
Exploring the Role of Anionic Lipid Nanodomains in the Membrane Disruption and Protein Folding of Human Islet Amyloid Polypeptide Oligomers on Lipid Membrane Surfaces Using Multiscale Molecular Dynamics Simulations


Abstract
1. Introduction
2. Results
2.1. Initial Structures of [hIAPP]n and Raft Membranes in Solution
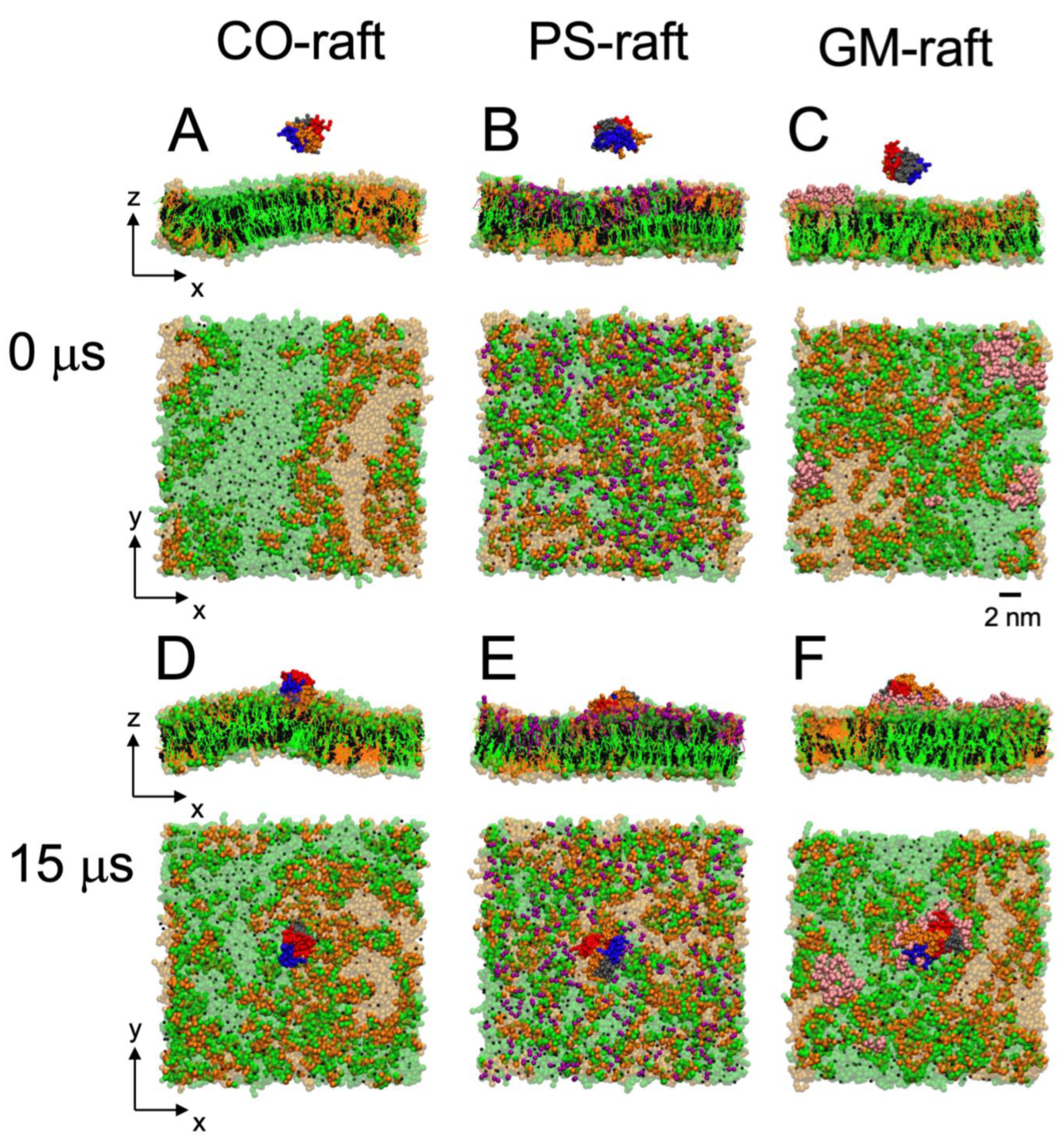
2.2. Kinetics and Lipid Domain Preference in [hIAPP]n Binding to Raft Membranes
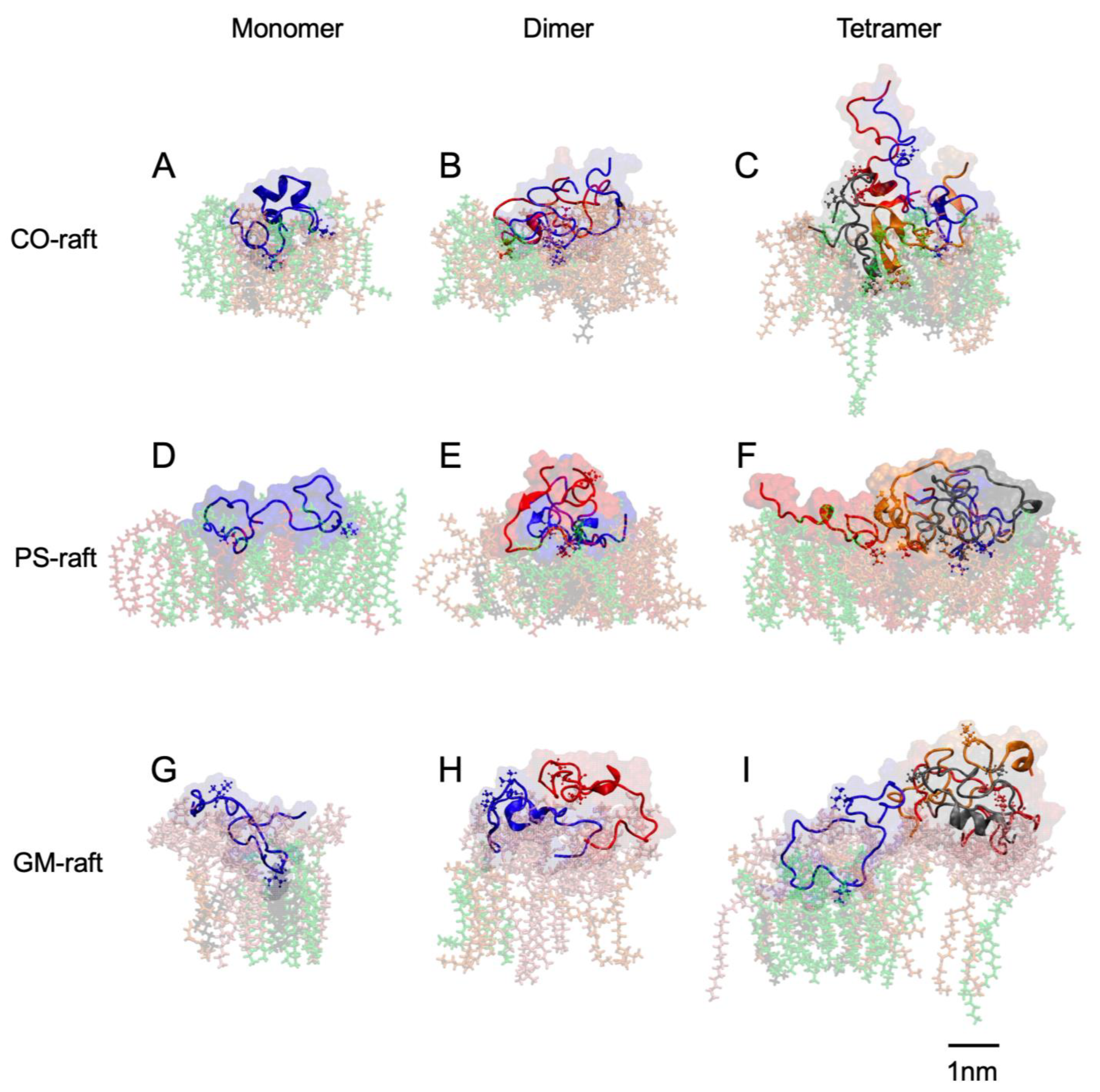
2.3. Residue-Specific Protein–Lipid Binding Pattern of [hIAPP]n on Raft Membrane Surfaces
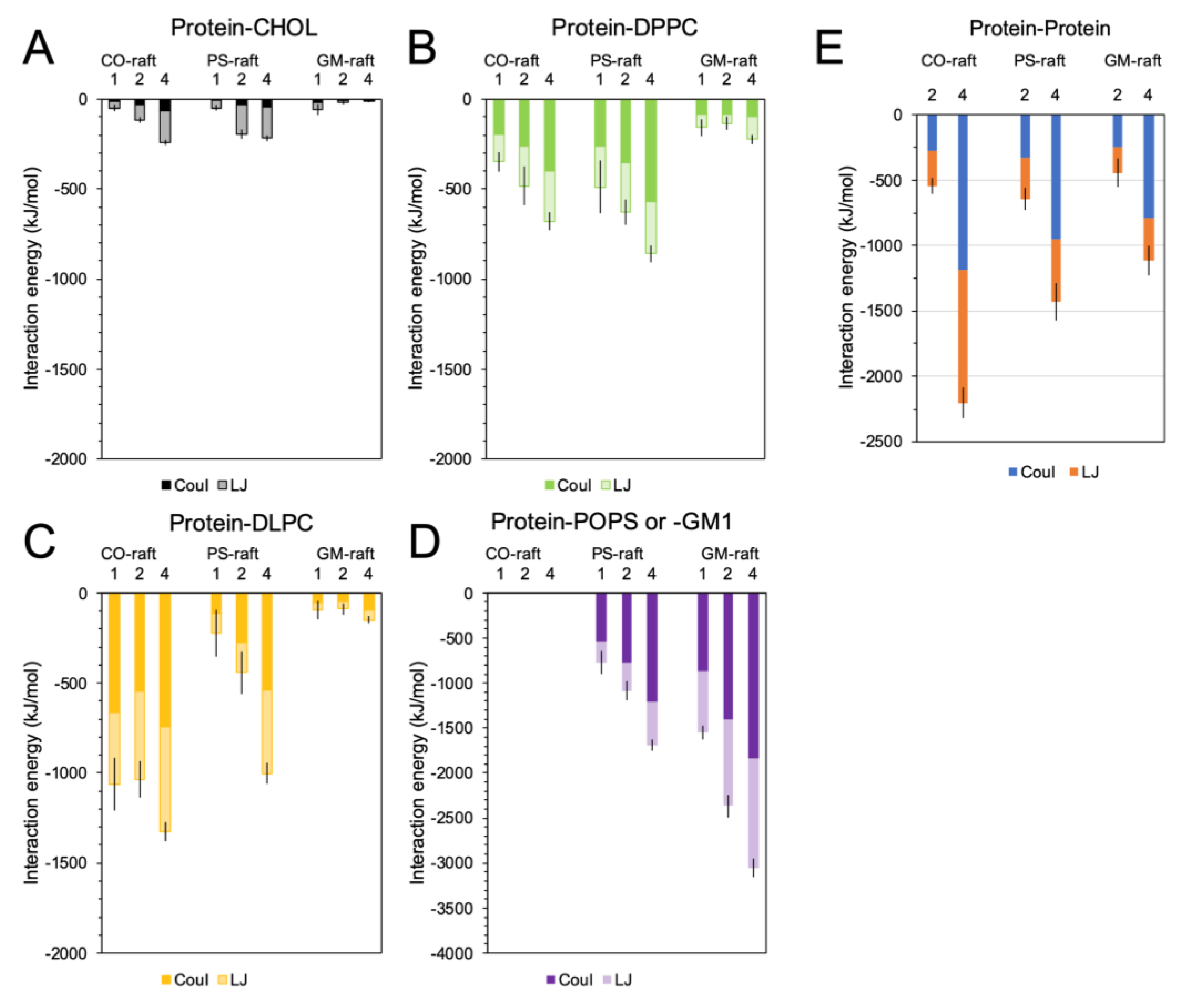
2.4. Binding Energies of Membrane-Bound [hIAPP]n
2.5. Membrane Disruption Behavior of [hIAPP]n
2.6. Surface-Induced Protein Folding of [hIAPP]n
3. Discussion
4. Materials and Methods
4.1. Raft Membranes and hIAPP Oligomers
4.2. Multiscale Simulations of Oligomer Binding to Raft Membranes
4.3. Classifications of Lipid Nanodomains and Annular Lipids
4.4. Membrane Binding Behaviors of Oligomers
4.5. Characterization of Membrane Disruption by Oligomer Binding to Raft Membrane
4.6. Secondary Structures of Membrane-Bound Oligomers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| hIAPP | human Islet Amyloid Polypeptide |
| CG | coarse-grained |
| PC | phosphatidylcholine |
| PS | phosphatidylserine |
| GM | ganglioside |
| CHOL | cholesterol |
| DPPC | dipalmitoyl-PC |
| DLPC | dilinoleoyl-PC |
| Lo | liquid-ordered |
| Ld | liquid-disordered |
| Lod | mixed Lo/Ld |
| MD | molecular dynamics |
| AA | all-atom |
| AL | annular lipid |
| nAL | non-annular lipid |
| mindist | minimum distance |
| APL | area per lipid |
| POPS | 1-palmitoyl-2-oleoyl-PS |
References
- Sciacca, M.F.M.; La Rosa, C.; Milardi, D. Amyloid-Mediated Mechanisms of Membrane Disruption. Biophysica 2021, 1, 137–156. [Google Scholar] [CrossRef]
