
Machine Learning Generation of Dynamic Protein Conformational Ensembles


{kind=link}
{kind=link}
Abstract
1. Introduction
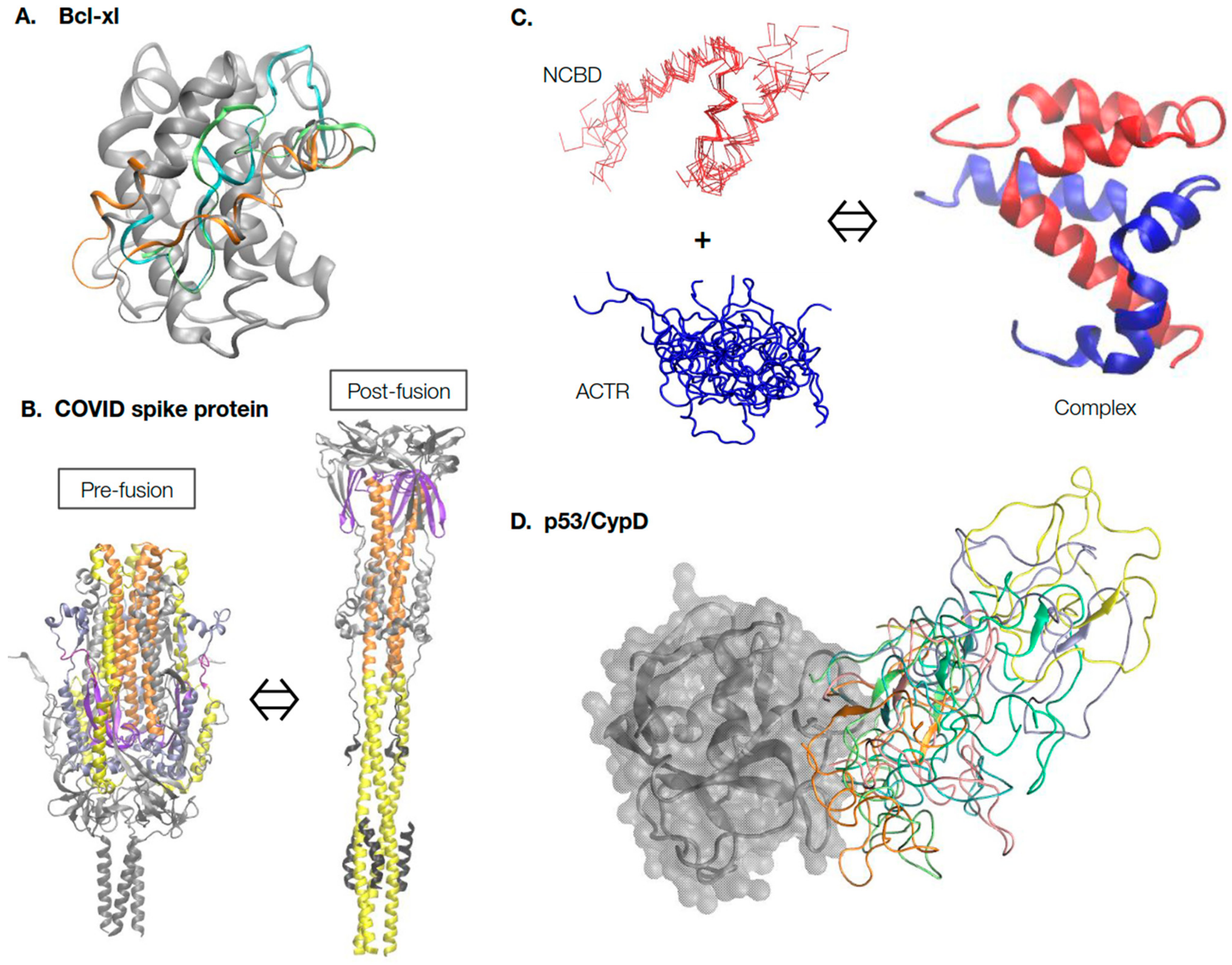
2. A Rich Continuum of Protein Structures and Dynamics for Function

3. Generative Deep Learning for Biomolecular Modeling
4. ML Approaches to Identify CVs and Drive Enhanced Sampling in MD Simulations
5. Directly Sampling Conformational Space using ML-Derived Latent Representation
6. One-Shot Generation of Dynamic Protein Conformational Ensembles
7. Conclusions and Future Directions
- Deeper and more systematic understanding of if and how the complexity of the high-dimensional conformational space of proteins can be mapped to a smooth and continuous space of sufficiently low dimensionality. This may be investigated using ultra-long trajectories of a large set of folded and unfolded proteins, such as those from Shaw and co-workers, using the latest balanced atomistic protein force field [49], as well as those generated using extensive enhanced sampling simulations [124,125,135]. A challenge here is to ensure the quality and convergence of the structural data, particularly for IDPs.
