
PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies



Abstract
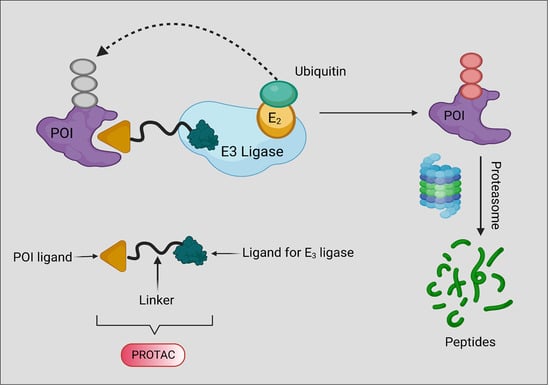
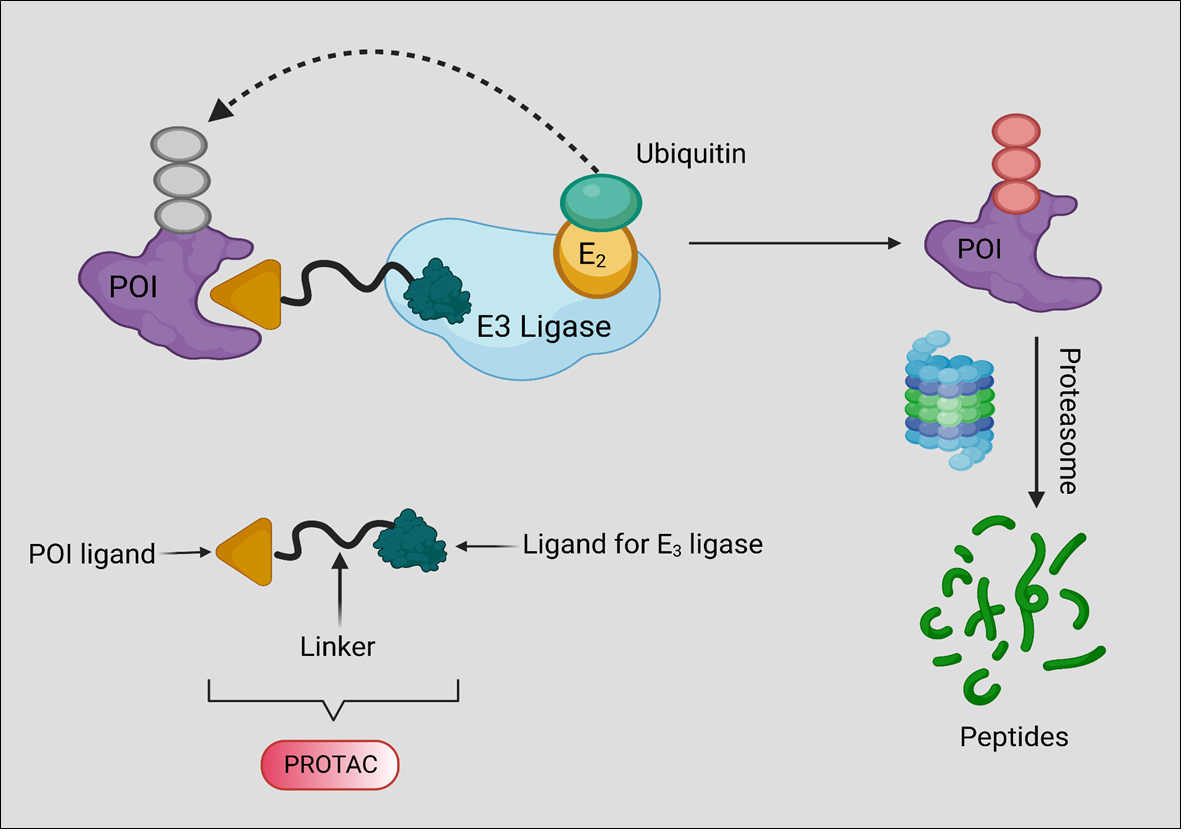
1. Introduction
2. Background, Composition, and Mechanism of Function
3. PROTACs Promote Targeted Cell or Tissue Selectivity
3.1. Photochemically Controllable PROTACs (PHOTACs)
3.1.1. Photocaged PROTACs
3.1.2. Photo Switchable PROTACs
3.1.3. Radiotherapy-Triggered Proteolysis
3.2. Hypoxia-Activated PROTACs
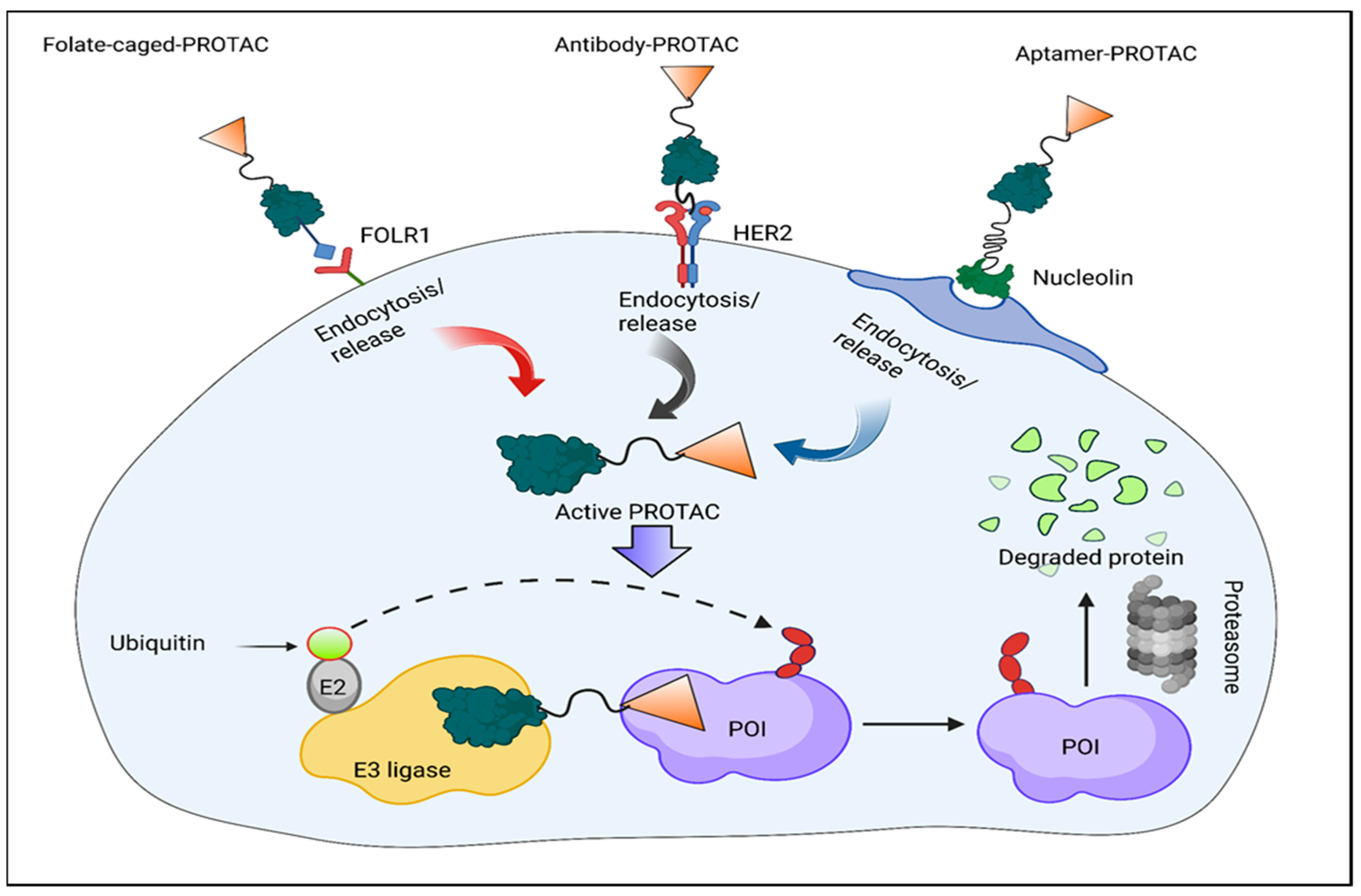
3.3. Folate-Caged PROTACs
3.4. Antibody–PROTAC Conjugates (Ab-PROTACs)
3.5. Conjugates of Aptamer–PROTACs (APCs)
3.6. Minimal Platelet Toxicity with BCL-XL PROTACs
4. Past, Present, and Future Advancement
4.1. The History of PROTAC (2001–2016)
4.2. Current: Target Scope for the PROTAC
5. Future Perspectives: Developing PROTAC Technology and Expedite PROTAC Discovery
6. Turning PROTAC into Medications
7. PROTACs: Targeted Management Strategy for Several Diseased Conditions
7.1. PROTACs in the Cancer Treatment
7.2. PROTACs in Immune System Diseases
7.3. PROTACs in Viral Infection
7.4. PROTACs in Metabolic Disease
8. Outlook for the Next 20 Years of Targeted Protein Degradation (TPD)
9. Targeted Protein Degradation: LYTAC and AUTAC
9.1. Lysosome-Targeting Chimaera (LYTAC)
9.2. Autophagy-Targeting Chimera (AUTAC)
9.3. GlueTAC
9.4. Antibody-Based PROTAC (AbTAC)
10. Therapeutic Application of PROTAC
11. Clinical Research on Proteolysis-Targeting Chimeras
12. Some Challenges of PROTAC Remain to Be Addressed
13. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef]
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun. Biol. 2020, 3, 140. [Google Scholar] [CrossRef]
- Guo, J.; Liu, J.; Wei, W. Degrading proteins in animals: “PROTAC” tion goes in vivo. Cell Res. 2019, 29, 179–180. [Google Scholar] [CrossRef]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Yan, G.; Zhong, X.; Yue, L.; Pu, C.; Shan, H.; Lan, S.; Zhou, M.; Hou, X.; Yang, J.; Li, R. Discovery of a PROTAC targeting ALK with in vivo activity. Eur. J. Med. Chem. 2021, 212, 113150. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.J.; Neklesa, T.K. Targeting nuclear receptors with PROTAC degraders. Mol. Cell. Endocrinol. 2019, 493, 110452. [Google Scholar] [CrossRef] [PubMed]
- Keen, A.C.; Jörg, M.; Halls, M.L. The application of targeted protein degradation technologies to G protein-coupled receptors. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.; Craigon, C.; Ciulli, A. Targeting epigenetic modulators using PROTAC degraders: Current status and future perspective. Bioorg. Med. Chem. Lett. 2022, 63, 128653. [Google Scholar] [CrossRef]
- Au, Y.Z.; Wang, T.; Sigua, L.H.; Qi, J. Peptide-based PROTAC: The predator of pathological proteins. Cell Chem. Biol. 2020, 27, 637–639. [Google Scholar] [CrossRef]
- Kumar, D.F.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The contribution of the 20S proteasome to proteostasis. Biomolecules 2019, 9, 5190. [Google Scholar]