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res. 2012, 45, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. How do membranes initiate Alzheimer’s Disease? Formation of toxic amyloid fibrils by the amyloid beta-protein on ganglioside clusters. Acc. Chem. Res. 2014, 47, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.K.; Ramamoorthy, A. Two-step mechanism of membrane disruption by Abeta through membrane fragmentation and pore formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef]
- Zhang, X.; St Clair, J.R.; London, E.; Raleigh, D.P. Islet Amyloid Polypeptide Membrane Interactions: Effects of Membrane Composition. Biochemistry 2017, 56, 376–390. [Google Scholar] [CrossRef]
- Sezgin, E.; Gutmann, T.; Buhl, T.; Dirkx, R.; Grzybek, M.; Coskun, U.; Solimena, M.; Simons, K.; Levental, I.; Schwille, P. Adaptive lipid packing and bioactivity in membrane domains. PLoS ONE 2015, 10, e0123930. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Cebecauer, M.; Amaro, M.; Jurkiewicz, P.; Sarmento, M.J.; Sachl, R.; Cwiklik, L.; Hof, M. Membrane Lipid Nanodomains. Chem. Rev. 2018, 118, 11259–11297. [Google Scholar] [CrossRef]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Rog, T.; Vattulainen, I. Multiscale Simulations of Biological Membranes: The Challenge to Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef]
- Jacobson, K.; Liu, P.; Lagerholm, B.C. The Lateral Organization and Mobility of Plasma Membrane Components. Cell 2019, 177, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid rafts and neurodegeneration: Structural and functional roles in physiologic aging and neurodegenerative diseases. J. Lipid Res. 2020, 61, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid rafts as a therapeutic