Abstract
Indole-containing acyloins are either key intermediates of many antimicrobial/antiviral natural products or building blocks in the synthesis of biologically active molecules. As such, access to structurally diverse indole-containing acyloins has attracted considerable attention. In this report, we present a pilot study of using biotransformation to provide acyloins that contain various indole substituents. The biotransformation system contains the tryptophan synthase standalone β-subunit variant, PfTrpB6, generated from directed evolution in the literature; a commercially available L-amino acid oxidase (LAAO); and the thiamine-diphosphate (ThDP)-dependent enzyme NzsH, encoded in the biosynthetic gene cluster (nzs) of the bacterial carbazole alkaloid natural product named neocarazostatin A. The utilization of the first two enzymes, the PfTrpB variant and LAAO, is designed to provide structurally diverse indole 3-pyruvate derivatives as donor substrates for NzsH-catalysed biotransformation to provide acyloin derivatives. Our results demonstrate that NzsH displays a considerable substrate profile toward donor substrates for production of acyloins with different indole ring systems, suggesting that NzsH could be further explored as a potential biocatalyst via directed evolution to improve the catalytic efficiency in the future.
1. Introduction
Indole-containing acyloins are key precursor of antimicrobial/antiviral agents, such as sattazolins 1–5 (Figure 1A) [1,2,3]. They are also useful intermediates in synthesis of biologically active molecules such as carbazoles, β-carbolines and other indole-containing molecules [4]. However, access to structurally diverse indole-containing acyloins has presented a synthetic challenge.
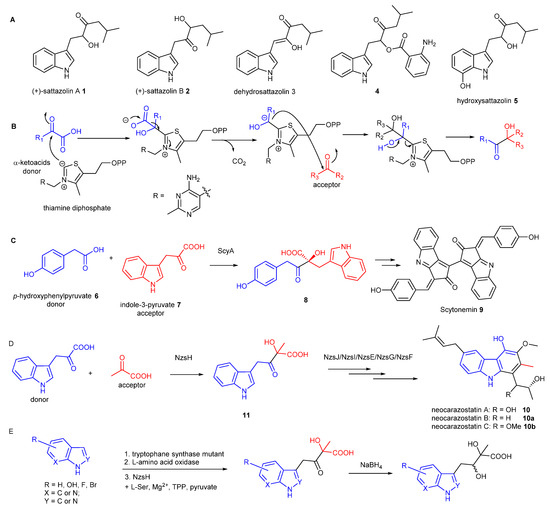
Figure 1.
Indole-containing acyloins generated from ThDP-dependent enzymes. (A) indole-containing antimicrobial/antiviral acyloin natural products. (B) a generic mechanism of caboligation catalysed by ThDP-dependent enzymes. (C) The key biotransformation to generate indole-containing acyloin intermediate during the biosynthesis of scytonemin 9. (D) The key biotransformation to generate indole-containing acyloin intermediate during the biosynthesis of neocarazostatin 10. (E) The biotransformation to generate structurally diverse acyloins in this study.
Thiamine diphosphate (ThDP)-dependent enzymes are widely found in the biological systems and have been demonstrated to be involved in diverse biotransformation, including C–C, C–N, C–S, and C–O bond cleavage and formation [5]. The general mechanism underlying all these reactions is that the enzyme’s active site-mediated dissociation of the C2–H proton from the thiazolium ring of ThDP will generate the C2 anion ylid which is covalently bound to the donor substrate (Figure 1B). Subsequent decarboxylation results in the ThDP-bound enolate intermediate with nucleophilic reactivity which readily react with electrophilic acceptors, such as aldehydes, ketones or α-ketoacids to form the α-hydroxyl ketone (an acyloin) [6].