- Consistent and rigorous evaluation of the performance of generative ML models that is widely accepted and adopted by the community. At present, most developments are benchmarked using different custom examples and special test-cases. There is only minimal cross-comparison needed to rigorously establish the strengths and weaknesses of various approaches, such as over-fitting, computational cost, and interpretability. Again, ultra-long MD trajectories or highly-converged conformational ensembles generated from enhanced sampling simulations of proteins of different sizes, topologies and dynamic properties could be used as a standard benchmark set. Similar practices have been instrumental in the development of methods for predicting protein structure [8], protein-protein interaction [136], and protein-ligand interaction [137].
- New ML approaches for integrating information from diverse datasets, such as protein structures, sequence alignment, and MD trajectories, and incorporating physical principles of molecular interactions, such as various empirical protein energy functions. ML has been under accelerated development in recent years, with many exciting ideas emerging, such as large language models (LLMs) [12], deep transfer learning [138], and diffusion models [139]. Furthermore, the computational infrastructure now allows much larger and increasingly complex models to be trained with extremely large datasets [140].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bushweller, J.H. Targeting transcription factors in cancer-from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins 2021, 89, 1607–1617. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst. 2021, 12, 654–669.e3. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021, 27, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Borkakoti, N.; Thornton, J.M. AlphaFold2 protein structure prediction: Implications for drug discovery. Curr. Opin. Struct. Biol. 2023, 78, 102526. [Google Scholar] [CrossRef]
- Pearce, R.; Zhang, Y. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021, 297, 100870. [Google Scholar] [CrossRef]
- Lane, T.J. Protein structure prediction has reached the single-structure frontier. Nat. Methods 2023, 20, 170–173. [Google Scholar] [CrossRef]
- Moore, P.B.; Hendrickson, W.A.; Henderson, R.; Brunger, A.T. The protein-folding problem: Not yet solved. Science 2022, 375, 507. [Google Scholar] [CrossRef]
- Ourmazd, A.; Moffat, K.; Lattman, E.E. Structural biology is solved—Now what? Nat. Methods 2022, 19, 24–26. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.D.; Phillips, G.N., Jr. Moving beyond static snapshots: Protein dynamics and the Protein Data Bank. J. Biol. Chem. 2021, 296, 100749. [Google Scholar] [CrossRef]
- Sugase, K.; Konuma, T.; Lansing, J.C.; Wright, P.E. Fast and accurate fitting of relaxation dispersion data using the flexible software package GLOVE. J. Biomol. NMR 2013, 56, 275–283. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; Depristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef]
- Lane, T.J.; Shukla, D.; Beauchamp, K.A.; Pande, V.S. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013, 23, 58–65. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, Y.; Chen, J. Advanced Sampling Methods for Multiscale Simulation of Disordered Proteins and Dynamic Interactions. Biomolecules 2021, 11, 1416. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; Izadi, S.; et al. AMBER 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2012, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kal, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Zhang, W.H.; Chen, J.H. Accelerate Sampling in Atomistic Energy Landscapes Using Topology-Based Coarse-Grained Models. J. Chem. Theory Comput. 2014, 10, 918–923. [Google Scholar] [CrossRef]