target. J. Lipid Res. 2020, 61, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 2020, 27, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Pham, T.; Nguyen, N.; Graf, A.; Cheng, K.H. Lipid domain boundary triggers membrane damage and protein folding of human islet amyloid polypeptide in the early pathogenesis of amyloid diseases. Biophys. Chem. 2023, 296, 106993. [Google Scholar] [CrossRef]
- Cheng, K.H.; Graf, A.; Lewis, A.; Pham, T.; Acharya, A. Exploring Membrane Binding Targets of Disordered Human Tau Aggregates on Lipid Rafts Using Multiscale Molecular Dynamics Simulations. Membranes 2022, 12, 1098. [Google Scholar] [CrossRef]
- Pham, T.; Cheng, K.H. Exploring the binding kinetics and behaviors of self-aggregated beta-amyloid oligomers to phase-separated lipid rafts with or without ganglioside-clusters. Biophys. Chem. 2022, 290, 106874. [Google Scholar] [CrossRef]
- Press-Sandler, O.; Miller, Y. Molecular mechanisms of membrane-associated amyloid aggregation: Computational perspective and challenges. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1889–1905. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Cornish, J.; Chamberlain, S.G.; Owen, D.; Mott, H.R. Intrinsically disordered proteins and membranes: A marriage of convenience for cell signalling? Biochem. Soc. Trans. 2020, 48, 2669–2689. [Google Scholar] [CrossRef]
- Rudajev, V.; Novotny, J. The Role of Lipid Environment in Ganglioside GM1-Induced Amyloid beta Aggregation. Membranes 2020, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Reid, K.R.; Kennedy, R.T.; Ramamoorthy, A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry 2008, 47, 12680–12688. [Google Scholar] [CrossRef] [PubMed]