In many cases, pyruvate is the donor in the ThDP-dependent reactions. However, some of ThDP-dependent enzymes utilise a range of α-ketoacids to initiate carboligation reactions. For example, the ThDP dependent enzyme, ScyA, was found to couple p-hydroxyphenylpyruvate 6 as the donor and indole-3-pyruvate 7 as the acceptor to afford the β-ketoacid product 8, the key intermediate during the biosynthesis of scytonemin 9, a pigment produced by cyanobacteria [7]. During our biosynthetic studies of the bacterial carbazole alkaloid, neocarazostatin A 10 [8,9,10,11], we found that the ThDP-dependent enzyme NzsH catalyses an unusual carboligation between indole-3-pyruvate 7 as the donor and pyruvate as the acceptor to generate an indole-containing acyloin intermediate 11 [9], followed by further modifications to finally decorate 10 [10,11], consistent with a parallel study in the biosynthetic pathway of the bacterial carbazole analogue, carbazomycin [12]. NzsH only accepts 2-oxobutyrate, an analogue of pyruvate, as the acceptor, unlike other ThDP enzymes that normally display a considerable range of aldehyde and ketones [9]. Phylogenetic analysis demonstrated that NzsH formed a distinct clade with other ThDP-dependent enzymes, suggesting that NzsH represents a unique group of ThDP-dependent enzymes that utilises indole-3-pyruvate as the donor for the carboligation reaction [9]. However, the donor substrate plasticity of NzsH has remain elusive. More recently, two NzsH homologues, CbeiHKI805_0381 and Cbei2730, were shown to be responsible to produce the indole-containing acyloin core of antimicrobial/antiviral agents, sattazolins 1-5 and its derivatives, isolated from various strains of Clostridium beijerinckii, a bacterium used for industrial solvent production [13]. Interestingly, the enzymes can accept branched chain α-ketoacids as acceptor substrates and both pyruvate and indole-3-pyruvate as donor substrates [13].
Here, we report a pilot study of new three-enzyme coupled biotransformation to access indole-containing acyloin derivatives with structural diversity. The reaction mixture includes an engineered tryptophane synthase β-subunit, a commercially available L-amino acid oxidase (LAAO), and NzsH. This coupled reaction is initiated from commercially available indole or indole derivatives with L-Ser catalysed by the engineered tryptophane synthetase to provide tryptophane derivatives, followed by addition of LAAO for oxidation reactions to give the corresponding indole-3-pyruvate analogues. Inclusion of NzsH in the reactions finally allow access of a new range of indole-containing acyloin derivatives. This newly introduced functionalised indole-3-pyruvate analogues substantially broaden the donor substrate range of the ThDP-dependent NzsH enzyme, thus holding potential to generate new antiviral/antimicrobial acyloins with different indole ring systems.
2. Results and Discussion
To generate structurally diverse indole-containing acyloin derivatives, indole-3-pyruvate derivatives with structural diversity to evaluate the substrate plasticity of NzsH is key. However, most of indole-3-pyruvate derivatives are not commercially available. In nature, indole-3-pyruvate is directly descended from L-tryptophane via deamination reactions. There has been a considerable interest in the application of tryptophane synthase to generate structurally diverse tryptophane derivatives due to its importance as a key building block for many bioactive molecules [14]. Tryptophan synthase is a heterodimeric complex consisting of two subunits, TrpA (α-subunit) catalysing the cleavage of indole glycerol phosphate to indole, and TrpB (β-subunit) mediating the coupling between indole and L-Ser to provide L-Tryptophan using pyridoxal phosphate (PLP) as cofactors (Supplementary Scheme S1A). The activities of both subunits are required for wild type tryptophan synthases, although only the reactivity of TrpB, coupling L-Ser and indole is necessary and desirable for L-tryptophan synthesis. Recent studies reported an engineered β-subunit of tryptophan synthase (PfTrpB) from Pyrococcus furiosus as standalone function that restore the catalytic efficiency and surpass the activity of the native complex (Supplementary Scheme S1B) [15,16,17]. One variant (PfTrpB6) with six amino acid site mutations displayed the best kinetics towards L-Ser and indole and a wide range of indole derivatives. To this end, we synthesized the gene coding PfTrpB6 for overexpression in E. coli. PfTrpB6 was expressed as an N-terminal pHis6 recombinant protein and was purified to near homogenesis by Ni-NTA chromatography, giving estimated molecular weight of ~45 kDa as previously reported (Supplementary Figure S1A). Incubation of PfTrpB6 with indole, L-Ser and PLP resulted in the accumulation of L-tryptophan 13 as evidenced in our LC-MS and tandem MS analyses (Figure 2 and Supplementary Figure S2).