- Moritsugu, K.; Terada, T.; Kidera, A. Scalable free energy calculation of proteins via multiscale essential sampling. J. Chem. Phys. 2010, 133, 224105. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef]
- Mittal, A.; Lyle, N.; Harmon, T.S.; Pappu, R.V. Hamiltonian Switch Metropolis Monte Carlo Simulations for Improved Conformational Sampling of Intrinsically Disordered Regions Tethered to Ordered Domains of Proteins. J. Chem. Theory Comput. 2014, 10, 3550–3562. [Google Scholar] [CrossRef]
- Peter, E.K.; Shea, J.E. A hybrid MD-kMC algorithm for folding proteins in explicit solvent. Phys. Chem. Chem. Phys. 2014, 16, 6430–6440. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, J. Enhanced sampling and applications in protein folding in explicit solvent. J. Chem. Phys. 2010, 132, 244101. [Google Scholar] [CrossRef]
- Zheng, L.Q.; Yang, W. Practically Efficient and Robust Free Energy Calculations: Double-Integration Orthogonal Space Tempering. J. Chem. Theory Comput. 2012, 8, 810–823. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the SC’14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Gkeka, P.; Stoltz, G.; Barati Farimani, A.; Belkacemi, Z.; Ceriotti, M.; Chodera, J.D.; Dinner, A.R.; Ferguson, A.L.; Maillet, J.B.; Minoux, H.; et al. Machine Learning Force Fields and Coarse-Grained Variables in Molecular Dynamics: Application to Materials and Biological Systems. J. Chem. Theory Comput. 2020, 16, 4757–4775. [Google Scholar] [CrossRef]
- Chen, M. Collective variable-based enhanced sampling and machine learning. Eur. Phys. J. B 2021, 94, 211. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, P.; Zhao, L.; Shehu, A. Generative deep learning for macromolecular structure and dynamics. Curr. Opin. Struct. Biol. 2021, 67, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Kragelund, B.B. On the Potential of Machine Learning to Examine the Relationship Between Sequence, Structure, Dynamics and Function of Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167196. [Google Scholar] [CrossRef]
- Ramanathan, A.; Ma, H.; Parvatikar, A.; Chennubhotla, S.C. Artificial intelligence techniques for integrative structural biology of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2021, 66, 216–224. [Google Scholar] [CrossRef]
- Wang, Y.; Lamim Ribeiro, J.M.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145. [Google Scholar] [CrossRef]
- Allison, J.R. Computational methods for exploring protein conformations. Biochem. Soc. Trans. 2020, 48, 1707–1724. [Google Scholar] [CrossRef]
- Noe, F.; De Fabritiis, G.; Clementi, C. Machine learning for protein folding and dynamics. Curr. Opin. Struct. Biol. 2020, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Liu, X.; Beugelsdijk, A.; Chen, J. Dynamics of the BH3-Only Protein Binding Interface of Bcl-xL. Biophys. J. 2015, 109, 1049–1057. [Google Scholar] [CrossRef]
- Liu, X.R.; Jia, Z.G.; Chen, J.H. Enhanced Sampling of Intrinsic Structural Heterogeneity of the BH3-Only Protein Binding Interface of Bcl-xL. J. Phys. Chem. B 2017, 121, 9160–9168. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L. Large-Scale Conformational Changes and Protein Function: Breaking the in silico Barrier. Front. Mol. Biosci. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Kay, L.E. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res. 2008, 41, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Noe, F.; Fischer, S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008, 18, 154–162. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Rangl, M.; Schmandt, N.; Perozo, E.; Scheuring, S. Real time dynamics of Gating-Related conformational changes in CorA. Elife 2019, 8, e47322. [Google Scholar] [CrossRef]
- Chung, H.S.; Eaton, W.A. Protein folding transition path times from single molecule FRET. Curr. Opin. Struct. Biol. 2018, 48, 30–39. [Google Scholar] [CrossRef]
