
Discovery of Bispecific Lead Compounds from Azadirachta indica against ZIKA NS2B-NS3 Protease and NS5 RNA Dependent RNA Polymerase Using Molecular Simulations

 , ,
, ,  ,
,  and
and 
Abstract
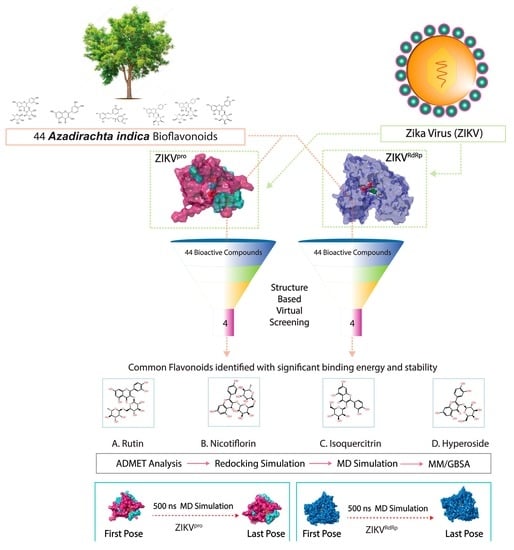
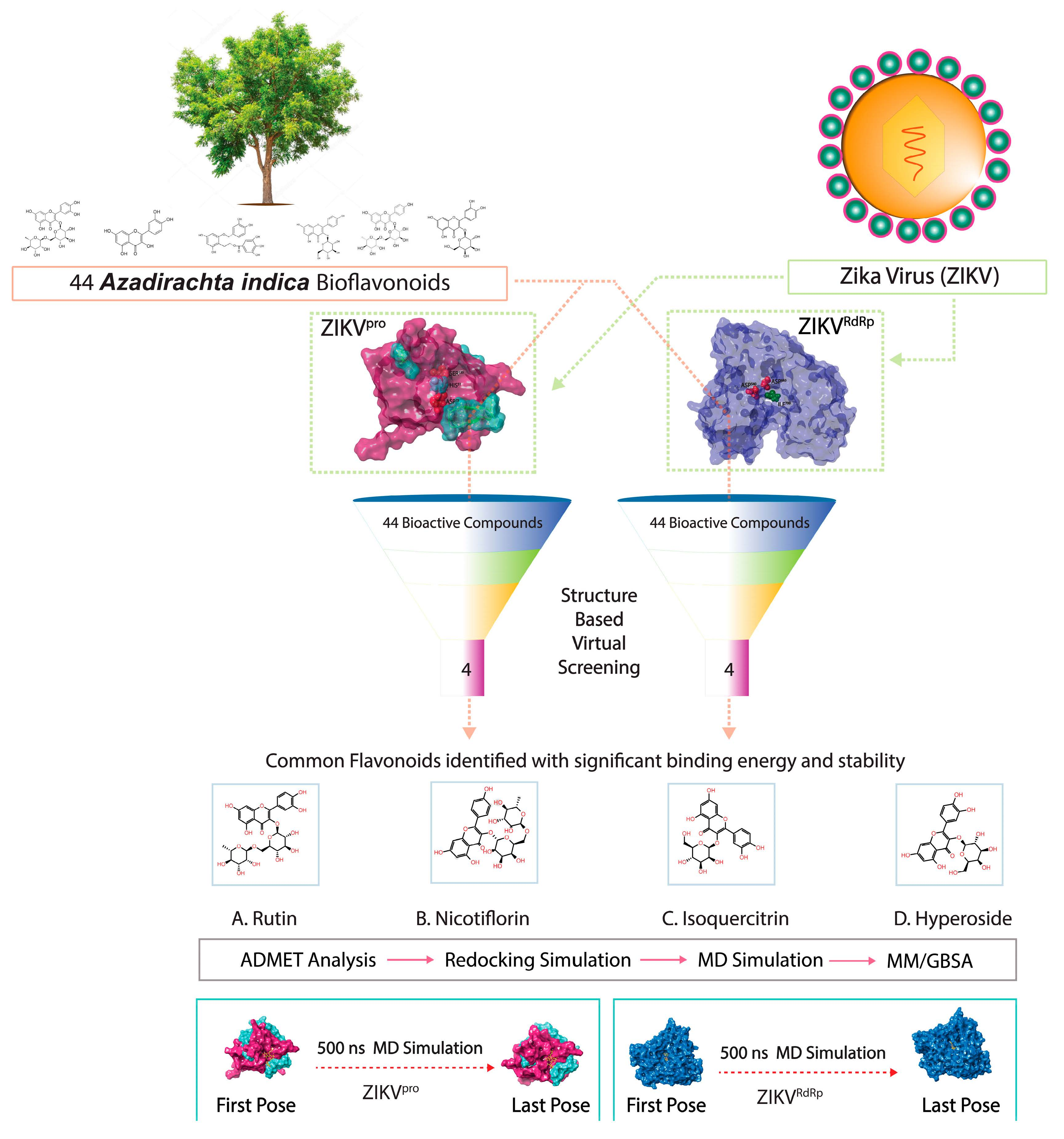
:
1. Introduction
2. Computational Methods
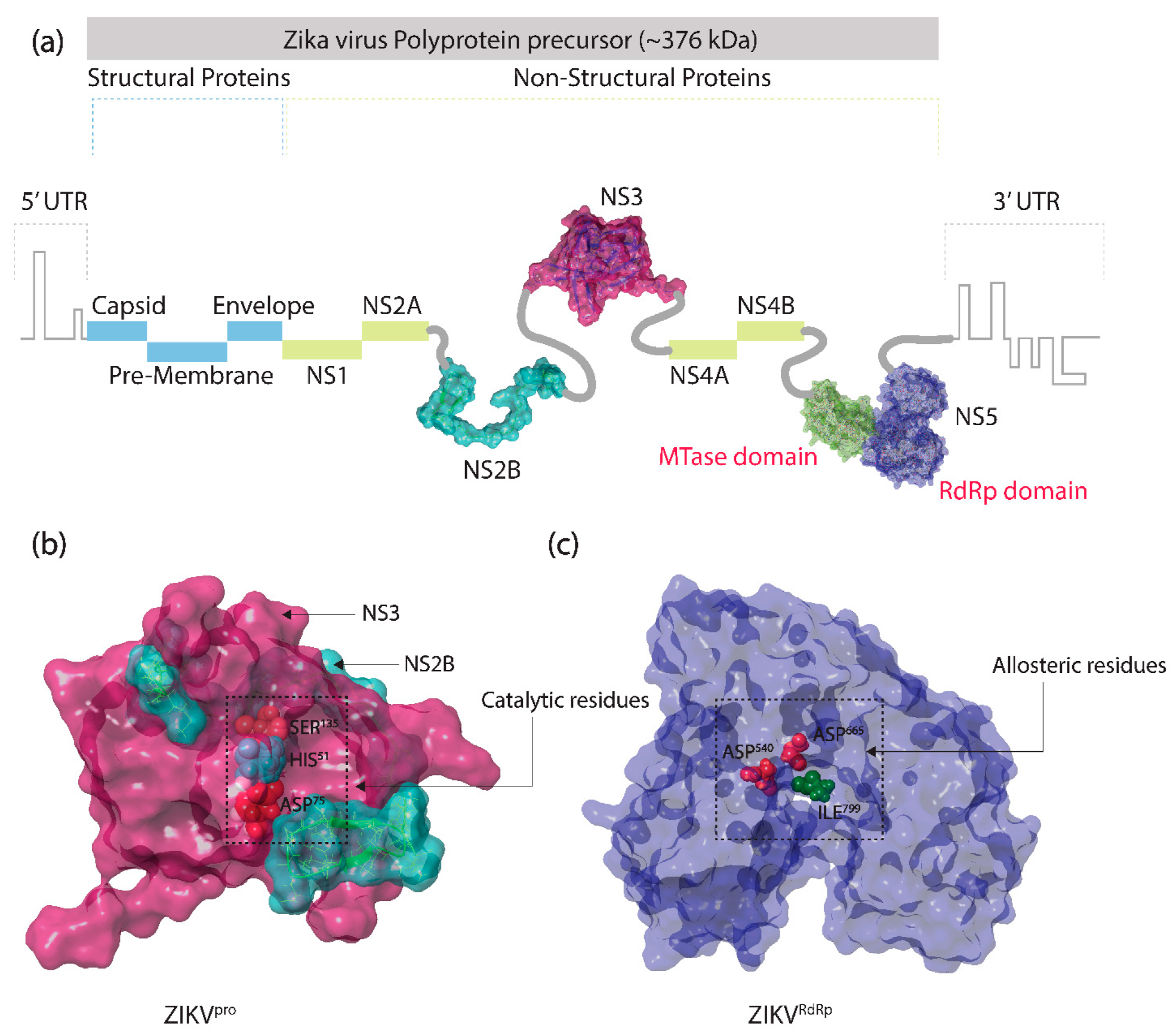
2.1. Receptors and Bioflavonoids
2.2. Structure-Based Virtual Screening and ADMET Analysis
2.3. Redocking and Intermolecular Interaction Profiling
2.4. Molecular Dynamics Simulation Analysis
2.5. Endpoint Free Binding Energy Calculation
3. Results and Discussion
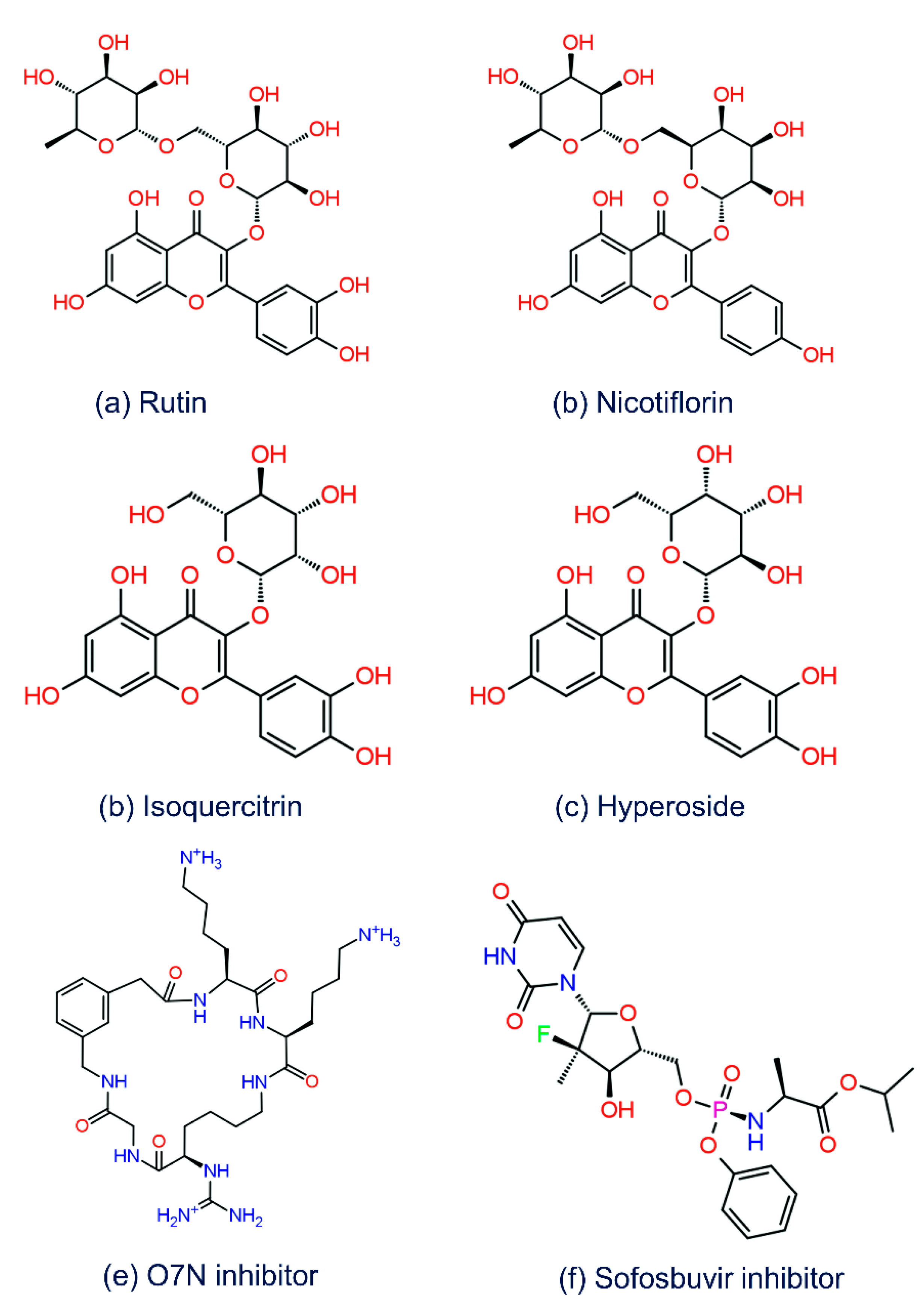
3.1. Structure-Based Virtual Screening
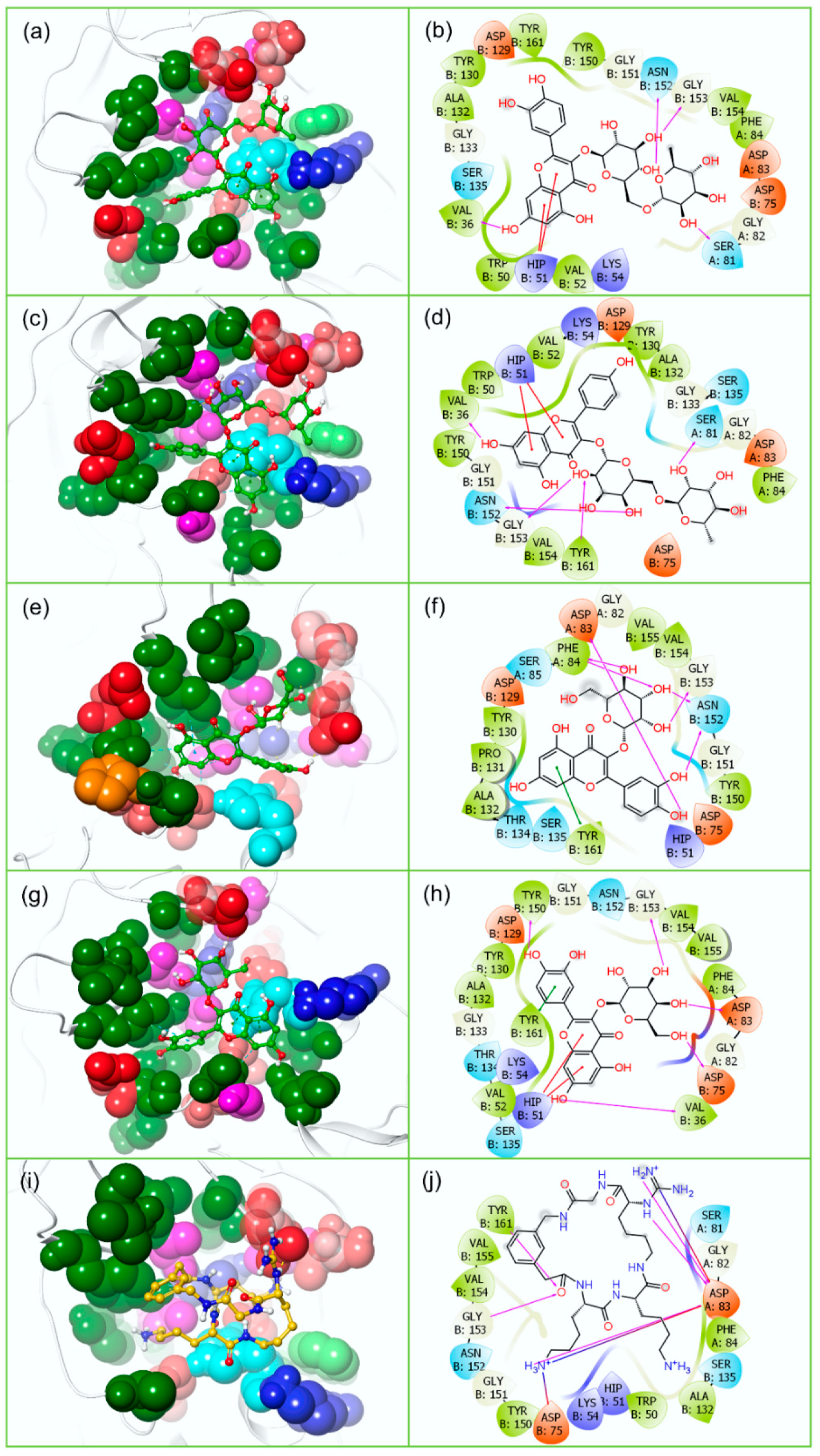
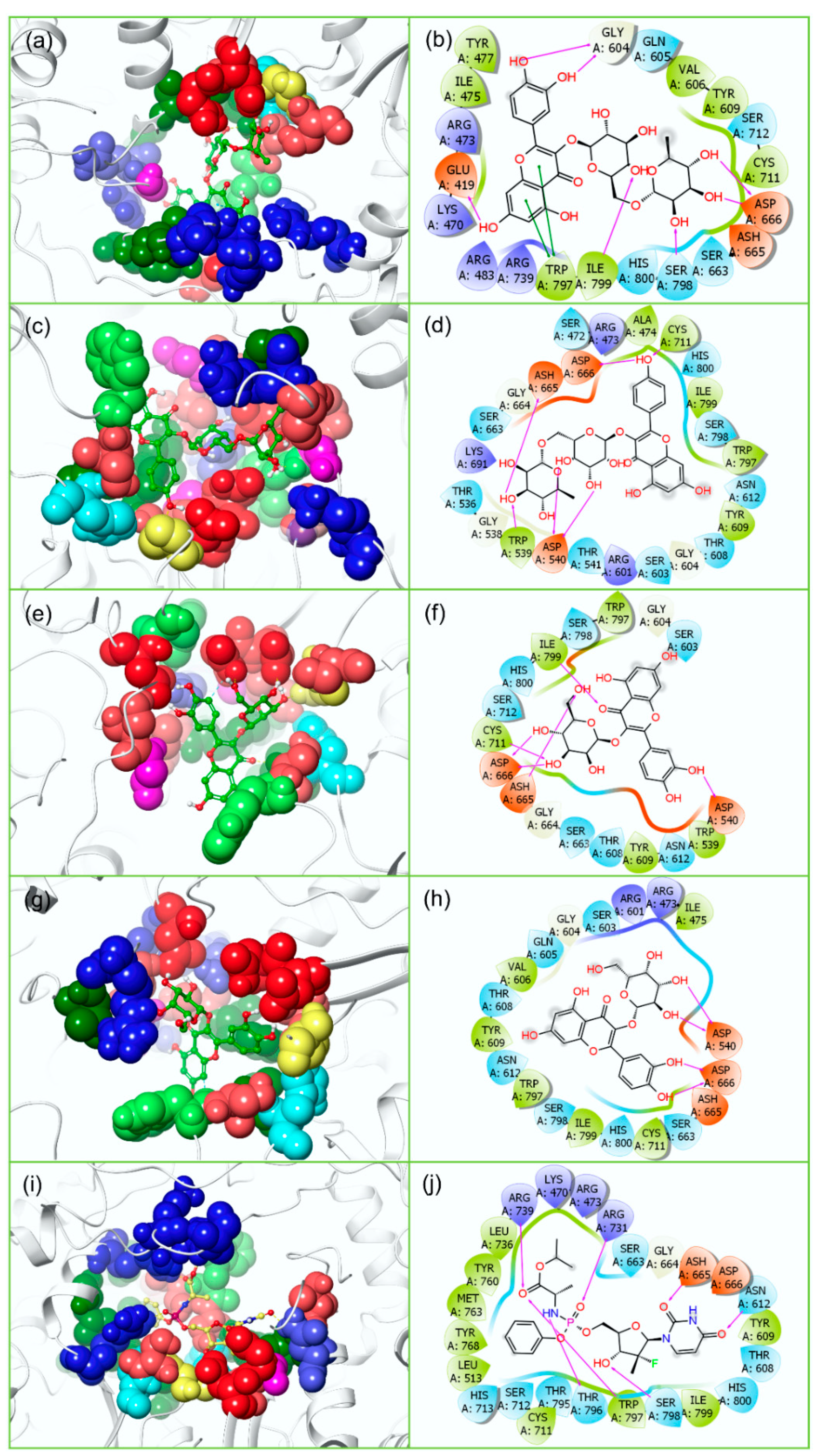
3.2. Redocking and Intermolecular Interaction Analysis
3.3. ADMET and Drug-Likeliness Analysis
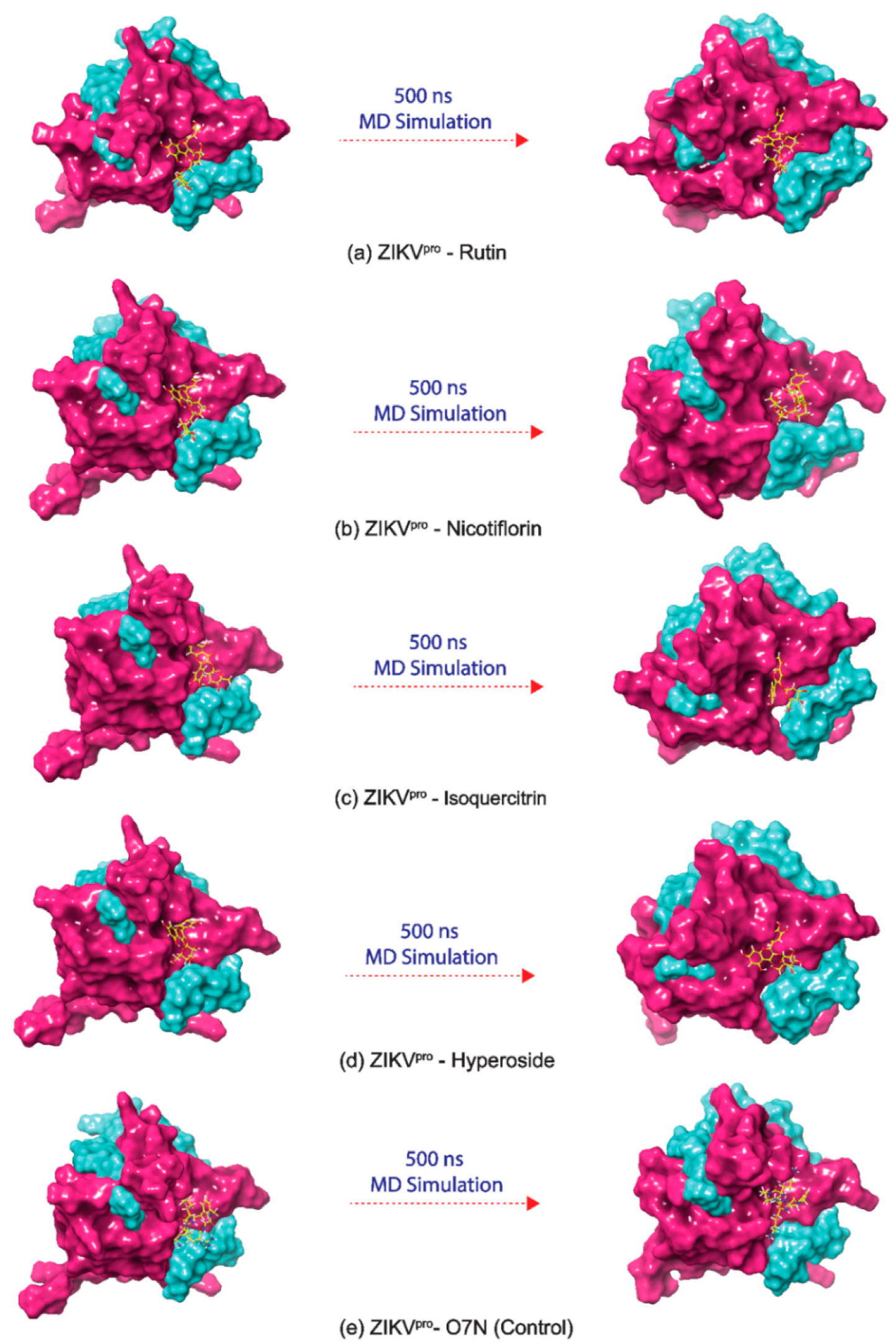
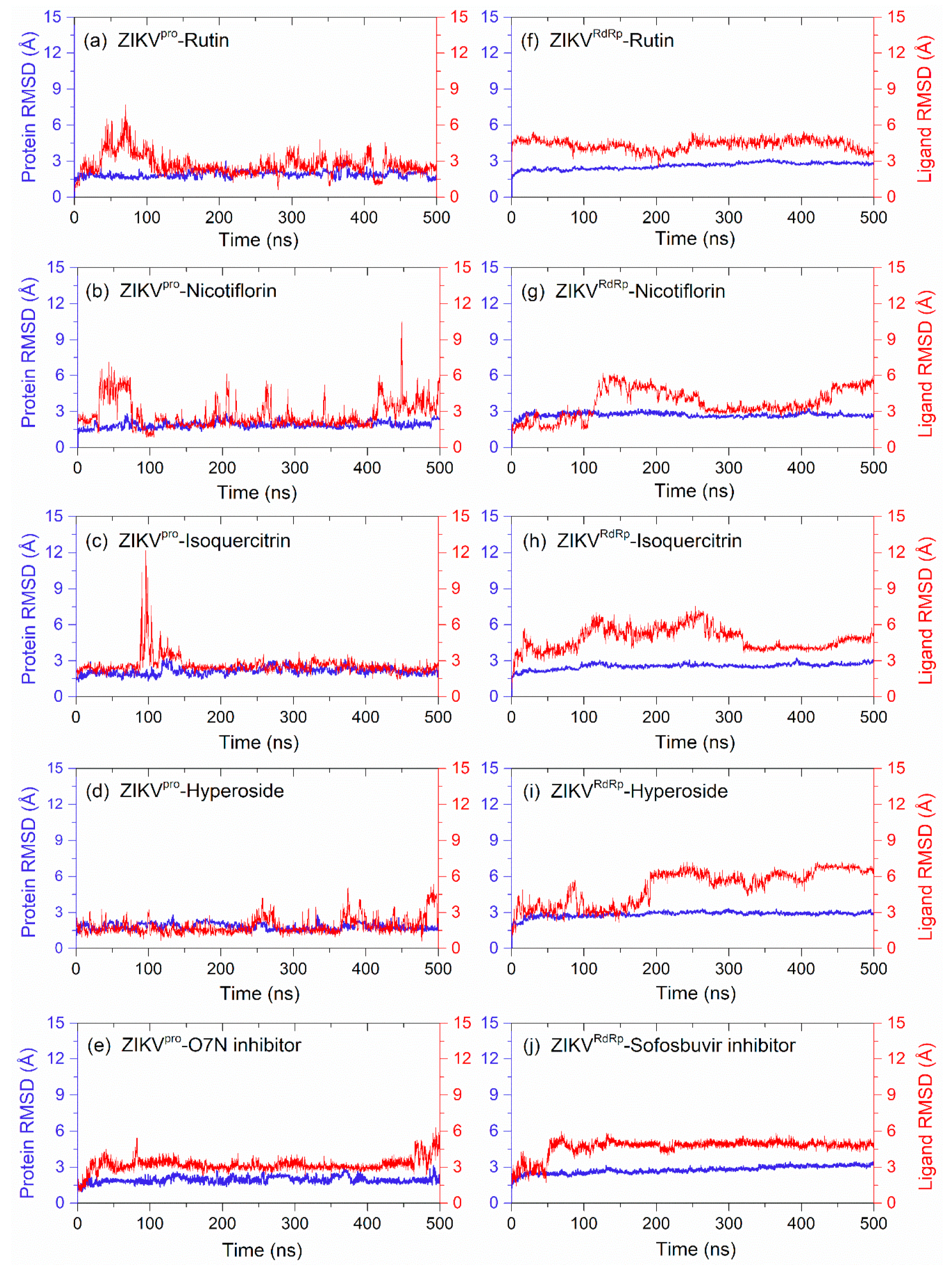
3.4. Long Interval Molecular Dynamics Simulation
3.4.1. RMSD and RMSF Analysis
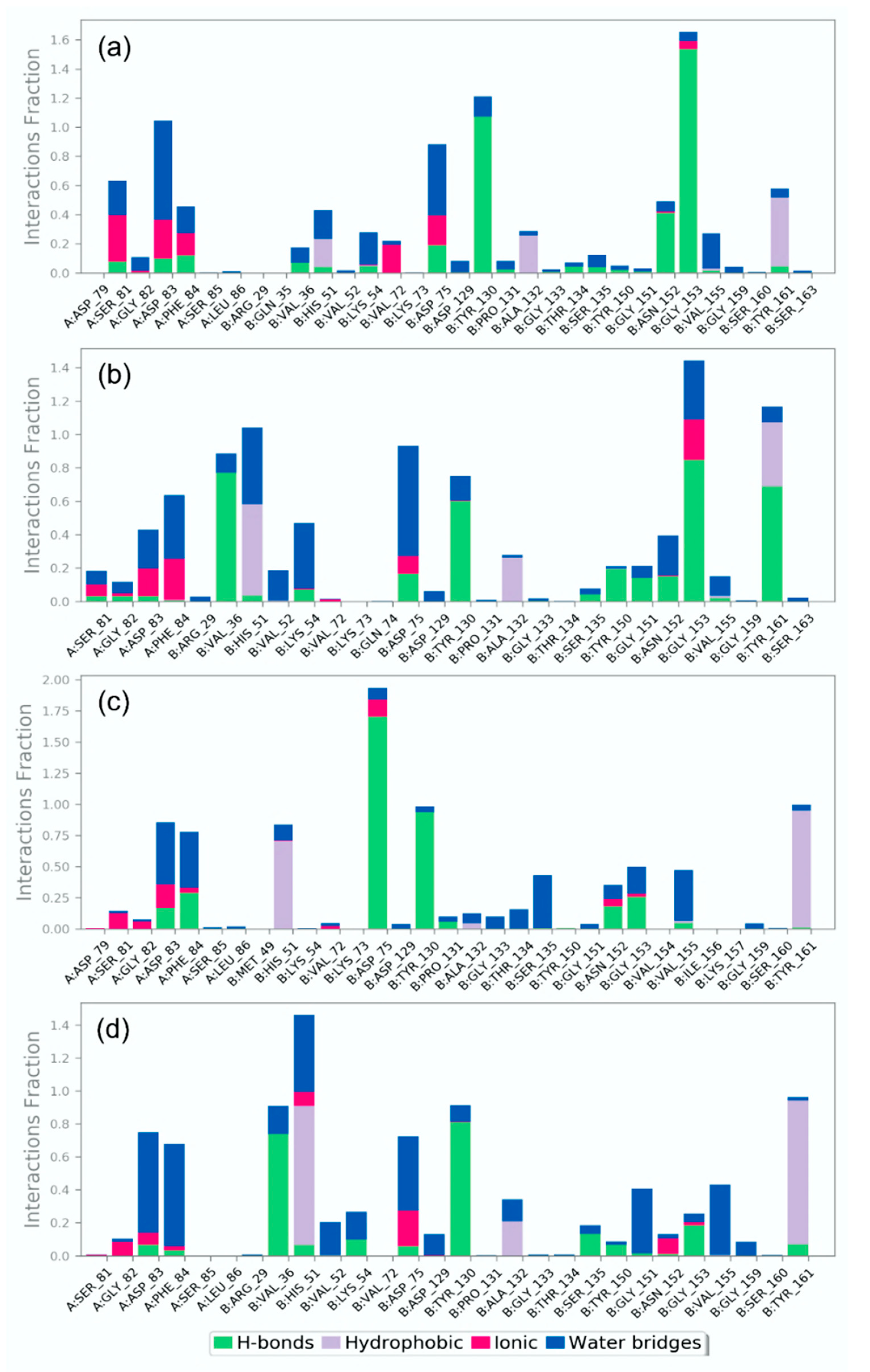
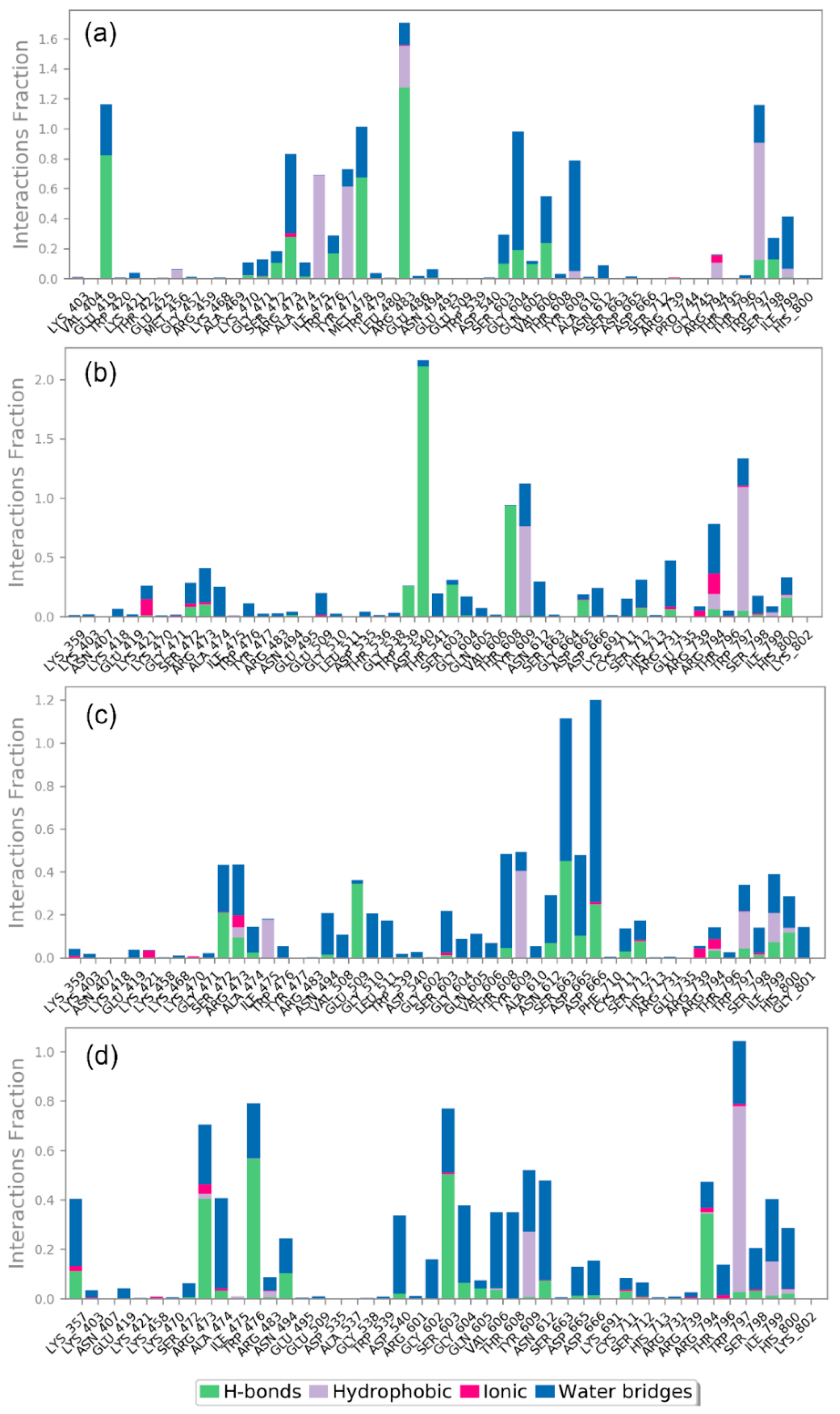
3.4.2. Protein-Ligand Interaction Profiling
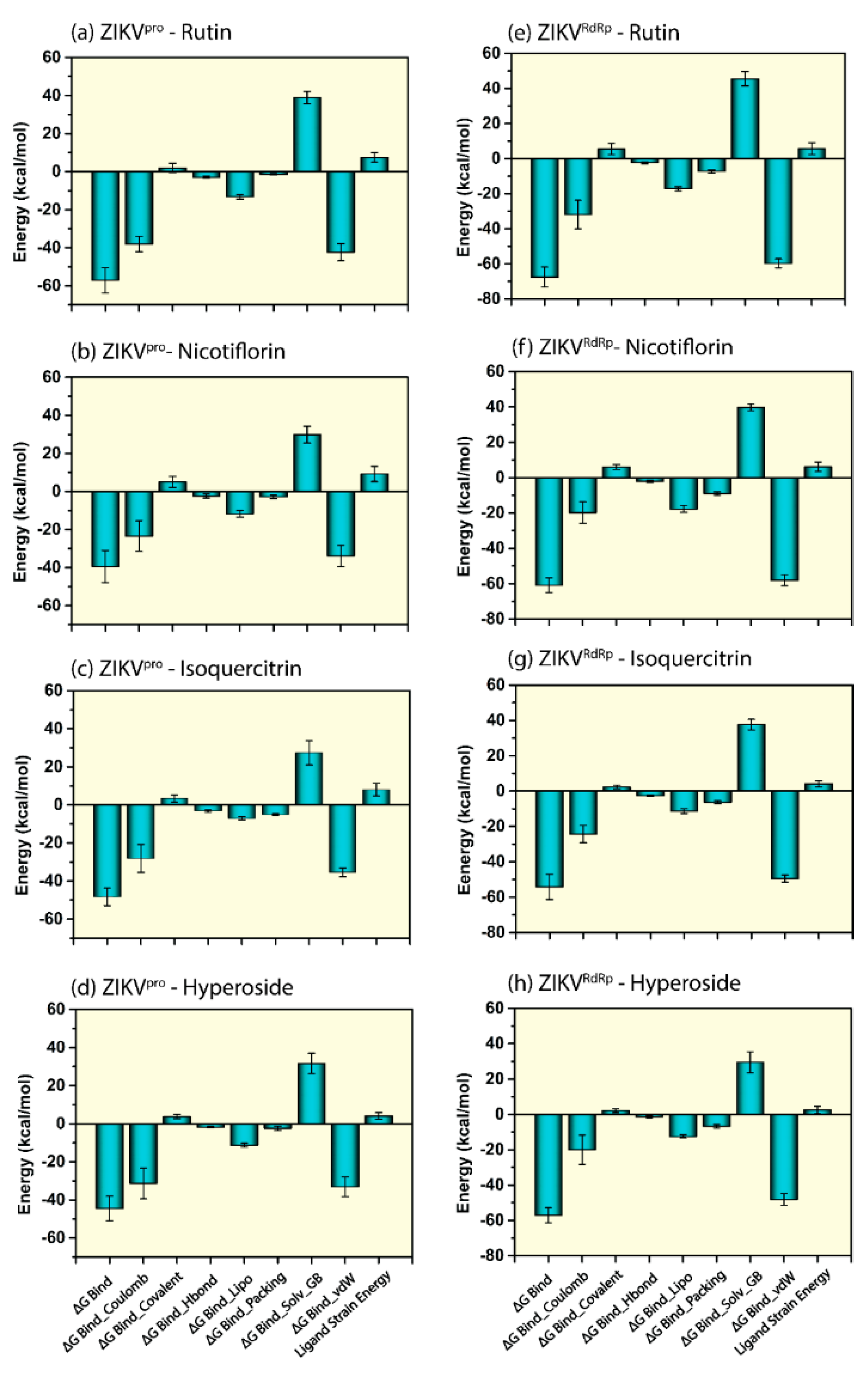