- Bondeson, D.P.; Crews, C.M. Targeted protein degradation by small molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. 2005, 44, 5932–5943. [Google Scholar] [CrossRef]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Xin, M.; Zhang, S.Q. Progress of small molecules for targeted protein degradation: PROTACs and other technologies. Drug Devel. Res. 2023, 84, 337–394. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, e41305. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Coen, M.; Zhang, A.X.; Pachl, F.; Castaldi, M.P.; Dahl, G.; Boyd, H.; Scott, C.; Newham, P. Proteolysis-targeting chimeras in drug development: A safety perspective. Br. J. Pharmacol. 2020, 177, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Ishoey, M.; Chorn, S.; Singh, N.; Jaeger, M.G.; Brand, M.; Paulk, J.; Bauer, S.; Erb, M.A.; Parapatics, K.; Müller, A.C. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 2018, 13, 553–560. [Google Scholar] [CrossRef]
- Reynders, M.; Trauner, D. Optical control of targeted protein degradation. Cell Chem. Biol. 2021, 28, 969–986. [Google Scholar] [CrossRef]
- Kounde, C.S.; Shchepinova, M.M.; Saunders, C.N.; Muelbaier, M.; Rackham, M.D.; Harling, J.D.; Tate, E.W. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun. 2020, 56, 5532–5535. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.-F. Targeting hypoxia: Hypoxia-activated prodrugs in cancer therapy. Front. Oncol. 2021, 11, 700407. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, Y.; Li, Y.; Ni, Q.; Li, J. Radiotherapy-Triggered Proteolysis Targeting Chimera Prodrug Activation in Tumors. J. Am. Chem. Soc. 2022, 145, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Zhang, Y.; Gao, Q.; Neumann, K.; Dong, H.; Porter, H.; Potter, M.; Ren, H.; Argyle, D.; Bradley, M. Switching on prodrugs using radiotherapy. Nat. Chem. 2021, 13, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Wouters, B.G.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personalised medicine. Br. J. Cancer 2016, 114, 1071–1077. [Google Scholar] [CrossRef]
- Cheng, W.; Li, S.; Wen, X.; Han, S.; Wang, S.; Wei, H.; Song, Z.; Wang, Y.; Tian, X.; Zhang, X. Development of hypoxia-activated PROTAC exerting a more potent effect in tumor hypoxia than in normoxia. Chem. Commun. 2021, 57, 12852–12855. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Du, Y.; Zou, Y.; Niu, J.; Cai, Z.; Wang, X.; Qiu, F.; Ding, Y.; Yang, G.; Wu, Y. Rational design for nitroreductase (NTR)-responsive proteolysis targeting chimeras (PROTACs) selectively targeting tumor tissues. J. Med. Chem. 2022, 65, 5057–5071. [Google Scholar] [CrossRef]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the folate receptor α in oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Liu, Y.; Shen, Y.; Meng, F.; Kaniskan, H.U.; Jin, J.; Wei, W. Cancer selective target degradation by folate-caged PROTACs. J. Am. Chem. Soc. 2021, 143, 7380–7387. [Google Scholar] [CrossRef]
- Chen, H.; Liu, J.; Kaniskan, H.U.; Wei, W.; Jin, J. Folate-guided protein degradation by immunomodulatory imide drug-based molecular glues and proteolysis targeting chimeras. J. Med. Chem. 2021, 64, 12273–12285. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive ReviewAntibody–Drug Conjugates in Cancer Immunotherapy. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody–PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Tao, X.; Zhai, X.; Zhu, Y.; Li, Y.; Chen, Y.; Dong, D.; Yang, S.; Lv, L. Application of aptamer-drug delivery system in the therapy of breast cancer. Biomed. Pharmacoth. 