- Sands, Z.; Grottesi, A.; Sansom, M.S. Voltage-gated ion channels. Curr. Biol. 2005, 15, R44–R47. [Google Scholar] [CrossRef]
- Jensen, M.O.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys.-Lausanne 2019, 7, 10. [Google Scholar] [CrossRef]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D-2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef]
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Smock, R.G.; Gierasch, L.M. Sending signals dynamically. Science 2009, 324, 198. [Google Scholar] [CrossRef]
- White, J.T.; Li, J.; Grasso, E.; Wrabl, J.O.; Hilser, V.J. Ensemble allosteric model: Energetic frustration within the intrinsically disordered glucocorticoid receptor. Philos. Trans. R. Soc. B-Biol. Sci. 2018, 373, 20170175. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/Function Implications in a Dynamic Complex of the Intrinsically Disordered Sic1 with the Cdc4 Subunit of an SCF Ubiquitin Ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef] [PubMed]
- McDowell, C.; Chen, J.; Chen, J. Potential Conformational Heterogeneity of p53 Bound to S100B(betabeta). J. Mol. Biol. 2013, 425, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 18, 494–506. [Google Scholar] [CrossRef]
- Csizmok, V.; Orlicky, S.; Cheng, J.; Song, J.H.; Bah, A.; Delgoshaie, N.; Lin, H.; Mittag, T.; Sicheri, F.; Chan, H.S.; et al. An allosteric conduit facilitates dynamic multisite substrate recognition by the SCFCdc4 ubiquitin ligase. Nat. Commun. 2017, 8, 13943. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Myers, J.B.; King, A.; Fiala, R.; Novacek, J.; Pearce, G.; Heierhorst, J.; Reichow, S.L.; Barbar, E.J. Multivalency regulates activity in an intrinsically disordered transcription factor. eLife 2018, 7, e36258. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Blayney, A.; Zhang, Y.; Gandy, L.; Mirsky, P.O.; Smith, N.; Zhang, F.; Linhardt, R.J.; Chen, J.; et al. Intrinsically Disordered N-terminal Domain (NTD) of p53 Interacts with Mitochondrial PTP Regulator Cyclophilin D. J. Mol. Biol. 2022, 434, 167552. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness in Protein Interactions-A Historical Perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef]
- Weng, J.; Wang, W. Dynamic multivalent interactions of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2019, 62, 9–13. [Google Scholar] [CrossRef]
- Miskei, M.; Antal, C.; Fuxreiter, M. FuzDB: Database of fuzzy complexes, a tool to develop stochastic structure-function relationships for protein complexes and higher-order assemblies. Nucleic Acids Res. 2017, 45, D228–D235. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S.; Birol, M.; Rhoades, E.; Miranker, A.D.; Levine, Z.A. Targeting the Intrinsically Disordered Proteome Using Small-Molecule Ligands. In Intrinsically Disordered Proteins; Rhoades, E., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 611, p. 703. [Google Scholar]
- Ruan, H.; Sun, Q.; Zhang, W.L.; Liu, Y.; Lai, L.H. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Chen, J. Targeting intrinsically disordered proteins through dynamic interactions. Biomolecules 2020, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.W.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Chen, J. Residual Structure Accelerates Binding of Intrinsically Disordered ACTR by Promoting Efficient Folding upon Encounter. J. Mol. Biol. 2019, 431, 422–432. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Pinheiro Cinelli, L.; Araújo Marins, M.; Barros da Silva, E.A.; Lima Netto, S. Variational Autoencoder. In Variational Methods for Machine Learning with Applications to Deep Networks; Cinelli, L.P., Marins, M.A., Barros da Silva, E.A., Netto, S.L., Eds.; Springer International Publishing: Cham, Switzerland, 2021; p. 111. [Google Scholar]
