 (m, 4H). 13C NMR (151 MHz, DMSO) δ 170.81, 169.02, 143.13, 126.50 (q, J = 4.0 Hz), 124.85 (q, J = 271.0 Hz), 123.51 (q, J = 31.9 Hz), 119.43, 48.37, 43.81, 35.27, 32.47, 23.28, 21.39. HRMS (ESI) m/z [M + H]+ calculated for C15H17F3N2O2: 315.1315, found: 315.1315.
- 3-((2-Methylphenyl)sulfonamido)-N-(4-(trifluoromethyl)phenyl)propanamide (7). Compound purified by crystallization from ACN: white solid. Yield: 27%. 1H NMR (600 MHz, DMSO) δ 10.31 (s, 1H), 7.83 (dd, J = 8.2, 1.1 Hz, 1H), 7.76 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.50 (td, J = 7.5, 1.2 Hz, 1H), 7.41–7.35 (m, 2H), 3.08 (t, J = 7.0 Hz, 2H), 2.57 (s, 3H), 2.54 (t, J = 7.0 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 169.97, 143.06 (d, J = 1.4 Hz), 138.94, 137.02, 132.97, 132.84, 128.89, 126.64, 126.44 (q, J = 3.7 Hz), 124.86 (q, J = 271.3 Hz), 123.57 (q, J = 31.9 Hz), 119.40, 38.92, 37.17, 20.23. HRMS (ESI) m/z [M + H]+ calculated for C17H17F3N2O3S: 387.0985, found: 387.0986.
- 3-((4-Methylphenyl)sulfonamido)-N-(4-(trifluoromethyl)phenyl)propanamide (8). Compound was purified by washing with ACN: white solid. Yield: 36%. 1H NMR (600 MHz, DMSO) δ 10.37 (s, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.72–7.63 (m, 4H), 7.39 (d, J = 8.0 Hz, 2H), 3.03 (t, J = 7.0 Hz, 2H), 2.53 (t, J = 7.1 Hz, 2H), 2.37 (s, 3H). 13C NMR (151 MHz, DMSO) δ 169.93, 143.10, 143.07, 137.87, 130.11, 127.03, 126.46 (q, J = 4.0 Hz), 124.85 (q, J = 271.3 Hz), 123.59 (q, J = 31.9 Hz), 119.40, 39.13, 37.14, 21.41. HRMS (ESI) m/z [M + H]+ calculated for C17H17F3N2O3S: 387.0985, found: 387.0986.
- 2-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-(4-(trifluoromethyl)phenyl)acetamide (9). The compound precipitated after cooling down the reaction mixture, then purified by washing with ACN: beige solid. Yield: 37%. 1H NMR (600 MHz, DMSO) δ 10.70 (s, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.70 (d, J = 8.5 Hz, 2H), 7.44 (dd, J = 7.8, 1.5 Hz, 1H), 7.28 (ddd, J = 8.5, 7.4, 1.6 Hz, 1H), 7.16 (dd, J = 8.3, 1.2 Hz, 1H), 7.08 (td, J = 7.5, 1.2 Hz, 1H), 4.78 (s, 2H), 3.59 (s, 2H). 13C NMR (151 MHz, DMSO) δ 167.19, 166.12, 142.81, 140.20, 128.46, 127.81, 126.62 (q, J = 4.0 Hz), 124.84 (q, J = 269.5 Hz), 123.91, 123.86 (q, J = 31.5 Hz), 123.08, 119.50, 118.54, 48.86, 30.66. HRMS (ESI) m/z [M + H]+ calculated for C17H13F3N2O2S: 367.0723, found: 367.0724.
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-phenylpropanamide (10). Compound was purified by washing with ACN: white solid. Yield: 47%. 1H NMR (600 MHz, DMSO) δ 10.00 (s, 1H), 7.59–7.53 (m, 2H), 7.42 (ddd, J = 10.9, 8.0, 1.3 Hz, 2H), 7.34–7.27 (m, 3H), 7.09–7.01 (m, 2H), 4.29–4.20 (m, 2H), 3.51 (s, 2H), 2.69–2.61 (m, 2H). 13C NMR (151 MHz, DMSO) δ 168.17, 164.20, 138.54, 138.40, 128.05, 127.54, 126.79, 122.74, 122.67, 122.58, 118.56, 117.48, 40.32, 33.88, 29.87. HRMS (ESI) m/z [M + H]+ calculated for C17H16N2O2S: 313.1005, found: 313.1005.