2023, 161, 114444. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.-Y.; Dent, P.; Reed, J.C.; Pellecchia, M. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A. Bcl-xL–inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood J. Am. Soc. Hematol. 2011, 118, 1663–1674. [Google Scholar] [CrossRef]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Lee, H.; Puppala, D.; Choi, E.Y.; Swanson, H.; Kim, K.B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. Chembiochem 2007, 8, 2058–2062. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin− Ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small Molecule Inhibitors of the Interaction Between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. Engl. 2012, 51, 11463. [Google Scholar] [CrossRef]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef]
- Jiang, H.; Xiong, H.; Gu, S.X.; Wang, M. E3 ligase ligand optimization of Clinical PROTACs. Front. Chem. 2023, 11, 17. [Google Scholar] [CrossRef]
- Li, W.; Gao, C.; Zhao, L.; Yuan, Z.; Chen, Y.; Jiang, Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur. J. Med. Chem. 2018, 151, 237–247. [Google Scholar] [CrossRef]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- Tan, L.; Gray, N.S. When kinases meet PROTACs. Chin. J. Chem. 2018, 36, 971–977. [Google Scholar] [CrossRef]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.K.; Varghese, J.O.; Das, S.; Nag, A.; Tang, G.; Tang, K.; Sutherland, A.M.; Heath, J.R. Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci. 2016, 22, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.L.; Phillips, A.J. Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol. 2018, 44, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 1–9. [Google Scholar] [CrossRef]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef]
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? J. Med. Chem. 2018, 61, 444–452. [Google Scholar] [CrossRef]
- Riching, K.M.; Mahan, S.; Corona, C.R.; McDougall, M.; Vasta, J.D.; Robers, M.B.; Urh, M.; Daniels, D.L. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem. Biol. 2018, 13, 2758–2770. [Google Scholar] [CrossRef]
- Bunnage, M.E.; Gilbert, A.M.; Jones, L.H.; Hett, E.C. Know your target, know your molecule. Nat. Chem. Biol. 2015, 11, 368–372. [Google Scholar] [CrossRef]
- Tomoshige, S.; Hashimoto, Y.; Ishikawa, M. Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg. Med. Chem. 2016, 24, 3144–3148. [Google Scholar] [CrossRef]
- Okitsu, K.; Hattori, T.; Misawa, T.; Shoda, T.; Kurihara, M.; Naito, M.; Demizu, Y. Development of a small hybrid molecule that mediates degradation of His-Tag fused proteins. J. Med. Chem. 2018, 61, 576–582. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Moreau, P.; Pylypenko, H.; Grosicki, S.; Karamanesht, I.; Leleu, X.; Grishunina, M.; Rekhtman, G.; Masliak, Z.; Robak, T.; Shubina, A. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: A randomised, phase 3, non-inferiority study. Lancet Oncol. 2011, 12, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Nakai, Y.; Matsumoto, Y.; Kagebayashi, Y.; Samma, S. Investigation of incidence and risk factors of subcutaneous granulomas induced by injection of leuprorelin acetate. Hinyokika Kiyo. Acta Urol. Jpn. 2015, 61, 55–59. [Google Scholar]