- Fatafta, H.; Kav, B.; Bundschuh, B.F.; Loschwitz, J.; Strodel, B. Disorder-to-order transition of the amyloid-beta peptide upon lipid binding. Biophys. Chem. 2022, 280, 106700. [Google Scholar] [CrossRef]
- Khemtemourian, L.; Fatafta, H.; Davion, B.; Lecomte, S.; Castano, S.; Strodel, B. Structural Dissection of the First Events Following Membrane Binding of the Islet Amyloid Polypeptide. Front. Mol. Biosci. 2022, 9, 849979. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Lolicato, F.; Di Mauro, G.; Milardi, D.; D’Urso, L.; Satriano, C.; Ramamoorthy, A.; La Rosa, C. The Role of Cholesterol in Driving IAPP-Membrane Interactions. Biophys. J. 2016, 111, 140–151. [Google Scholar] [CrossRef]
- Calamai, M.; Pavone, F.S. Partitioning and confinement of GM1 ganglioside induced by amyloid aggregates. FEBS Lett. 2013, 587, 1385–1391. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Matsuzaki, K. Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett. 2009, 583, 2854–2858. [Google Scholar] [CrossRef]
- Risselada, H.J.; Marrink, S.J. The molecular face of lipid rafts in model membranes. Proc. Natl. Acad. Sci. USA 2008, 105, 17367–17372. [Google Scholar] [CrossRef]
- Ingolfsson, H.I.; Bhatia, H.; Zeppelin, T.; Bennett, W.F.D.; Carpenter, K.A.; Hsu, P.C.; Dharuman, G.; Bremer, P.T.; Schiott, B.; Lightstone, F.C.; et al. Capturing Biologically Complex Tissue-Specific Membranes at Different Levels of Compositional Complexity. J. Phys. Chem. B 2020, 124, 7819–7829. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Pluhackova, K.; Bockmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, G.M.; Greig, N.H.; Khan, T.A.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.A.; Zaidi, S.K.; Akram, M.; Jabir, N.R.; et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Christensen, M.; Schiott, B. Revealing a Dual Role of Ganglioside Lipids in the Aggregation of Membrane-Associated Islet Amyloid Polypeptide. J. Membr. Biol. 2019, 252, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 2008, 130, 6424–6429. [Google Scholar] [CrossRef] [PubMed]