3.5. Endpoint Free Binding Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika Virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; DuBray, C.; et al. Zika Virus Outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Nilles, E.J.; Cao-Lormeau, V.-M. Rapid spread of emerging Zika virus in the Pacific area. Clin. Microbiol. Infect. 2014, 20, O595–O596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao-Lormeau, V.-M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.-L.; Mallet, H.-P.; Sall, A.A.; Musso, D. Zika Virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1084–1086. [Google Scholar] [CrossRef]
- Hennessey, M.; Fischer, M.; Staples, J.E. Zika Virus Spreads to New Areas—Region of the Americas, May 2015-January 2016. Am. J. Transplant. 2016, 16, 1031–1034. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Zika Virus, Microcephaly and Guillain–Barre’ Syndrome; WHO: Geneva, Switzerland, 2016; pp. 1–15. Available online: https://www.who.int/emergencies/zikavirus/situation-report/1-september-2016/en/ (accessed on 1 September 2016).
- Torres, J.R.; Murillo, J.; Bofill, L. The ever changing landscape of Zika virus infection. Learning on the fly. Int. J. Infect. Dis. 2016, 51, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Tavernise, S. CDC Investigating 14 New Reports of Zika Transmission Through Sex. The New York Times, 23 February 2016. [Google Scholar]