- Link, J.O.; Taylor, J.G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.; Kirschberg, T.; Sun, J.; Squires, N. Discovery of ledipasvir (GS-5885): A potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 2014, 57, 2033–2046. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.D.; Ciulli, A. Driving e3 ligase substrate specificity for targeted protein degradation: Lessons from nature and the laboratory. Annu. Rev. Biochem. 2022, 91, 295–319. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, J.; Zhang, Q.; Pan, X.; Zhang, J. Novel strategies and promising opportunities for targeted protein degradation: An innovative therapeutic approach to overcome cancer resistance. Pharmacol. Ther. 2023, 244, 108371. [Google Scholar] [CrossRef]
- Wu, J.; Wang, W.; Leung, C.H. Computational strategies for PROTAC drug discovery. Acta Mater. Med. 2023, 2, 42–53. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.-Y.; Zhao, Y. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef]
- Dogheim, G.M.; Amralla, M.T. Proteolysis Targeting Chimera (PROTAC) as a promising novel therapeutic modality for the treatment of triple-negative breast cancer (TNBC). Drug Dev. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Zheng, G.; Kaye, F.J.; Wu, L. PROTAC therapy as a new targeted therapy for lung cancer. Mol. Ther. 2022, 31, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Lospinoso Severini, L.; Bufalieri, F.; Infante, P.; Di Marcotullio, L. Proteolysis-Targeting Chimera (PROTAC): Is the Technology Looking at the Treatment of Brain Tumors? Front. Cell Devel. Biol. 2022, 10, 291. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Deng, J.; Liang, B.; Xing, D. Developments of PROTACs technology in immune-related diseases. Europ. J. Med. Chem. 2023, 249, 115127. [Google Scholar] [CrossRef]
- Shukla, S.K.; Singh, G.; Ahmad, S.; Pant, P. Infections, genetic and environmental factors in pathogenesis of autoimmune thyroid diseases. Microb. Pathog. 2018, 116, 279–288. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Degradation of IRAK4 for the Treatment of Cancer. ACS. Med. Chem. Lett. 2019, 10, 1370–1371. [Google Scholar] [CrossRef]
- Nunes, J.; McGonagle, G.A.; Eden, J.; Kiritharan, G.; Touzet, M.; Lewell, X.; Emery, J.; Eidam, H.; Harling, J.D.; Anderson, N.A. Targeting IRAK4 for degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. [Google Scholar] [CrossRef]
- Humphreys, P.G.; Bamborough, P.; Chung, C.W.; Craggs, P.D.; Gordon, L.; Grandi, P.; Hayhow, T.G.; Hussain, J.; Jones, K.L.; Lindon, M.; et al. Discovery of a potent, cell penetrant, and selective p300/CBP-associated factor (PCAF)/general control nonderepressible 5 (GCN5) bromodomain chemical probe. J. Med. Chem. 2017, 60, 695–709. [Google Scholar] [CrossRef]
- Choi, W.M.; Choi, J.; Lim, Y.S. Hepatitis B: Epidemiology, natural history, and diagnosis. In Comprehensive Guide to Hepatitis Advances; Academic Press: Cambridge, MA, USA, 2023; pp. 183–203. [Google Scholar]
- Kar, A.; Samanta, A.; Mukherjee, S.; Barik, S.; Biswas, A. The HBV web: An insight into molecular interactomes between the hepatitis B virus and its host en route to hepatocellular carcinoma. J. Med. Virol. 2023, 95, e28436. [Google Scholar] [CrossRef]