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-(2-(trifluoromethyl)phenyl)propanamide (11). Compound was purified by washing with ACN: white solid. Yield: 42%. 1H NMR (300 MHz, DMSO) δ 9.71 (s, 1H), 7.76–7.62 (m, 2H), 7.49–7.36 (m, 4H), 7.36–7.28 (m, 1H), 7.07 (td, J = 7.5, 1.3 Hz, 1H), 4.22 (t, J = 7.5 Hz, 2H), 3.51 (s, 2H), 2.67 (t, J = 7.6 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 170.29, 165.31, 139.59, 135.71, 133.39, 130.75, 128.66, 127.86, 127.25, 126.72 (q, J = 5.3 Hz), 125.43 (q, J = 30.6 Hz), 123.85 (q, J = 269.5 Hz), 123.85, 123.76, 118.47, 41.34, 34.13, 30.94. HRMS (ESI) m/z [M + H]+ calculated for C18H15F3N2O2S: 381.0879, found: 381.0878.
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-(o-tolyl)propanamide (12). Compound purified by crystallization from ACN: white solid. Yield: 37%. 1H NMR (600 MHz, DMSO) δ 9.37 (s, 1H), 7.43 (td, J = 5.3, 2.8 Hz, 2H), 7.35 (d, J = 7.9 Hz, 1H), 7.34–7.29 (m, 1H), 7.18 (d, J = 7.4 Hz, 1H), 7.14 (td, J = 7.6, 1.6 Hz, 1H), 7.07 (ddd, J = 10.5, 5.2, 2.9 Hz, 2H), 4.25 (t, J = 7.5 Hz, 2H), 3.51 (s, 2H), 2.68 (t, J = 7.5 Hz, 2H), 2.16 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.16, 164.22, 138.52, 135.55, 131.04, 129.61, 127.55, 126.78, 125.22, 124.51, 124.47, 122.74, 122.69, 117.54, 40.37, 33.33, 29.90, 17.19. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O2S: 327.1162, found: 327.1163.
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-(m-tolyl)propanamide (13). Compound purified by DCVC (hexane/ethyl acetate 1:1): white solid. Yield: 49%. 1H NMR (600 MHz, DMSO) δ 9.92 (s, 1H), 7.44–7.38 (m, 3H), 7.32 (ddd, J = 17.4, 8.2, 1.9 Hz, 2H), 7.16 (t, J = 7.8 Hz, 1H), 7.06 (td, J = 7.5, 1.1 Hz, 1H), 6.85 (d, J = 7.5 Hz, 1H), 4.28–4.18 (m, 2H), 3.51 (s, 2H), 2.64 (dd, J = 8.5, 6.6 Hz, 2H), 2.26 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.10, 164.20, 138.55, 138.32, 137.19, 127.89, 127.54, 126.79, 123.29, 122.74, 122.66, 119.13, 117.47, 115.79, 40.34, 33.89, 29.87, 20.58. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O2S: 327.1162, found: 327.1161.
- 3-(3-Oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)-N-(p-tolyl)propanamide (14). Compound purified by DCVC (hexane/ethyl acetate 7:3): white solid. Yield: 38%. 1H NMR (600 MHz, DMSO) δ 9.91 (s, 1H), 7.46–7.39 (m, 4H), 7.31 (ddd, J = 8.4, 7.4, 1.6 Hz, 1H), 7.09 (d, J = 8.2 Hz, 2H), 7.06 (td, J = 7.5, 1.1 Hz, 1H), 4.29–4.17 (m, 2H), 3.51 (s, 2H), 2.70–2.58 (m, 2H), 2.24 (s, 3H). 13C NMR (151 MHz, DMSO) δ 167.91, 164.18, 138.55, 135.90, 131.47, 128.43, 127.54, 126.79, 122.74, 122.65, 118.59, 117.47, 40.37, 33.82, 29.86, 19.82. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O2S: 327.1162, found: 327.1170.
- N-(2-Chlorophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (15). Compound purified by DCVC (hexane/ethyl acetate 7:3): white solid. Yield: 42%. 1H NMR (600 MHz, DMSO) δ 9.63 (s, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.45–7.39 (m, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.19 (t, J = 7.8 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 4.24 (t, J = 7.5 Hz, 2H), 3.51 (s, 2H), 2.73 (t, J = 7.6 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 168.62, 164.21, 138.52, 134.15, 128.81, 127.55, 126.78, 126.70, 126.01, 125.82, 125.74, 122.74, 122.66, 117.48, 40.27, 33.33, 29.88. HRMS (ESI) m/z [M + H]+ calculated for C17H15ClN2O2S: 347.0616, found: 347.0616.