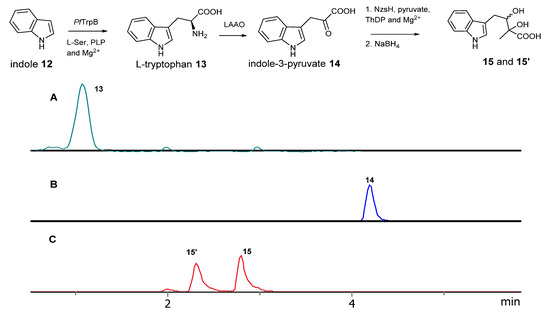
Figure 2.
The reaction scheme of the step-wise coupled reactions leading the formation of indole-containing acyloins. (A) the extracted ion chromatography of L-tryptophan catalysed by the engineered PfTrpB6. (B) the extracted ion chromatography of indole-3-pyruvate catalysed by a coupled reaction of the engineered PfTrpB6 and L-AAO. (C) the extracted ion chromatography of reduced indole-containing acyloin derivative catalysed by a three-enzyme reaction of the engineered PfTrpB6, L-AAO and NzsH, followed by NaBH4 reduction.
To further in situ generate indole-3-pyruvate from L-tryptophan, we decided to use commercially available L-amino acid oxidase (LAAO) from snake venom (Sigma Aldrich catalogue number: A5147) because this enzyme displays broad substrate tolerance toward amino acids and does not require any additional cofactors (Supplementary Scheme S1C). It also can efficiently shift the reaction equilibrium during the coupled reaction as observed in our previous report when the fluorination enzyme, FlA, from Streptomyces cattleya, was investigated for a chlorination reaction [18]. Addition of LAAO into the aforementioned enzyme system led to the formation of indole-3-pyruvate 14 as evidenced in our MS and tandem MS analysis (Figure 2 and Supplementary Figure S3). We next overexpressed the synthetic construct containing the gene nzsH in E coli BL-21 (DE3). NzsH was expressed as an N-terminal pHis6 recombinant protein and was purified to near homogenesis by Ni-NTA chromatography, giving estimated molecular weight of ~64 kDa as previously reported [9] (Supplementary Figure S1B). Inclusion of NzsH into the coupled reaction of PfTrpB6 and LAAO together with TPP and Mg2+ resulted in the formation of the indole-containing acyloin scaffold. However, this molecule is not stable and is subjected to facile decarboxylation in the LC-MS analysis [9]. We used NaBH4 to reduce the β-ketone of the molecule to generate two diastereomeric diols, 15 and 15′, as evidenced in our LC-MS and tandem MS analyses (Figure 2 and Supplementary Figure S4).
Encouraged by the results of this three-enzyme coupling system, we then acquired four commercially available indole derivatives including 4-fluoroindole 16, 5-fluoroindole 17, 4-bromoindole 18 and 6-hydroxyl-indole 19 (Table 1). Our LC-MS and tandem MS analysis demonstrated that all of these indole derivatives can be efficiently transformed into the corresponding indole-3-pyruvate derivatives, 26–29 as well as the corresponding diols, 31–35, (Table 1), in the corresponding coupled chemoenzymatic systems.

Table 1.
Structures of indole derivatives and related indole-containing products generated from the step-wise coupled reaction. Addition of NzsH together with ThDP and Mg2+ provided the corresponding indole-containing acyloin derivatives, 31–35 (Table 1), as evidenced in our LC-MS and tandem MS analyses (Supplementary Figure S5–S19).
We also monitored the biotransformation of the formation of fluorinated indole-containing acyloins in 19F NMR. As shown in Figure 3A,E, the reaction was initiated by addition of 4-fluoroindole 16 (−122.8 ppm) and 5-fluoroindole 17 (−125.2 ppm), respectively, in the PfTrpB6 -mediated systems to provide 4-fluoro-tryptophan 21 (−124.3 ppm) and 5-fluoro-tryptophan 22 (−125.4 ppm), respectively, in 19FNMR spectrum (Figure 3B,F). When LAAO was added to the reactions, 4-fluoro-indole-3-pyruvate 22 (−124.3 ppm) and 5-fluoroindole-3-pyruvate 27 (−124.8 ppm), respectively, appeared (Figure 3C,G). Finally, after inclusion of NzsH, the corresponding 4-fluoro-acyloin 31 (−124.4 ppm) and 5-fluoro-acyloin 32 (−125.6 ppm) was formed (Figure 3D,H), respectively.