- Musso, D.; Roche, C.; Robin, E.; Nhan, T.; Teissier, A.; Cao-Lormeau, V.-M. Potential Sexual Transmission of Zika Virus. Emerg. Infect. Dis. 2015, 21, 359–361. [Google Scholar] [CrossRef]
- Vasilakis, N.; Weaver, S.C. Flavivirus transmission focusing on Zika. Curr. Opin. Virol. 2017, 22, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Driggers, R.; Ho, C.-Y.; Korhonen, E.; Kuivanen, S.; Jääskeläinen, A.; Smura, T.; Rosenberg, A.; Hill, D.; DeBiasi, R.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. Obstet. Anesthesia Dig. 2017, 37, 51. [Google Scholar] [CrossRef]
- Petersen, E.; Wilson, M.E.; Touch, S.; McCloskey, B.; Mwaba, P.; Bates, M.; Dar, O.; Mattes, F.; Kidd, M.; Ippolito, G.; et al. Rapid Spread of Zika Virus in The Americas—Implications for Public Health Preparedness for Mass Gatherings at the 2016 Brazil Olympic Games. Int. J. Infect. Dis. 2016, 44, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Nogueira, R.M.R.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.-A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.-J.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Appl. Microbiol. 2003, 59, 23–61. [Google Scholar] [CrossRef]
- Yu, I.-M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the Immature Dengue Virus at Low pH Primes Proteolytic Maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Thurmond, S.; Islas, L.; Hui, K.; Hai, R. Zika virus genome biology and molecular pathogenesis. Emerg. Microbes Infect. 