- N-(3-Chlorophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (16). Compound purified by washing with ACN: white solid. Yield: 28%. 1H NMR (600 MHz, DMSO) δ 10.19 (s, 1H), 7.77 (t, J = 2.0 Hz, 1H), 7.44–7.38 (m, 3H), 7.34–7.28 (m, 2H), 7.09 (ddd, J = 7.9, 2.1, 1.0 Hz, 1H), 7.06 (td, J = 7.6, 1.2 Hz, 1H), 4.27–4.20 (m, 2H), 3.51 (s, 2H), 2.69–2.62 (m, 2H). 13C NMR (151 MHz, DMSO) δ 168.61, 164.24, 139.80, 138.50, 132.39, 129.77, 127.55, 126.78, 122.76, 122.72, 122.30, 118.04, 117.49, 116.91, 40.15, 33.95, 29.88. HRMS (ESI) m/z [M + H]+ calculated for C17H15ClN2O2S: 347.0616, found: 347.0616.
- N-(4-Chlorophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (17). Compound purified by crystallization from MeOH: white solid. Yield: 21%. 1H NMR (600 MHz, DMSO) δ 10.14 (s, 1H), 7.61–7.55 (m, 2H), 7.42 (td, J = 7.3, 1.4 Hz, 2H), 7.38–7.33 (m, 2H), 7.33–7.27 (m, 1H), 7.06 (td, J = 7.6, 1.3 Hz, 1H), 4.24 (dd, J = 8.5, 6.7 Hz, 2H), 3.51 (s, 2H), 2.65 (dd, J = 8.6, 6.5 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 168.35, 164.22, 138.51, 137.34, 127.97, 127.54, 126.78, 126.12, 122.75, 122.70, 120.08, 117.49, 40.21, 33.90, 29.87. HRMS (ESI) m/z [M + H]+ calculated for C17H15ClN2O2S: 347.0616, found: 347.0617.
- N-(2-Methoxyphenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (18). Compound purified by washing with ACN: white solid. Yield: 41%. 1H NMR (600 MHz, DMSO) δ 9.25 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.42 (dd, J = 8.2, 4.2 Hz, 2H), 7.31 (t, J = 7.9 Hz, 1H), 7.09–7.00 (m, 3H), 6.89 (t, J = 7.7 Hz, 1H), 4.21 (t, J = 7.5 Hz, 2H), 3.80 (s, 3H), 3.51 (s, 2H), 2.73 (t, J = 7.5 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 168.38, 164.13, 149.11, 138.55, 127.51, 126.77, 126.50, 123.83, 122.70, 122.59, 121.65, 119.53, 117.49, 110.53, 54.99, 40.40, 33.63, 29.86. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O3S: 343.1111, found: 343.1112.
- N-(3-Methoxyphenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (19). Compound purified by DCVC (hexane/ethyl acetate 7:3): white solid. Yield: 37%. 1H NMR (600 MHz, DMSO) δ 9.99 (s, 1H), 7.42 (td, J = 6.1, 5.6, 3.2 Hz, 2H), 7.36–7.30 (m, 1H), 7.27 (t, J = 2.3 Hz, 1H), 7.19 (t, J = 8.1 Hz, 1H), 7.07 (q, J = 7.8 Hz, 2H), 6.62 (dd, J = 8.3, 2.6 Hz, 1H), 4.23 (t, J = 7.6 Hz, 2H), 3.72 (s, 3H), 3.51 (s, 2H), 2.64 (t, J = 7.6 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 168.23, 164.20, 158.85, 139.56, 138.53, 128.85, 127.54, 126.79, 122.75, 122.66, 117.46, 110.86, 108.04, 104.40, 54.34, 40.29, 33.92, 29.87. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O3S: 343.1111, found: 343.1111.
- N-(4-Methoxyphenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (20). Compound purified by DCVC (hexane/ethyl acetate 7:3): white solid. Yield: 41%. 1H NMR (600 MHz, DMSO) δ 9.86 (s, 1H), 7.50–7.44 (m, 2H), 7.42 (t, J = 7.3 Hz, 2H), 7.31 (t, J = 7.8 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.93–6.82 (m, 2H), 4.22 (t, J = 7.6 Hz, 2H), 3.71 (s, 3H), 3.51 (s, 2H), 2.61 (t, J = 7.5 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 167.63, 164.17, 154.56, 138.56, 131.56, 127.54, 126.79, 122.74, 122.64, 120.14, 117.47, 113.18, 54.53, 40.45, 33.75, 29.87. HRMS (ESI) m/z [M + H]+ calculated for C18H18N2O3S: 343.1111, found: 343.1113.