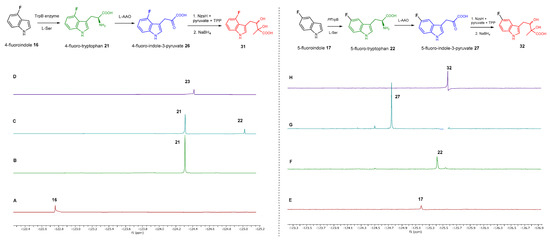
Figure 3.
13F NMR results for the coupled reaction leading to the formation of 4-fluoroindole (left column) and 5-fluoroindole (right column) containing acyloins. (A) 4-fluoroindole (−122.8 ppm) as starting material. (B) 4-fluoro-L-tryptophan (−124.3 ppm) yielded from the PfTrpB6 and 4-fluoroindole system. (C) 4-fluoroindole-3-pyruvate (−125.0 ppm) obtained when LAAO was added to the reaction. (D) 4-fluoro-acyloin (−124.4 ppm) generated by three-enzyme system. (E) 5-fluoroindole (−125.2 ppm) as starting material. (F) 5-fluoro-L-tryptophan (−125.4 ppm) yielded from the PfTrpB6 and 4-fluoroindole system. (G) 5-fluoroindole-3-pyruvate (−124.8 ppm) obtained when LAAO was added to the reaction. (H) 5-fluoro-acyloin (−125.6 ppm) generated by the three-enzyme system.Azaindoles are structurally bioisosteric chemical structures to indoles in biological materials [19]. They display distinct physiochemical properties, aqueous solubility and total polar surface area due to the presence of extra nitrogen atom within the six membered ring in comparison to indoles. As such, azaindole-containing building blocks have attracted medicinal chemists to derive a number of pharmacological agents [20]. Among these, 7-azaindole 20 has appeared in a plethora of biologically active molecules [21], such as potent anticancer natural products, variolins [22] and their synthetic derivatives, meriolins [23]. To this end, we tested commercially available 7-azaindole 20 as the starting material in our biotransformation system. Again, the formation of aza-tryptophan 25, azaindole-3-pyruvate 30 and azaindole-containing acyloin 35 were observed in the corresponding PfTrpB6, PfTrpB6 + LAAO and PfTrpB6 + LAAO +NzsH systems, respectively, as evidenced in our LC-MS and MS tandem analyses (Table 1 and Supplementary Figures S9, S14 and S19). Taken together, NzsH display considerable substrate tolerance toward modified benzene rings of indole moieties when indole-3-pyruvate derivatives were used as substrates.
Next, we acquired three commercially available indole derivatives, 2-methyl-indole 36, indoline 37, and indazole 38 that contain modified indole rings (Figure 4). Consistent with the previous report [15,16,17], the biotransformation catalysed by PfTrpB6 gave the corresponding tryptophan derivatives, 39–41, respectively. In all three cases, addition of LAAO provided the corresponding indole-pyruvate derivatives, 42–44, respectively, further demonstrating the considerable substrate promiscuity of LAAO. To our surprise, these indole-pyruvate derivatives can be further utilized as the donor substrates in the NzsH-mediated enzymatic reaction to provide the final indole-containing acyloins, 45–47, respectively, as observed in our LC-MS and tandem MS analyses of the corresponding reduced forms (Supplementary Figure S20–S28). Taken together, our studies demonstrated that NzsH, unlike its activities towards acceptor substrates, displays considerable substrate tolerances toward its donor substrates. However, further studies are required to improve the reactivities of NzsH enzyme toward the unnatural indole-3-pyruvate derivatives. This can be achieved via either identification of new NzsH homologues through comparative genomics or directed evolution to provide an engineered NzsH with the aim of finding a suitable biocatalyst with better kinetics in order to efficiently generate structurally diverse indole-containing acyloin derivatives.