- Schollmeier, A.; Glitscher, M.; Hildt, E. Relevance of HBx for Hepatitis B Virus-Associated Pathogenesis. Int. J. Mol. Sci. 2023, 24, 4964. [Google Scholar] [CrossRef]
- Ahmad, H.; Zia, B.; Husain, H.; Husain, A. Recent Advances in PROTAC-Based Antiviral Strategies. Vaccines 2023, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Kong, B.; Zhu, Z.; Huang, F.; Zhang, L.; Lu, T.; Chen, Y.; Zhang, Y.; Jiang, Y. Recent advances in targeted protein degraders as potential therapeutic agents. Mol. Divers. 2023, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ruan, Y.; Wang, D.; Fan, J.; Luo, N.; Chen, H.; Li, X.; Chen, W.; Wang, X. VHL-recruiting PROTAC attenuates renal fibrosis and preserves renal function via simultaneous degradation of Smad3 and stabilization of HIF-2α. Cell. Biosci. 2022, 12, 203. [Google Scholar] [CrossRef]
- Boeckmans, J.; Gatzios, A.; Schattenberg, J.M.; Koek, G.H.; Rodrigues, R.M.; Vanhaecke, T. PNPLA3 I148M and response to treatment for hepatic steatosis. A systematic review. Liver Int. 2023, 43, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, K.T.; Crews, C.M. Targeted protein degradation: A promise for undruggable proteins. Cell Chem. Biol. 2021, 28, 934–951. [Google Scholar] [CrossRef] [PubMed]
- Tamatam, R.; Shin, D. Emerging Strategies in Proteolysis-Targeting Chimeras (PROTACs): Highlights from 2022. Int. J. Mol. Sci. 2023, 24, 5190. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021, 17, 937–946. [Google Scholar] [CrossRef]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–213. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther. 2017, 25, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Park, E.I.; Mi, Y.; Unverzagt, C.; Gabius, H.J.; Baenziger, J.U. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acidα2, 6GalNAc. Proc. Nat. Acad. Sci. USA 2005, 102, 17125–17129. [Google Scholar] [CrossRef]
- Steirer, L.M.; Park, E.I.; Townsend, R.R.; Baenziger, J.U. The asialoglycoprotein receptor regulates levels of plasma glycoproteins terminating with sialic acid α2, 6-galactose. J. Biol. Chem. 2009, 284, 3777–3783. [Google Scholar] [CrossRef]
- Takahashi, D.; Arimoto, H. Targeting selective autophagy by AUTAC degraders. Autophagy 2020, 16, 765–766. [Google Scholar] [CrossRef]
- Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.; Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [Google Scholar] [CrossRef]
- Zhang, H.; Han, Y.; Yang, Y.; Lin, F.; Li, K.; Kong, L.; Liu, H.; Dang, Y.; Lin, J.; Chen, P.R. Covalently engineered nanobody chimeras for targeted membrane protein degradation. J. Am. Chem. Soc. 2021, 143, 16377–16382. [Google Scholar] [CrossRef]
- Verdurmen, W.P.; Mazlami, M.; Plückthun, A. A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep. 2017, 7, 13194. [Google Scholar] [CrossRef]
- Han, Y.; Da, Y.; Yu, M.; Cheng, Y.; Wang, X.; Xiong, J.; Guo, G.; Li, Y.; Jiang, X.; Cai, X. Protein labeling approach to improve lysosomal targeting and efficacy of antibody–drug conjugates. Org. Biomol. Chem. 2020, 18, 3229–3233. [Google Scholar] [CrossRef]
- Story, C.M.; Mikulska, J.E.; Simister, N.E. A major histocompatibility complex class I-like Fc receptor cloned from human placenta: Possible role in transfer of immunoglobulin G from mother to fetus. J. Exp. Med. 1994, 180, 2377–2381. [Google Scholar] [CrossRef]