- N-(2-Nitrophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (21). Compound purified by DCVC (hexane/ethyl acetate 7:3): yellow solid. Yield: 37%. 1H NMR (600 MHz, DMSO) δ 10.36 (s, 1H), 7.94 (dd, J = 8.2, 1.5 Hz, 1H), 7.70 (ddd, J = 8.7, 7.4, 1.5 Hz, 1H), 7.60 (dd, J = 8.1, 1.4 Hz, 1H), 7.42 (dd, J = 7.7, 1.5 Hz, 1H), 7.37 (td, J = 8.4, 7.9, 1.4 Hz, 2H), 7.32 (ddd, J = 8.4, 7.3, 1.5 Hz, 1H), 7.07 (td, J = 7.5, 1.2 Hz, 1H), 4.25–4.16 (m, 2H), 3.51 (s, 2H), 2.74–2.62 (m, 2H). 13C NMR (151 MHz, DMSO) δ 169.55, 165.35, 142.97, 139.52, 134.39, 131.36, 128.65, 127.88, 125.94, 125.80, 125.32, 123.88, 123.78, 118.46, 41.12, 34.53, 30.93. HRMS (ESI) m/z [M + H]+ calculated for C17H15N3O4S: 358.0856, found: 358.0856.
- N-(3-Nitrophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (22). Compound purified by DCVC (hexane/ethyl acetate 7:3): yellow solid. Yield: 41%. 1H NMR (600 MHz, DMSO) δ 10.49 (s, 1H), 8.59 (t, J = 2.1 Hz, 1H), 7.90 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H), 7.85 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H), 7.60 (t, J = 8.2 Hz, 1H), 7.42 (ddd, J = 14.3, 8.0, 1.3 Hz, 2H), 7.32 (ddd, J = 8.5, 7.3, 1.6 Hz, 1H), 7.06 (td, J = 7.5, 1.1 Hz, 1H), 4.34–4.24 (m, 2H), 3.51 (s, 2H), 2.75–2.66 (m, 2H). 13C NMR (151 MHz, DMSO) δ 170.09, 165.36, 148.39, 140.53, 139.55, 130.62, 128.63, 127.85, 125.57, 123.85, 123.82, 118.57, 118.22, 113.71, 41.17, 35.10, 30.95. HRMS (ESI) m/z [M + H]+ calculated for C17H15N3O4S: 358.0856, found: 358.0855.
- N-(4-Nitrophenyl)-3-(3-oxo-2,3-dihydro-4H-benzo[b][1,4]thiazin-4-yl)propanamide (23). Compound purified by DCVC (hexane/ethyl acetate 7:3): yellow solid. Yield: 33%. 1H NMR (600 MHz, DMSO) δ 10.60 (s, 1H), 8.26–8.19 (m, 2H), 7.83–7.76 (m, 2H), 7.42 (ddd, J = 11.3, 8.0, 1.4 Hz, 2H), 7.31 (ddd, J = 8.4, 7.3, 1.5 Hz, 1H), 7.06 (td, J = 7.5, 1.2 Hz, 1H), 4.36–4.21 (m, 2H), 3.51 (s, 2H), 2.76–2.68 (m, 2H). 13C NMR (151 MHz, DMSO) δ 170.37, 165.37, 145.61, 142.62, 139.53, 128.64, 127.86, 125.43, 123.86, 123.83, 119.24, 118.60, 41.03, 35.19, 30.95. HRMS (ESI) m/z [M + H]+ calculated for C17H15N3O4S: 358.0856, found: 358.0855.