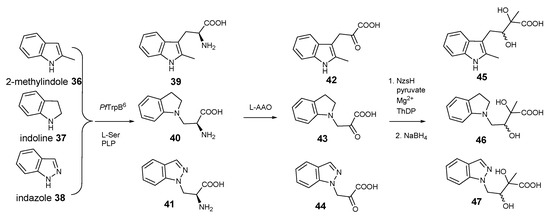
Figure 4.
Coupled reactions yielding 2-methyl-indole, indoline and indazole-containing acyloins 45–47, respectively, starting from three commercially available indole derivatives 36–38.
In conclusion, we developed a coupled biotransformation system including a genetically modified β-subunit of tryptophan synthase (PfTrpB6) from Pyrococcus furiosus, and a commercially available L-amino acid oxidase (LAAO) from snake venom and the ThDP-dependent NzsH enzyme from the biosynthetic pathway of neocarazostatins to access structurally diverse (aza)indole-containing acyloin derivatives. The combination of PfTrpB6 and LAAO allowed generation of (aza)indole-3-pyruvate derivatives, starting from commercially available (aza)indole derivatives, which were subsequently accessed by NzsH-mediated reaction. Our results demonstrated that NzsH displays considerably good substrate tolerance, suggesting that this three-enzyme coupled system holds potential as a means of new biotransformation to produce indole-containing acyloins, important precursors of many biologically relevant molecules.
3. Methods and Materials
3.1. General Chemicals, Reagents, and Analytical Methods
All starting materials and reagents were bought from commercial sources and used as received. All biochemical reactions apart from where noted were carried out in triplicate. Before every set of measurements, triplicate control reactions were performed to ensure that the assay were functioning correctly. All flash column chromatography was carried out using silica purchased from Sigma Aldrich using the solvent system noted. 19F NMR spectra were recorded at 298 K on Bruker Avance III 400 using CFCl3 as an external reference. Chemical shifts are reported in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz).
Enzymatic assays were analyzed on a Bruker MaXis II ESI-Q-TOF-MS connected to an Agilent 1290 Infinity II UHPLC fitted with a Phenomenex Kinetex XB-C18 (2.6 μM, 100 × 2.1 mm) column. The column was eluted with a linear gradient of 5–100% MeCN containing 0.1% formic acid over 15 min. The mass spectrometer was operated in positive ion mode with a scan range of 200–3000 m/z. Source conditions were: end plate offset at −500 V; capillary at −4500 V; nebulizer gas (N2) at 4.0 bar; dry gas (N2) at 9.0 L min−1; dry temperature at 200 °C. Ion transfer conditions were: ion funnel RF at 400 Vpp; multiple RF at 200 Vpp; quadrupole low mass at 200 m/z; collision energy at 8.0 eV; collision RF at 2000 Vpp; transfer time at 110.0 μs; pre-pulse storage time at 10.0 μs. MS data were analysed using Bruker DataAnalysis or Thermo Xcalibur.
3.2. General Methods of Protein Expression and Purification
The synthetic constructs encoding PfTrpB6 and NzsH proteins were purchased from Genscript Ltd. and were individually transformed into E. coli BL21 (DE3). Single colonies from each transformation were grown overnight in LB media (5 mL) containing kanamycin (50 µg/mL) and chloramphenicol (25 µg/mL). The overnight culture was transferred to fresh LB medium (500 mL) supplemented with kanamycin (50 µg/mL) and cultivated at 37 °C until the cell density reached an OD600 of 0.6. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM to induce protein expression. Cells were grown for 16–20 h at 16 °C and then harvested by centrifugation at 4 °C. The cells pellets were resuspended in ice-cold lysis buffer (20 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole, pH 8.0), and further disrupted by Ultrasonic Homogenizer JY92-IIN. Then, the supernatant of cell debris was loaded onto Ni-NTA-affinity column. Bound proteins were eluted with the same Tris-HCl buffer containing different concentrations of imidazole. The desired elution fractions were combined and concentrated using a Centrifugal Filter Unit (Millipore). The final yields of PfTrpB6 and NzsH were estimated to be 10 mg/100 mL culture and 5 mg/100 mL culture, respectively.