- West, A.P.; Bjorkman, P.J. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry 2000, 39, 9698–9708. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 2013, 4, 2787. [Google Scholar] [CrossRef]
- Bond, M.J.; Crews, C.M. Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol. 2021, 2, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Vignali, V.; Hines, P.A.; Cruz, A.G.; Ziętek, B.; Herold, R. Health horizons: Future trends and technologies from the European Medicines Agency’s horizon scanning collaborations. Front. Med. 2022, 9, 3588. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Treatment of Cancer and Alzheimer’s Disease by PROTAC Degradation of EGFR. ACS Med. Chem. Lett. 2019, 10, 1098–1099. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, Q.; Jiang, T.; Li, S.; Ye, J.; Zheng, J.; Wang, X.; Liu, Y.; Deng, M.; Ke, D.; et al. A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics 2021, 11, 5279. [Google Scholar] [CrossRef]
- Tonali, N.; Nencetti, S.; Orlandini, E.; Ciccone, L. Application of PROTAC strategy to TTR-Aβ protein-protein interaction for the development of Alzheimer’s disease drugs. Neural Regen. Res. 2021, 8, 1554. [Google Scholar]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 1–9. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC-Mediated Degradation of Janus Kinase as a Therapeutic Strategy for Cancer and Rheumatoid Arthritis. ACS Med. Chem. Lett. 2021, 12, 945–946. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Degradation of IRAK4 for the Treatment of Neurodegenerative and Cardiovascular Diseases. ACS Med. Chem. Lett. 2019, 10, 1251–1252. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. PROTAC Compounds Targeting Androgen Receptor for Cancer Therapeutics: Prostate Cancer and Kennedy’s Disease. ACS Med. Chem. Lett. 2020, 11, 1092–1093. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Wu, J.; Cao, Y.; He, Y.; Zhang, Y.; Gao, X.; Xue, Y.; Fang, M.; Wu, Z. The synthesis of PROTAC molecule and new target KAT6A identification of CDK9 inhibitor iCDK9. Chi. Chem. Lett. 2023, 34, 107741. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Wu, Y.; Xing, D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 2021, 225, 113749. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Li, Y.; Wang, Y.; et al. Attenuation of NLRP3 inflammasome activation by indirubin-derived PROTAC targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody–drug conjugates for cancer treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-protac conjugates (apcs) for tumor-specific targeting in breast cancer. Angew. Chem. 2021, 133, 23487–23493. [Google Scholar]
- Available online: https://clinicaltrials.gov/ct2/results?cond=Cancer&term=PROTAC (accessed on 17 April 2023).