- Welling, D.P.K.a.M. An Introduction to Variational Autoencoders. Found. Trends Mach. Learn. 2019, 12, 307–392. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. Adv. Neural Inf. Process. Syst. 2014, 27, 1–9. [Google Scholar]
- Parshakova, T.; Andreoli, J.-M.; Dymetman, M. Global Autoregressive Models for Data-Efficient Sequence Learning. arXiv 2019, arXiv:1909.07063. [Google Scholar]
- Noe, F.; Tkatchenko, A.; Muller, K.R.; Clementi, C. Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Mardt, A.; Pasquali, L.; Wu, H.; Noe, F. VAMPnets for deep learning of molecular kinetics. Nat. Commun. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Advanced Sampling for Molecular Simulation is Coming of Age. J. Comput. Chem. 2016, 37, 549. [Google Scholar] [CrossRef]
- Tribello, G.A.; Gasparotto, P. Using Dimensionality Reduction to Analyze Protein Trajectories. Front. Mol. Biosci. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Salawu, E.O. DESP: Deep Enhanced Sampling of Proteins’ Conformation Spaces Using AI-Inspired Biasing Forces. Front. Mol. Biosci. 2021, 8, 587151. [Google Scholar] [CrossRef]
- Kukharenko, O.; Sawade, K.; Steuer, J.; Peter, C. Using Dimensionality Reduction to Systematically Expand Conformational Sampling of Intrinsically Disordered Peptides. J. Chem. Theory Comput. 2016, 12, 4726–4734. [Google Scholar] [CrossRef]
- Smith, Z.; Ravindra, P.; Wang, Y.; Cooley, R.; Tiwary, P. Discovering Protein Conformational Flexibility through Artificial-Intelligence-Aided Molecular Dynamics. J. Phys. Chem. B 2020, 124, 8221–8229. [Google Scholar] [CrossRef]
- Kleiman, D.E.; Shukla, D. Multiagent Reinforcement Learning-Based Adaptive Sampling for Conformational Dynamics of Proteins. J. Chem. Theory Comput. 2022, 18, 5422–5434. [Google Scholar] [CrossRef]
- Tian, H.; Jiang, X.; Trozzi, F.; Xiao, S.; Larson, E.C.; Tao, P. Explore Protein Conformational Space With Variational Autoencoder. Front. Mol. Biosci. 2021, 8, 781635. [Google Scholar] [CrossRef]
- Moritsugu, K. Multiscale Enhanced Sampling Using Machine Learning. Life 2021, 11, 1076. [Google Scholar] [CrossRef]
- Kaelbling, L.P.; Littman, M.L.; Moore, A.W. Reinforcement Learning: A Survey. J. Artif. Intell. Res. 1996, 4, 237–285. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Liang, X.; Zhao, D.; Huang, J.; Xu, X.; Dai, B.; Miao, Q. Deep Reinforcement Learning: A Survey. IEEE Trans. Neural. Netw. Learn Syst. 2022. early access. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, D.M.; Chong, L.T. Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys. 2017, 46, 43–57. [Google Scholar] [CrossRef]
- Degiacomi, M.T. Coupling Molecular Dynamics and Deep Learning to Mine Protein Conformational Space. Structure 2019, 27, 1034–1040.e1033. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.K.; Musson, S.C.; Willcocks, C.G.; Degiacomi, M.T. Deep Learning Protein Conformational Space with Convolutions and Latent Interpolations. Phys. Rev. X 2021, 11, 011052. [Google Scholar] [CrossRef]
- Jin, Y.; Johannissen, L.O.; Hay, S. Predicting new protein conformations from molecular dynamics simulation conformational landscapes and machine learning. Proteins 2021, 89, 915–921. [Google Scholar] [CrossRef]
- Tatro, N.J.; Das, P.; Chen, P.-Y.; Chenthamarakshan, V.; Lai, R. ProGAE: A Geometric Autoencoder-Based Generative Model for Disentangling Protein Conformational Space. In Proceedings of the International Conference on Learning Representations, Vienna, Austria, 4 May 2021. [Google Scholar]
- Gupta, A.; Dey, S.; Hicks, A.; Zhou, H.-X. Artificial intelligence guided conformational mining of intrinsically disordered proteins. Commun. Biol. 2022, 5, 610. [Google Scholar] [CrossRef]
- Schrag, L.G.; Liu, X.; Thevarajan, I.; Prakash, O.; Zolkiewski, M.; Chen, J. Cancer-Associated Mutations Perturb the Disordered Ensemble and Interactions of the Intrinsically Disordered p53 Transactivation Domain. J. Mol. Biol. 2021, 433, 167048. [Google Scholar] [CrossRef]