3.3. Biochemical Reactions
A sample of PfTrpB6 (20 µM) was incubated with indole or indole derivatives (1 mM), L-Ser (1 mM) and PLP (1 mM) in phosphate buffer (50 mM, pH 7.5) to the final volume of 100 µL at 28 °C for 3 h and then quenched by addition of 100 µL of acetonitrile. The mixture was centrifuged at 13,000 rpm for 10 min to remove protein precipitates.
For the production of indole-3-pyruvate derivatives, a sample of PfTrpB6 (20 µM) was incubated with indole or indole derivatives (1 mM), L-Ser (1 mM) and PLP (1 mM) in phosphate buffer (50 mM, pH 7.5) to a final volume of 50 µL at 28 °C for 3 h. To this mixture were added LAAO (20 µM) to a final volume of 100 µL at 28 °C for another 1 h. The reaction mix was then quenched by addition of 100 µL of acetonitrile. The mixture was centrifuged at 13,000 rpm for 10 min to remove protein precipitates.
For the production of indole-containing acyloin derivatives, a sample of PfTrpB6 (20 µM) was incubated with indole or indole derivatives (1 mM), L-Ser (1 mM) and PLP (1 mM) in phosphate buffer (50 mM, pH 7.5) to a final volume of 100 µL at 28 °C for 3 h. To this mixture were added LAAO (20 µM) to a volume of 100 µL at 28 °C for another 1 h. Inclusion of NzsH (25 µM), TPP (1 mM) and Mg2+ (1 mM) into the mixture was perform to the final volume of 100 µL at 28 °C for 3 h. Because of the instability of acyloins, overdose NaBH4 was treated to the three-enzyme systems. Finally, the reaction mix was quenched by addition of 100 µL of acetonitrile. The mixture was centrifuged at 13,000 rpm for 10 min to remove protein precipitates.
All the supernatants from the above reaction systems were then analyzed by Bruker MaXis II QTOF in tandem with an Agilent 1290 Infinity UHPLC. Samples were separated on a Phenomenex Kinetex XB-C18 (2.6 μM, 100 × 2.1 mm) column with a mobile phase of 5% ACN + 0.1% formic acid to 100% ACN + 0.1% formic acid in 15 min.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28010354/s1, Figure S1 SDS Page electrophoresis analysis of purifies PfTrpB6(A) and NzsH(B); Figure S2. LC-MS (A) and MS/MS data (B) of L-tryptophan 13.;Figure S3. LC-MS (A) and MS/MS data (B) of indole-3-pyruvate 14. Figure S4. LC-MS (A) and MS/MS data(B) of indole-containing acyloin 15.Figure S5. LC-MS (A) and MS/MS data (B) of 4-fluoro-indole-tryptophan 21. Figure S6. LC-MS (A) and MS/MS data (B) of 5-fluoro-indole-tryptophan 22. Figure S7. LC-MS (A) and MS/MS data (B) of 4-bromo-indole-tryptophan 23. Figure S8. LC-MS (A) and MS/MS data (B) of 6-hydroxyl-tryptophan 24.Figure S9. LC-MS (A) and MS/MS data (B) of 7-azaindole-tryptophan 25.Figure S10. LC-MS (A) and MS/MS data (B) of 4-fluoro-3-indole pyruvate 26; Figure S11. LC-MS (A) and MS/MS data (B) of 5-fluoro-3-indole pyruvate 27. Figure S12. LC-MS (A) and MS/MS data (B) of 4-bromo-3-indole pyruvate 28. Figure S13. LC-MS (A) and MS/MS data (B) of 6-hydroxyl-3-indole pyruvate 29. Figure S14. LC-MS (A) and MS/MS data (B) of 7-azaindole-3-pyruvate 30. Figure S15. LC-MS (A) and MS/MS data (B) of 4-fluoro-indole-containing acyloin 31. Figure S16. LC-MS (A) and MS/MS data (B) of 5-fluoro-indole-containing acyloin 32.Figure S17. LC-MS (A) and MS/MS data (B) of 4-bromo-indole-containing acyloin 33; Figure S18. LC-MS (A) and MS/MS data (B) of 6-hydroxyl-indole-containing acyloin 34.Figure S19. LC-MS (A) and MS/MS data (B) of 7-azaindole-containing-acyloin 35.Figure S20. LC-MS (A) and MS/MS data (B) of 2-methyl-indole-tryptophan 39.Figure S21. LC-MS (A) and MS/MS data (B) of indoline-tryptophan 40. Figure S22. LC-MS (A) and MS/MS data (B) of indazole-tryptophan 41. Figure S23. LC-MS (A) and MS/MS data (B) of 2-methyl-indole-3-pyruvate 42.Figure S24. LC-MS (A) and MS/MS data (B) of indoline-3-pyruvate 43.Figure S25. LC-MS (A) and MS/MS data (B) of indazole-3-pyruvate 44.Figure S26. LC-MS (A) and MS/MS data (B) of 2-methyl-indole-containing-acyloin 45.Figure S27. LC-MS (A) and MS/MS data (B) of indoline-containing-acyloin 46.Figure S28. LC-MS (A) and MS/MS data (B) of indazole-containing-acyloin 47.