- Dicke, S.S.; Maj, M.; Fields, C.R.; Zanni, M.T. Metastable intermediate during hIAPP aggregation catalyzed by membranes as detected with 2D IR spectroscopy. RSC Chem. Biol. 2022, 3, 931–940. [Google Scholar] [CrossRef]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef]
- Sepehri, A.; Nepal, B.; Lazaridis, T. Distinct Modes of Action of IAPP Oligomers on Membranes. J. Chem. Inf. Model. 2021, 61, 4645–4655. [Google Scholar] [CrossRef]
- Pilkington, E.H.; Gurzov, E.N.; Kakinen, A.; Litwak, S.A.; Stanley, W.J.; Davis, T.P.; Ke, P.C. Pancreatic beta-Cell Membrane Fluidity and Toxicity Induced by Human Islet Amyloid Polypeptide Species. Sci. Rep. 2016, 6, 21274. [Google Scholar] [CrossRef]
- Zhu, H.; Tao, Q.; Ang, T.F.A.; Massaro, J.; Gan, Q.; Salim, S.; Zhu, R.Y.; Kolachalama, V.B.; Zhang, X.; Devine, S.; et al. Association of Plasma Amylin Concentration with Alzheimer Disease and Brain Structure in Older Adults. JAMA Netw. Open 2019, 2, e199826. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.; Arnarez, C.; Wassenaar, T.A.; Schafer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, D.; Grater, F.; Daday, C. CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps. Biophys. J. 2018, 114, 1267–1273. [Google Scholar] [CrossRef]
- Han, B.; Tashjian, A.H., Jr. User-friendly and versatile software for analysis of protein hydrophobicity. Biotechniques 1998, 25, 256–263. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Blumer, M.; Harris, S.; Li, M.; Martinez, L.; Untereiner, M.; Saeta, P.N.; Carpenter, T.S.; Ingolfsson, H.I.; Bennett, W.F.D. Simulations of Asymmetric Membranes Illustrate Cooperative Leaflet Coupling and Lipid Adaptability. Front. Cell Dev. Biol. 2020, 8, 575. [Google Scholar] [CrossRef]