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; et al. ARV-110: An androgen receptor PROTAC degrader for prostate cancer. Cancer Res. 2018, 78, 5236. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Mao, S.; Xu, P.; Xu, X.; Zhao, R.; Wu, Z.; Zhong, K.; Yu, G.; Wang, X. Discovery of A031 as effective proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader for the treatment of prostate cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Liao, Y.Y.; Cheng, G.; Chen, J.; Mo, S.; Yuan, L.; Cheng, X.D.; Qin, J.J.; Shao, Z. Aspeterreurone A, a cytotoxic dihydrobenzofuran—Phenyl acrylate hybrid from the deep-sea-derived fungus Aspergillus terreus CC-S06–18. J. Nat. Prod. 2020, 83, 1998–2003. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 1200. [Google Scholar] [CrossRef]
- Zeng, S.; Huang, W.; Zheng, X.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, J.; Yao, X.; Zheng, W.; Mao, Y.; Lan, T.; Wang, L.; Sun, Y.; Zhang, X.; Zhao, Q.; et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 2019, 5, 10. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Li, J.; Zhang, B.; Wang, Z.; Shaabani, S.; Ter, B.F.; Essa, K.; Dömling, A. PROTACs—A game-changing technology. Exp. Opin. Drug Discov. 2019, 14, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Tsuji, G.; Shoda, T.; Fujisato, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. ACS Chem. Biol. 2019, 14, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Exp. Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of PROTAC | Target | Purpose | E3 Ligase | Target Receptor | Target Site | Name of PROTAC | Target | Purpose |
|---|---|---|---|---|---|---|---|---|
| ARV-110, Phase III | AR | Prostate cancer | CRBN | Nuclear receptor | Cytosol, nucleus | ARV-110, Phase III | AR | Prostate cancer |
| ARV-471, Phase II | ER | Breast cancer | CRBN | Nuclear receptor | Cytosol, nucleus | ARV-471, Phase II | ER | Breast cancer |
| AC682, Phase I | ER | Breast cancer | CRBN | Nuclear receptor | Cytosol, nucleus | AC682, Phase I | ER | Breast cancer |
| ARV-766, Phase I | AR | Prostate cancer | - | Nuclear receptor | Cytosol, nucleus | ARV-766, Phase I | AR | Prostate cancer |
| CC-94676, Phase I | AR | Prostate cancer | CRBN | Nuclear receptor | Cytosol, nucleus | CC-94676, Phase I | AR | Prostate cancer |
| DT2216, Phase I | BCL-XL | Liquid and solid tumors | VHL | Anti-Apoptotic protein | Cytosol, nucleus, MOM | DT2216, Phase I | BCL-XL | Liquid and solid tumors |
| FHD-609, Phase I | BRD9 | Synovial sarcoma | - | Nuclear factor | Nucleus | FHD-609, Phase I | BRD9 | Synovial sarcoma |
| KT-474, Phase I | IRAK4 | Autoimmune diseases | - | Kinase | Cytosol | KT-474, Phase I | IRAK4 | Autoimmune diseases |
| KT-413, Phase I | IRAK4 | Diffuse large B cell lymphoma | CRBN | Kinase | Cytosol | KT-413, Phase I | IRAK4 | Diffuse large B cell lymphoma |
| KT-333, Phase I | STAT3 | Liquid and solid tumors | - | Nuclear factor | Cytosol, nucleus | KT-333, Phase I | STAT3 | Liquid and solid tumors |
| NX-2127, Phase I | BTK | B cell malignancies | CRBN | Tyrosine kinase | Cytosol | NX-2127, Phase I | BTK | B cell malignancies |
| NX-5948, Phase I | BTK | B cell malignancies and Autoimmune diseases | CRBN | Tyrosine kinase | Cytosol | NX-5948, Phase I | BTK | B cell malignancies and Autoimmune diseases |
| CFT8634, IND-e | BRD9 | Synovial sarcoma | CRBN | Nuclear factor | Nucleus | CFT8634, IND-e | BRD9 | Synovial sarcoma |
| CFT8919, IND-e | EGFR L858R | NSLC | CRBN | Cell surface receptor | Cell membrane | CFT8919, IND-e | EGFR L858R | NSLC |