4.2. Receptor Radioligand Binding Assays
Radioligand Binding Data Analysis
4.3. Functional Assays of cAMP Signalling at D2 Receptors
4.4. Molecular Modeling
4.5. X-ray Studies
4.6. Behavioral Studies
4.6.1. Animals
4.6.2. Drugs
4.6.3. Motor Coordination Evaluated by Chimney and Rotarod Tests
4.6.4. Spontaneous Locomotor Activity and Amphetamine-Induced Hyperactivity
4.6.5. Elevated Plus-Maze (EPM) Test
4.6.6. Forced Swim Test (FST)
4.6.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Alhosaini, K.; Azhar, A.; Alonazi, A.; Al-Zoghaibi, F. GPCRs: The Most Promiscuous Druggable Receptor of the Mankind. Saudi Pharm. J. 2021, 29, 539–551. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-Target Approach for Drug Discovery against Schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Silva, A.G.; Loza, M.I.; Kolb, P.; Castro, M.; Poso, A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. ChemMedChem 2016, 11, 718–729. [Google Scholar] [CrossRef]
- Free, R.B.; Nilson, A.N.; Boldizsar, N.M.; Doyle, T.B.; Rodriguiz, R.M.; Pogorelov, V.M.; Machino, M.; Lee, K.H.; Bertz, J.W.; Xu, J.; et al. Identification and Characterization of ML321: A Novel and Highly Selective D2 Dopamine Receptor Antagonist with Efficacy in Animal Models That Predict Atypical Antipsychotic Activity. ACS Pharmacol. Transl. Sci. 2023, 6, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Free, R.B.; Barnaeva, E.; Conroy, J.L.; Doyle, T.; Miller, B.; Bryant-Genevier, M.; Taylor, M.K.; Hu, X.; Dulcey, A.E.; et al. Discovery, Optimization, and Characterization of Novel D2 Dopamine Receptor Selective Antagonists. J. Med. Chem. 2014, 57, 3450–3463. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.; Verma, P.K.; Sharma, D.; Kumar, N.; Singh, B. Highly Efficient Water-Mediated Approach to Access Benzazoles: Metal Catalyst and Base-Free Synthesis of 2-Substituted Benzimidazoles, Benzoxazoles, and Benzothiazoles. Mol. Divers. 2015, 19, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Mithlesh, Y.; Monika, M.; Vaishali, A.K.; Devender, P. Synthesis and Antimicrobial Activity of Some 1, 4-Benzothiazine Derivatives. J. Pharm. Res. 2009, 8, 142–145. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 Dopamine Receptor Bound to the Atypical Antipsychotic Drug Risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Fan, L.; Tan, L.; Chen, Z.; Qi, J.; Nie, F.; Luo, Z.; Cheng, J.; Wang, S. Haloperidol Bound D2 Dopamine Receptor Structure Inspired the Discovery of Subtype Selective Ligands. Nat. Commun. 2020, 11, 1074. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the Dopamine D2 Receptor in Complex with the Antipsychotic Drug Spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Won Han, G.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D4 Dopamine Receptor High-Resolution Structures Enable the Discovery of Selective Agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Baud, M.G.J.; Wilcken, R.; Boeckler, F.M.; Fersht, A.R.; Joerger, A.C.; Spencer, J. Harnessing Fluorine-Sulfur Contacts and Multipolar Interactions for the Design of P53 Mutant Y220C Rescue Drugs. ACS Chem. Biol. 2016, 11, 2265–2274. [Google Scholar] [CrossRef]
- Allen, F.H.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Typical Interatomic Distances: Organic Compounds. In International Tables for Crystallography Volume C: Mathematical, Physical and Chemical Tables; Springer: Dordrecht, The Netherlands, 2006; pp. 790–811. ISBN 978-1-4020-5408-2. [Google Scholar] [CrossRef]
- Ellouz, M.; Sebbar, N.K.; Ouzidan, Y.; Essassi, E.M.; Mague, J.T. 4-Methyl-3,4-Di hydro-2H-1,4-Benzo thia zin-3-One. IUCrData 2017, 2, x170097. [Google Scholar] [CrossRef]