- Grote, F.; Lyubartsev, A.P. Optimization of Slipids Force Field Parameters Describing Headgroups of Phospholipids. J. Phys. Chem. B 2020, 124, 8784–8793. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Buchoux, S. FATSLiM: A fast and robust software to analyze MD simulations of membranes. Bioinformatics 2017, 33, 133–134. [Google Scholar] [CrossRef]
- Cheng, K.H.; Qiu, L.; Cheng, S.Y.; Vaughn, M.W. Lipid insertion domain unfolding regulates protein orientational transition behavior in a lipid bilayer. Biophys. Chem. 2015, 206, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulation | n | Raft | Binding Time (μs) | CHOL% * | DPPC% * | DLPC% * | POPS% * or GM1% * | Number of Lipids * | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CG | 1 | CO | 3.84 | 8.41 | 0.26 | 17 ± 1 | 34 ± 2 | 49 ± 2 | 9.0 ± 0.2 | |
| PS | 1.19 | 0.91 | 0.29 | 20 ± 1 | 25 ± 2 | 27 ± 3 | 29 ± 3 | 10.3 ± 0.4 | ||
| GM | 0.94 | 0.60 | 0.09 | 8 ± 4 | 8 ± 3 | 4 ± 1 | 80 ± 1 | 15.6 ± 0.8 | ||
| 2 | CO | 7.85 | 0.61 | 4.03 | 19 ± 1 | 36 ± 3 | 45 ± 1 | 11.9 ± 0.6 | ||
| PS | 2.99 | 0.19 | 0.70 | 22 ± 1 | 25 ± 2 | 23 ±1 | 30 ± 1 | 14.2 ± 0.3 | ||
| GM | 0.40 | 0.16 | 1.50 | 4 ± 3 | 7 ± 3 | 6 ± 3 | 83 ± 16 | 16.7 ± 2.8 | ||
| 4 | CO | 8.34 | 10.55 | 1.02 | 20 ± 2 | 34 ± 2 | 47 ± 3 | 16.3 ± 0.7 | ||
| PS | 1.77 | 3.75 | 3.10 | 22 ± 1 | 22 ± 1 | 25 ± 1 | 31 ± 1 | 20.0 ± 0.7 | ||
| GM | 0.44 | 0.96 | 1.20 | 4 ± 3 | 9 ± 3 | 6 ± 3 | 81± 8 | 24.2 ± 2.2 | ||
| AA | 1 | CO | 15 ± 4 | 27 ± 5 | 58 ± 9 | 18.2 ± 2.0 | ||||
| PS | 14 ± 2 | 36 ± 10 | 19 ± 12 | 30 ± 9 | 19.2 ± 3.3 | |||||
| GM | 17 ± 7 | 31 ± 10 | 12 ± 3 | 40 ± 7 | 15.7 ± 2.3 | |||||
| 2 | CO | 20 ± 10 | 32 ± 11 | 48 ± 14 | 23.6 ± 4.9 | |||||