Author Contributions
Conceptualization, H.D.; formal analysis, S.A., F.C. and A.A.; investigation, S.A.; writing—review and editing, H.D. and K.K.; All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the College of Science and Arts, Jouf University, King Khaled Road, Kingdom of Saudi Arabia, and the Royal Embassy of Saudi Arabia Cultural Bureau in the UK for the PhD scholarship (S.A.); the PhD studentship funded by IBioIC (F.C.), thank the financial supports of Leverhulme Trust-Royal Society Africa award (AA090088) and the jointly funded UK Medical Research Council UK Department for International Development (MRC/DFID) Concordat agreement African Research Leaders Award (MR/S00520X/1) (H.D. and K.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors after request.
References
- Lampis, G.; Deidda, D.; Maullu, C.; Madeddu, M.A.; Pompei, R.; Delle Monachie, F.; Satta, G. Sattabacins and sattazolins: New biologically active compounds with antiviral properties extracted from a Bacillus sp. J. Antibiot. 1995, 48, 967–972. [Google Scholar] [CrossRef]
- Li, J.; Chen, G.; Webster, J.M.; Czyzewska, E. Antimicrobial metabolites from a bacterial symbiont. J. Nat. Prod. 1995, 58, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Mancha, S.R.; Regnery, C.M.; Dahlke, J.R.; Miller, K.A.; Blake, D.J. Antiviral activity of (+)-sattabacin against varicella zoster. Bioorg. Med. Chem. Lett. 2013, 23, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Shiri, M. Indoles in multicomponent processes (MCPs). Chem. Rev. 2012, 112, 3508–3549. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Gocke, D.; Pohl, M. Thiamin diphosphate in biological chemistry: Exploitation of diverse thiamin diphosphate-dependent enzymes for asymmetric chemoenzymatic synthesis. FEBS J. 2009, 279, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 1797, 833. [Google Scholar] [CrossRef]
- Balskus, E.P.; Walsh, C.T. Investigating the initial steps in the biosynthesis of cyanobacterial sunscreen scytonemin. J. Am. Chem. Soc. 2008, 130, 15260–15261. [Google Scholar] [CrossRef]
- Huang, S.; Elsayed, S.S.; Lv, M.; Tabudravu, J.; Rateb, M.E.; Gyampoh, R.; Kyeremeh, K.; Ebel, R.; Jaspars, M.; Deng, Z.; et al. Biosynthesis of Neocarazostatin A Reveals the Sequential Carbazole Prenylation and Hydroxylation in the Tailoring Steps. Chem. Biol. 2015, 22, 1633–1642. [Google Scholar] [CrossRef]
- Su, L.; Lv, M.; Kyeremeh, K.; Deng, Z.; Deng, H.; Yu, Y. A ThDP-dependent enzymatic carboligation reaction involved in Neocarazostatin A tricyclic carbazole formation. Org. Biomol. Chem. 2016, 14, 8679–8684. [Google Scholar] [CrossRef]
- Su, L.; Zhang, R.; Kyeremeh, K.; Deng, Z.; Deng, H.; Yu, Y. Dissection of the neocarazostatin: A C4 alkyl side chain biosynthesis by in vitro reconstitution. Org. Biomol. Chem. 2017, 15, 3843–3848. [Google Scholar] [CrossRef]