| CG001419, IND-e | TRK | Cancer and other indications | CRBN | Tyrosine kinase | Cell membrane | CG001419, IND-e | TRK | Cancer and other indications |
| Therapeutics | Clinical Trial Number and Phase | Target | Toxicity Outcomes | Primary Efficacy Data |
|---|---|---|---|---|
| ARV110 | NCT03888612, Phase 2 | Prostate cancer | ARV-110 has an acceptable safety profile; however, co-administration of rosuvastatin with ARV-110 could produce toxic side effects. | The paragraph describes the results of a study or clinical trial involving a group of patients. Specifically, it states that out of 15 patients who received a 140 mg dose, 2 of them experienced a reduction in PSA (prostate-specific antigen) levels of more than 50%. Additionally, out of five patients who had either T878 or H875 mutations in AR (and received the same dose), two of them also had PSA reductions of over 50%. Finally, among the 15 patients who had wild-type AR, 2 of them (13%) had PSA reductions of over 50%. |
| ARV471 | NCT04072952, Phase 2 | Breast cancer | ARV-471 has been found to be well tolerated across all tested dosage levels with no reported treatment-related adverse events of grade 3 or 4. Additionally, no dose-limiting toxicities (DLTs) were reported during testing. These adverse events include nausea, which occurs in 24% of cases, arthralgia and fatigue, which occur in 19% of cases each, and decreased appetite, which occurs in 14% of cases. | In the ARV471 trial, which involved 21 adult patients, 1 patient achieved a confirmed partial response (PR) with a 51% reduction in the size of the target lesion. Two other patients had PRs, but they were not confirmed. Additionally, one patient had a stable disease with a reduction in target lesion size of over 50%. Overall, 42% of the 12 patients evaluated for clinical benefit response (CBR) achieved CBR. |
| KT474 | NCT04772885, Phase 1 | Autoimmune including AD, HS and RA | Not reported | Not reported |
| NX2127 | NCT04830137, Phase 1 | B cell malignancies | Not reported | Not reported |
| ARV-471, Drug: Ribociclib | NCT05573555, Phase 2 | Breast Cancer | Less toxicities in ARV-471 in combination with Ribociclib | Overall Survival |
| Drug: ARV-471Drug: Abemaciclib | NCT05548127, Phase 1, Phase 2 | Breast Cancer | The number of participants with dose-limiting toxicities; dose-limiting toxicity rate for ARV-471 in combination with Abemaciclib. | % of participants gaining clinical advantage enhances |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sincere, N.I.; Anand, K.; Ashique, S.; Yang, J.; You, C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules 2023, 28, 4014. https://doi.org/10.3390/molecules28104014
Sincere NI, Anand K, Ashique S, Yang J, You C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules. 2023; 28(10):4014. https://doi.org/10.3390/molecules28104014
Chicago/Turabian StyleSincere, Nuwayo Ishimwe, Krishnan Anand, Sumel Ashique, Jing Yang, and Chongge You. 2023. "PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies" Molecules 28, no. 10: 4014. https://doi.org/10.3390/molecules28104014
APA StyleSincere, N. I., Anand, K., Ashique, S., Yang, J., & You, C. (2023). PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules, 28(10), 4014. https://doi.org/10.3390/molecules28104014