| PS | 23 ± 5 | 28 ± 5 | 20 ± 9 | 29 ± 7 | 29.0 ± 6.5 | |||||
| GM | 4 ± 4 | 24 ± 7 | 26 ± 1 | 46 ± 5 | 15.0 ± 1.4 | |||||
| 4 | CO | 26 ± 3 | 29 ± 3 | 45 ± 5 | 31.4 ± 2.2 | |||||
| PS | 25 ± 5 | 24 ± 3 | 22 ± 7 | 30 ± 1 | 29.7 ± 1.0 | |||||
| GM | 11 ± 6 | 34 ± 3 | 18 ± 4 | 37 ± 1 | 22.6 ± 1.7 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.; Lewis, A.; Pham, T.; Sikazwe, D.; Cheng, K.H. Exploring the Role of Anionic Lipid Nanodomains in the Membrane Disruption and Protein Folding of Human Islet Amyloid Polypeptide Oligomers on Lipid Membrane Surfaces Using Multiscale Molecular Dynamics Simulations. Molecules 2023, 28, 4191. https://doi.org/10.3390/molecules28104191
Nguyen N, Lewis A, Pham T, Sikazwe D, Cheng KH. Exploring the Role of Anionic Lipid Nanodomains in the Membrane Disruption and Protein Folding of Human Islet Amyloid Polypeptide Oligomers on Lipid Membrane Surfaces Using Multiscale Molecular Dynamics Simulations. Molecules. 2023; 28(10):4191. https://doi.org/10.3390/molecules28104191
Chicago/Turabian StyleNguyen, Ngoc, Amber Lewis, Thuong Pham, Donald Sikazwe, and Kwan H. Cheng. 2023. "Exploring the Role of Anionic Lipid Nanodomains in the Membrane Disruption and Protein Folding of Human Islet Amyloid Polypeptide Oligomers on Lipid Membrane Surfaces Using Multiscale Molecular Dynamics Simulations" Molecules 28, no. 10: 4191. https://doi.org/10.3390/molecules28104191
APA StyleNguyen, N., Lewis, A., Pham, T., Sikazwe, D., & Cheng, K. H. (2023). Exploring the Role of Anionic Lipid Nanodomains in the Membrane Disruption and Protein Folding of Human Islet Amyloid Polypeptide Oligomers on Lipid Membrane Surfaces Using Multiscale Molecular Dynamics Simulations. Molecules, 28(10), 4191. https://doi.org/10.3390/molecules28104191