- Fun, H.-K.; Loh, W.-S.; Janardhana, G.; Khader, A.M.A.; Kalluraya, B. 2-(3-Oxo-3,4-Dihydro-2H-1,4-Benzo-Thia-Zin-4-Yl)Acetic Acid Monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o2358–o2359. [Google Scholar] [CrossRef] [PubMed]
- Sebbar, N.K.; Ellouz, M.; Essassi, E.M.; Saadi, M.; El Ammari, L. 4-[(3-Phenyl-4,5-Di hydro isoxazol-5-Yl)Meth yl]-2H-Benzo[b][1,4]Thia zin-3(4H)-One. IUCrData 2016, 1, x161012. [Google Scholar] [CrossRef]
- Saeed, A.; Mahmood, Z.; Yang, S.; Salim, M.; Akhtar, M.S. 2-(3-Oxo-3,4-Dihydro-2H-1,4-Benzothia-Zin-4-Yl)Acetamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o2567. [Google Scholar] [CrossRef]
- Saeed, A.; Mahmood, Z.; Yang, S.; Ahmad, S.; Salim, M. 2-(3-Oxo-3,4-Dihydro-2H-1,4-Benzothia zin-4-Yl)Acetohydrazide. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o2289–o2290. [Google Scholar] [CrossRef]
- Fun, H.-K.; Rosli, M.M.; Gowda, J.; Khader, A.M.A.; Kalluraya, B. 4-(1H-Benzimidazol-2-Ylmeth yl)-2H-1,4-Benzothia zin-3(4H)-One. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o1495. [Google Scholar] [CrossRef]
- Sebbar, N.K.; Zerzouf, A.; Essassi, E.M.; Saadi, M.; El Ammari, L. 4-(Prop-2-Yn yl)-2H-1,4-Benzo thia zin-3(4H)-One. Acta Crystallogr. Sect. E Struct. Rep. Online 2014, 70, o641. [Google Scholar] [CrossRef] [PubMed]
- Sebbar, N.K.; El Fal, M.; Essassi, E.M.; Saadi, M.; El Ammari, L. 4-[(1-Benzyl-1,2,3-Triazol-5-Yl)Meth yl]-2H-1,4-Benzo thia zin-3(4H)-One. Acta Crystallogr. Sect. E Struct. Rep. Online 2014, 70, o363–o364. [Google Scholar] [CrossRef]
- Hursthouse, M.B.; Huth, L.S.; Threlfall, T.L. Why Do Organic Compounds Crystallise Well or Badly or Ever so Slowly? Why Is Crystallisation Nevertheless Such a Good Purification Technique? Org. Process Res. Dev. 2009, 13, 1231–1240. [Google Scholar] [CrossRef]
- Gowda, B.T.; Foro, S.; Saraswathi, B.S.; Terao, H.; Fuess, H. Methyl N-(4-Chlorophenyl)Succinamate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o388. [Google Scholar] [CrossRef]
- Saraswathi, B.S.; Foro, S.; Gowda, B.T. N-(4-Chloro phen yl)-N′-(3-Methyl phen yl)Succinamide Monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o2418. [Google Scholar] [CrossRef] [PubMed]
- Samai, S.; Biradha, K. Halogen⋯halogen Interactions in Assembling β-Sheets into 2D Layers in the Bis-(4-Halo-Phenylamido)Alkanes and Their Co-Crystals via Inter-Halogen Interactions. CrystEngComm 2009, 11, 482–492. [Google Scholar] [CrossRef]
- Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K.D.; Waldvogel, S.R. Insights into the Mechanism of Anodic N–N Bond Formation by Dehydrogenative Coupling. J. Am. Chem. Soc. 2017, 139, 12317–12324. [Google Scholar] [CrossRef]
- Demir, S.; Sarioğlu, A.O.; Güler, S.; Dege, N.; Sönmez, M. Synthesis, Crystal Structure Analysis, Spectral IR, NMR UV–Vis Investigations, NBO and NLO of 2-Benzoyl-N-(4-Chlorophenyl)-3-Oxo-3-Phenylpropanamide with Use of X-Ray Diffractions Studies along with DFT Calculations. J. Mol. Struct. 2016, 1118, 316–324. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Boeyens, J.C.A. The Conformation of Six-Membered Rings. J. Cryst. Mol. Struct. 1978, 8, 317–320. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G. The Conformation of Six-Membered Rings Described by Puckering Coordinates Derived from Endocyclic Torsion Angles. J. Am. Chem. Soc. 1992, 114, 882–887. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Vogel, H.G. Psychotropic and Neurotropic Activity. In Drug Discovery and Evaluation: Pharmacological Assays; Vogel, H.G., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 565–876. ISBN 978-3-540-70995-4. [Google Scholar]