2017, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chappell, K.J.; Stoermer, M.; Fairlie, D.; Young, P. West Nile Virus NS2B/NS3 Protease as An Antiviral Target. Curr. Med. Chem. 2008, 15, 2771–2784. [Google Scholar] [CrossRef]
- Yildiz, M.; Ghosh, S.; Bell, J.A.; Sherman, W.; Hardy, J.A. Allosteric Inhibition of the NS2B-NS3 Protease from Dengue Virus. ACS Chem. Biol. 2013, 8, 2744–2752. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, S.A.; Kozlov, I.A.; Ratnikov, B.I.; Smith, J.W.; Lebl, M.; Strongin, A.Y. Cleavage preference distinguishes the two-component NS2B–NS3 serine proteinases of Dengue and West Nile viruses. Biochem. J. 2007, 401, 743–752. [Google Scholar] [CrossRef]
- Leung, D.; Schroder, K.; White, H.; Fang, N.-X.; Stoermer, M.; Abbenante, G.; Martin, J.L.; Young, P.; Fairlie, D. Activity of Recombinant Dengue 2 Virus NS3 Protease in the Presence of a Truncated NS2B Co-factor, Small Peptide Substrates, and Inhibitors. J. Biol. Chem. 2001, 276, 45762–45771. [Google Scholar] [CrossRef] [Green Version]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Yusof, R.; Clum, S.; Wetzel, M.; Murthy, H.M.K.; Padmanabhan, R. Purified NS2B/NS3 Serine Protease of Dengue Virus Type 2 Exhibits Cofactor NS2B Dependence for Cleavage of Substrates with Dibasic Amino Acids in Vitro. J. Biol. Chem. 2000, 275, 9963–9969. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Loh, Y.R.; Hung, A.W.; Kang, C. Characterization of molecular interactions between Zika virus protease and peptides derived from the C-terminus of NS2B. Biochem. Biophys. Res. Commun. 2018, 503, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Clum, S.; Ebner, K.E.; Padmanabhan, R. Cotranslational Membrane Insertion of the Serine Proteinase Precursor NS2B-NS3(Pro) of Dengue Virus Type 2 Is Required for Efficient in Vitro Processing and Is Mediated through the Hydrophobic Regions of NS2B. J. Biol. Chem. 1997, 272, 30715–30723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktepe, T.E.; Mackenzie, J.M. Shaping the flavivirus replication complex: It is curvaceous! Cell. Microbiol. 2018, 20, e12884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescar, J.; Soh, S.; Lee, L.T.; Vasudevan, S.G.; Kang, C.; Lim, S.P. The Dengue Virus Replication Complex: From RNA Replication to Protein-Protein Interactions to Evasion of Innate Immunity. In BT—Dengue and Zika: Control and Antiviral Treatment Strategies; Hilgenfeld, R., Vasudevan, S.G., Eds.; Springer: Singapore, 2018; pp. 115–129. [Google Scholar] [CrossRef]
- Ngo, A.M.; Shurtleff, M.J.; Popova, K.D.; Kulsuptrakul, J.; Weissman, J.S.; Puschnik, A.S. The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. eLife 2019, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Li, R.; Yang, H.Y.; Jansson, A.E.; Hill, J.; Keller, T.; Nacro, K.; et al. Structural Insights into the Inhibition of Zika Virus NS2B-NS3 Protease by a Small-Molecule Inhibitor. Structure 2018, 26, 555–564.e3. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Wang, W.; Liu, S.; Chen, M.W.; Hung, A.W.; Keller, T.H.; Luo, D.; et al. Structural Dynamics of Zika Virus NS2B-NS3 Protease Binding to Dipeptide Inhibitors. Structure 2017, 25, 1242–1250.e3. [Google Scholar] [CrossRef] [Green Version]
- Quek, J.P.; Liu, S.; Zhang, Z.; Li, Y.; Ng, E.Y.; Loh, Y.R.; Hung, A.W.; Luo, D.; Kang, C. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antivir. Res. 2020, 175, 104707. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Phoo, W.W.; Loh, Y.R.; Zhang, Z.; Ng, E.Y.; Wang, W.; Keller, T.H.; Luo, D.; Kang, C. Structural characterization of the linked NS2B-NS3 protease of Zika virus. FEBS Lett. 2017, 591, 2338–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Kang, C. Structure and Dynamics of Zika Virus Protease and Its Insights into Inhibitor Design. Biomedicines 2021, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R.; Lei, J.; Zhang, L. The Structure of the Zika Virus Protease, NS2B/NS3pro. Adv. Exp. Med. Biol. 2018, 1062, 131–145. [Google Scholar] [CrossRef]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.