- Zhao, J.; Blayney, A.; Liu, X.; Gandy, L.; Jin, W.; Yan, L.; Ha, J.H.; Canning, A.J.; Connelly, M.; Yang, C.; et al. EGCG binds intrinsically disordered N-terminal domain of p53 and disrupts p53-MDM2 interaction. Nat. Commun. 2021, 12, 986. [Google Scholar] [CrossRef]
- Janson, G.; Valdes-Garcia, G.; Heo, L.; Feig, M. Direct generation of protein conformational ensembles via machine learning. Nat. Commun. 2023, 14, 774. [Google Scholar] [CrossRef]
- Noe, F.; Olsson, S.; Kohler, J.; Wu, H. Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning. Science 2019, 365, eaaw1147. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.; Tewari, A. RL Boltzmann Generators for Conformer Generation in Data-Sparse Environments. arXiv 2022, arXiv:2211.10771. [Google Scholar]
- Liu, X.; Chen, J. Residual Structures and Transient Long-Range Interactions of p53 Transactivation Domain: Assessment of Explicit Solvent Protein Force Fields. J. Chem. Theory Comput. 2019, 15, 4708–4720. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Perdikaris, P. Physics-informed deep generative models. arXiv 2018, arXiv:1812.03511. [Google Scholar]
- Raissi, M.; Perdikaris, P.; Karniadakis, G.E. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 2019, 378, 686–707. [Google Scholar] [CrossRef]
- Jagtap, A.D.; Kharazmi, E.; Karniadakis, G.E. Conservative physics-informed neural networks on discrete domains for conservation laws: Applications to forward and inverse problems. Comput. Methods Appl. Mech. Eng. 2020, 365, 113028. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, D.; Karniadakis, G.E. Physics-Informed Generative Adversarial Networks for Stochastic Differential Equations. SIAM J. Sci. Comput. 2020, 42, A292–A317. [Google Scholar] [CrossRef]
- Liang, C.; Savinov, S.N.; Fejzo, J.; Eyles, S.J.; Chen, J. Modulation of Amyloid-beta42 Conformation by Small Molecules Through Nonspecific Binding. J. Chem. Theory Comput. 2019, 15, 5169–5174. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Modeling protein assemblies: Critical Assessment of Predicted Interactions (CAPRI) 15 years hence: 6TH CAPRI evaluation meeting April 17–19 Tel-Aviv, Israel. Proteins 2017, 85, 357–358. [Google Scholar] [CrossRef]
- Mobley, D.L.; Gilson, M.K. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu. Rev. Biophys. 2017, 46, 531–558. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Sun, F.; Kong, T.; Zhang, W.; Yang, C.; Liu, C. A Survey on Deep Transfer Learning. arXiv 2018, arXiv:1808.01974. [Google Scholar]
- Jing, B.; Erives, E.; Pao-Huang, P.; Corso, G.; Berger, B.; Jaakkola, T. EigenFold: Generative Protein Structure Prediction with Diffusion Models. arXiv 2023, arXiv:2304.02198. [Google Scholar]
- Leiter, C.; Zhang, R.; Chen, Y.; Belouadi, J.; Larionov, D.; Fresen, V.; Eger, S. ChatGPT: A Meta-Analysis after 2.5 Months. arXiv 2023, arXiv:2302.13795. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, L.-E.; Barethiya, S.; Nordquist, E.; Chen, J. Machine Learning Generation of Dynamic Protein Conformational Ensembles. Molecules 2023, 28, 4047. https://doi.org/10.3390/molecules28104047
Zheng L-E, Barethiya S, Nordquist E, Chen J. Machine Learning Generation of Dynamic Protein Conformational Ensembles. Molecules. 2023; 28(10):4047. https://doi.org/10.3390/molecules28104047
Chicago/Turabian StyleZheng, Li-E, Shrishti Barethiya, Erik Nordquist, and Jianhan Chen. 2023. "Machine Learning Generation of Dynamic Protein Conformational Ensembles" Molecules 28, no. 10: 4047. https://doi.org/10.3390/molecules28104047
APA StyleZheng, L.-E., Barethiya, S., Nordquist, E., & Chen, J. (2023). Machine Learning Generation of Dynamic Protein Conformational Ensembles. Molecules, 28(10), 4047. https://doi.org/10.3390/molecules28104047