- Liu, Y.; Su, L.; Fang, Q.; Tabudravu, J.; Yang, X.; Rickaby, K.; Trembleau, L.; Kyeremeh, K.; Deng, Z.; Deng, H.; et al. Enzymatic Reconstitution and Biosynthetic Investigation of the Bacterial Carbazole Neocarazostatin A. J. Org. Chem. 2019, 24, 16323–16328. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Tomita, T.; Shin-Ya, K.; Nishiyama, M.; Kuzuyama, T. An Unprecedented Cyclization Mechanism in the Biosynthesis of Carbazole Alkaloids in Streptomyces. Angew. Chem. Int. Ed. Engl. 2019, 58, 13349–13353. [Google Scholar] [CrossRef]
- Schieferdecker, S.; Shabuer, G.; Letzel, A.C.; Urbansky, B.; Ishida-Ito, M.; Ishida, K.; Cyrulies, M.; Dahse, H.M.; Pidot, S.; Hertweck, C. Biosynthesis of Diverse Antimicrobial and Antiproliferative Acyloins in Anaerobic Bacteria. ACS Chem. Biol. 2019, 114, 1490–1497. [Google Scholar] [CrossRef]
- Watkins-Dulaney, E.; Straathof, S.; Arnold, F. Tryptophan Synthase: Biocatalyst Extraordinaire. Chembiochem 2021, 22, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Buller, A.R.; Brinkmann-Chen, S.; Romney, D.K.; Herger, M.; Murciano-Calles, J.; Arnold, F.H. Directed evolution of the tryptophan synthase β-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci. USA. 2015, 112, 14599–14604. [Google Scholar] [CrossRef]
- Murciano-Calles, J.; Romney, D.K.; Brinkmann-Chen, S.; Buller, A.R.; Arnold, F.H. A Panel of TrpB Biocatalysts Derived from Tryptophan Synthase through the Transfer of Mutations that Mimic Allosteric Activation. Angew Chem. Int. Ed. Engl. 2016, 55, 11577–11581. [Google Scholar] [CrossRef] [PubMed]
- Boville, C.E.; Scheele, R.A.; Koch, P.; Brinkmann-Chen, S.; Buller, A.R.; Arnold, F.H. Engineered Biosynthesis of β-Alkyl Tryptophan Analogues. Angew Chem. Int. Ed. Engl. 2018, 57, 14764–14768. [Google Scholar] [CrossRef]
- Deng, H.; Cobb, S.L.; McEwan, A.R.; McGlinchey, R.P.; Naismith, J.H.; O'Hagan, D.; Robinson, D.A.; Spencer, J.B. The fluorinase from Streptomyces cattleya is also a chlorinase. Angew. Chem. Int. Ed. Engl. 2006, 45, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Motati, D.R.; Amaradhi, R.; Ganesh, T. Azaindole therapeutic agents. Bioorg. Med. Chem. 2020, 28, 115830. [Google Scholar] [CrossRef]
- Kim, K.S.; Zhang, L.; Schmidt, R.; Cai, Z.W.; Wei, D.; Williams, D.K.; Lombardo, L.J.; Trainor, G.L.; Xie, D.; Zhang, Y.; et al. Discovery of pyrrolopyridine-pyridone based inhibitors of Met kinase: Synthesis, X-ray crystallographic analysis, and biological activities. J. Med. Chem. 2008, 51, 5330–5341. [Google Scholar] [CrossRef]
- Irie, T.; Sawa, M. 7-Azaindole: A Versatile Scaffold for Developing Kinase Inhibitors. Chem. Pharm. Bull. 2018, 66, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Tirado, O.M.; Marionneau-Lambot, S.; Ferandin, Y.; Lozach, O.; Morris, J.C.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.H.; et al. Meriolins, a new class of cell death inducing kinase inhibitors with enhanced selectivity for cyclin-dependent kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clément, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.C.; Endicott, J.A.; et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): Synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef] [PubMed]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).