- Gobira, P.H.; Ropke, J.; Aguiar, D.C.; Crippa, J.A.S.; Moreira, F.A. Animal Models for Predicting the Efficacy and Side Effects of Antipsychotic Drugs. Braz. J. Psychiatry 2013, 35, S132–S139. [Google Scholar] [CrossRef]
- Jones, C.; Watson, D.; Fone, K. Animal Models of Schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef]
- Brady, A.E.; Jones, C.K.; Bridges, T.M.; Kennedy, J.P.; Thompson, A.D.; Heiman, J.U.; Breininger, M.L.; Gentry, P.R.; Yin, H.; Jadhav, S.B.; et al. Centrally Active Allosteric Potentiators of the M4 Muscarinic Acetylcholine Receptor Reverse Amphetamine-Induced Hyperlocomotor Activity in Rats. J. Pharmacol. Exp. Ther. 2008, 327, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, P.; Woźniak, M.; Rook, J.M.; Tantawy, M.N.; Conn, P.J.; Acher, F.; Tokarski, K.; Kusek, M.; Pilc, A.; Wierońska, J.M. Mutual Activation of Glutamatergic MGlu4 and Muscarinic M4 Receptors Reverses Schizophrenia-Related Changes in Rodents. Psychopharmacology 2018, 235, 2897–2913. [Google Scholar] [CrossRef]
- Partyka, A.; Jarosz, J.; Wasik, A.; Jastrzębska-Więsek, M.; Zagórska, A.; Pawłowski, M.; Wesołowska, A. Novel Tricyclic [2,1-f]Theophylline Derivatives of LCAP with Activity in Mouse Models of Affective Disorders. J. Pharm. Pharmacol. 2014, 66, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Targowska-Duda, K.M.; Stępnicki, P.; Silva, A.G.; Koszła, O.; Kędzierska, E.; Grudzińska, A.; Kruk-Słomka, M.; Biała, G.; Castro, M. N-(3-{4-[3-(Trifluoromethyl)Phenyl]Piperazin-1-Yl}propyl)-1H-Indazole-3-Carboxamide (D2AAK3) as a Potential Antipsychotic: In Vitro, in Silico and in Vivo Evaluation of a Multi-Target Ligand. Neurochem. Int. 2021, 146, 105016. [Google Scholar] [CrossRef]
- Selent, J.; Marti-Solano, M.; Rodríguez, J.; Atanes, P.; Brea, J.; Castro, M.; Sanz, F.; Loza, M.I.; Pastor, M. Novel Insights on the Structural Determinants of Clozapine and Olanzapine Multi-Target Binding Profiles. Eur. J. Med. Chem. 2014, 77, 91–95. [Google Scholar] [CrossRef]
- Berg, K.A.; Maayani, S.; Goldfarb, J.; Scaramellini, C.; Leff, P.; Clarke, W.P. Effector Pathway-Dependent Relative Efficacy at Serotonin Type 2A and 2C Receptors: Evidence for Agonist-Directed Trafficking of Receptor Stimulus. Mol. Pharmacol. 1998, 54, 94–104. [Google Scholar] [CrossRef]
- Leff, P.; Dougall, I.G. Further Concerns over Cheng-Prusoff Analysis. Trends Pharmacol. Sci. 1993, 14, 110–112. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2021–4; Epik, Schrödinger, LLC: New York, NY, USA, 2021.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Stępnicki, P.; Targowska-Duda, K.M.; Martínez, A.L.; Zięba, A.; Wronikowska-Denysiuk, O.; Wróbel, M.Z.; Bartyzel, A.; Trzpil, A.; Wróbel, T.M.; Chodkowski, A.; et al. Discovery of Novel Arylpiperazine-Based DA/5-HT Modulators as Potential Antipsychotic Agents—Design, Synthesis, Structural Studies and Pharmacological Profiling. Eur. J. Med. Chem. 2023, 252, 115285. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- CrysAlis PRO Version 1.171.37.35g; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2014.
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Lister, R.G. The Use of a Plus-Maze to Measure Anxiety in the Mouse. Psychopharmacology 1987, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A New Animal Model Sensitive to Antidepressant Treatments. Nature 1977, 266, 730–732. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| R1 | R2 | n | huD2 | ||
| % inh. at 10 µM | Ki [nM] | ||||
| 1 |  | 4-CF3 | 2 | 35 ± 14 | |
| 2 |  | 4-CF3 | 2 | 11 ± 5 | |
| 3 |  | 4-CF3 | 2 | 8 ± 8 | |
| 4 |  | 4-CF3 | 2 | 3846 ± 35 | |
| 5 |  | 4-CF3 | 2 | 14 ± 10 | |
| 6 |  | 4-CF3 | 2 | 14 ± 11 | |
| 7 |  | 4-CF3 | 2 | 13 ± 13 | |
| 8 |  | 4-CF3 | 2 | 8 ± 7 | |
| 9 |  | 4-CF3 | 1 | 8 ± 8 | |
| 10 | – | 2 | 39 ± 4 | ||
| 11 | 2-CF3 | 2 | 9 ± 1 | ||
| 12 | 2-CH3 | 2 | 10 ± 0 | ||
| 13 | 3-CH3 | 2 | 32 ± 4 | ||
| 14 | 4-CH3 | 2 | 676 ± 134 | ||
| 15 | 2-Cl | 2 | 9 ± 3 | ||
| 16 | 3-Cl | 2 | 42 ± 8 | ||
| 17 | 4-Cl | 2 | 381 ± 48 | ||
| 18 | 2-OCH3 | 2 | 6 ± 0 | ||
| 19 | 3-OCH3 | 2 | 10 ± 4 | ||
| 20 | 4-OCH3 | 2 | 47 ± 1 | ||
| 21 | 2-NO2 | 2 | 5 ± 3 | ||
| 22 | 3-NO2 | 2 | 27 ± 2 | ||
| 23 | 4-NO2 | 2 | 724 ± 114 | ||
| D2AAK2 | 4-CF3 | 2 | 321 ± 25 [10] | ||
| Haloperidol | 6.56 ± 0.88 | ||||
| huD1 | huD3 | hu5-HT1A | hu5-HT2A | hu5-HT7 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % inh. at 10 µM | Ki (nM) | % inh. at 10 µM | Ki (nM) | % inh. at 10 µM | Ki (nM) | % inh. at 10 µM | Ki (nM) | % inh. at 10 µM | Ki (nM) | |
| 14 | 15 ± 2 | 8 ± 4 | 27 ± 2 | 15 ± 1 | 0 | |||||
| 17 | 21 ± 2 | 6 ± 3 | 29 ± 9 | 28 ± 1 | 5 ± 11 | |||||
| 23 | 12 ± 2 | 4 ± 3 | 22 ±3 | 23 ± 3 | 0 | |||||
| Haloperidol | 6.31 ± 0.79 | 4.95 ± 1.13 | ||||||||
| 5-CT | 0.12 ± 0.02 | |||||||||
| Risperidone | 0.42 ± 0.03 | |||||||||
| Methiothepin | 2.78 ± 1.37 | |||||||||
| D2 Signalling (cAMP) | ||
|---|---|---|
| pKb (Mean ± SEM) | Kb (nM) (Mean) | |
| 14 | 6.93 ± 0.11 | 119 |
| 17 | 7.57 ± 0.19 | 27 |
| 23 | 6.95 ± 0.19 | 113 |
| Haloperidol | 9.97 ± 0.22 | 0.11 |
| Hydrogen Bond [Å, º] | ||||
|---|---|---|---|---|
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠ DHA |
| N2–H2N···O1 i | 0.82(3) | 2.18(3) | 2.962(4) | 159(4) |
| C13–H13···O2 | 0.93 | 2.24 | 2.834(5) | 121 |
| Treatment | Distance Traveled (cm) after 10 min | Distance Traveled (cm) after 30 min |
|---|---|---|
| vehicle (control group) | 1002 ± 104.0 | 2529 ± 460.6 |
| 17 (25 mg/kg) | 910.1 ± 139.3 | 2003 ± 355.7 |
| 17 (50 mg/kg) | 855.9 ± 195.1 | 2345 ± 616.0 |
| 17 (75 mg/kg) | 1200 ± 216.0 | 2983 ± 595.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stępnicki, P.; Wośko, S.; Bartyzel, A.; Zięba, A.; Bartuzi, D.; Szałaj, K.; Wróbel, T.M.; Fornal, E.; Carlsson, J.; Kędzierska, E.; et al. Development and Characterization of Novel Selective, Non-Basic Dopamine D2 Receptor Antagonists for the Treatment of Schizophrenia. Molecules 2023, 28, 4211. https://doi.org/10.3390/molecules28104211
Stępnicki P, Wośko S, Bartyzel A, Zięba A, Bartuzi D, Szałaj K, Wróbel TM, Fornal E, Carlsson J, Kędzierska E, et al. Development and Characterization of Novel Selective, Non-Basic Dopamine D2 Receptor Antagonists for the Treatment of Schizophrenia. Molecules. 2023; 28(10):4211. https://doi.org/10.3390/molecules28104211
Chicago/Turabian StyleStępnicki, Piotr, Sylwia Wośko, Agata Bartyzel, Agata Zięba, Damian Bartuzi, Klaudia Szałaj, Tomasz M. Wróbel, Emilia Fornal, Jens Carlsson, Ewa Kędzierska, and et al. 2023. "Development and Characterization of Novel Selective, Non-Basic Dopamine D2 Receptor Antagonists for the Treatment of Schizophrenia" Molecules 28, no. 10: 4211. https://doi.org/10.3390/molecules28104211
APA StyleStępnicki, P., Wośko, S., Bartyzel, A., Zięba, A., Bartuzi, D., Szałaj, K., Wróbel, T. M., Fornal, E., Carlsson, J., Kędzierska, E., Poleszak, E., Castro, M., & Kaczor, A. A. (2023). Development and Characterization of Novel Selective, Non-Basic Dopamine D2 Receptor Antagonists for the Treatment of Schizophrenia. Molecules, 28(10), 4211. https://doi.org/10.3390/molecules28104211