-C.; Vaughan, R.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Soh, T.S.; Lim, S.P.; Chung, K.Y.; Swaminathan, K.; Vasudevan, S.G.; Shi, P.-Y.; Lescar, J.; Luo, D. Molecular basis for specific viral RNA recognition and 2′-O-ribose methylation by the dengue virus nonstructural protein 5 (NS5). Proc. Natl. Acad. Sci. USA 2015, 112, 14834–14839. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.K.; Cyr, M.; Longenecker, K.; Tripathi, R.; Sun, C.; Kempf, D.J. Crystal structure of full-lengthZika virusNS5 protein reveals a conformation similar toJapanese encephalitis virusNS5. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.P.; Moraes, A.H. Zika virus proteins at an atomic scale: How does structural biology help us to understand and develop vaccines and drugs against Zika virus infection? J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, e20190013. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tan, X.-F.; Thurmond, S.; Zhang, Z.-M.; Lin, A.; Hai, R.; Song, J. The structure of Zika virus NS5 reveals a conserved domain conformation. Nat. Commun. 2017, 8, 14763. [Google Scholar] [CrossRef]
- Wang, B.; Thurmond, S.; Hai, R.; Song, J. Structure and function of Zika virus NS5 protein: Perspectives for drug design. Cell. Mol. Life Sci. 2018, 75, 1723–1736. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sakamuru, S.; Huang, R.; Brecher, M.; Koetzner, C.A.; Zhang, J.; Chen, H.; Qin, C.-F.; Zhang, Q.-Y.; Zhou, J.; et al. Erythrosin B is a potent and broad-spectrum orthosteric inhibitor of the flavivirus NS2B-NS3 protease. Antivir. Res. 2018, 150, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brecher, M.; Deng, Y.-Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; A Koetzner, C.; Allen, C.; A Jones, S.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecher, M.; Li, Z.; Liu, B.; Zhang, J.; Koetzner, C.A.; Alifarag, A.; Jones, S.A.; Lin, Q.; Kramer, L.D.; Li, H. A conformational switch high-throughput screening assay and allosteric inhi-bition of the flavivirus NS2B-NS3 protease. PLoS Pathog. 2017, 13, e1006411. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Passioura, T.; Varava, P.; Mahawaththa, M.C.; Leuthold, M.M.; Klein, C.D.; Suga, H.; Otting, G. De Novo Discovery of Nonstandard Macrocyclic Peptides as Noncompetitive Inhibitors of the Zika Virus NS2B-NS3 Protease. ACS Med. Chem. Lett. 2019, 10, 168–174. [Google Scholar] [CrossRef]
- Millies, B.; Von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-Based Allosteric Inhibitors of Zika and Dengue Virus NS2B/NS3 Proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C.; Zhang, L.; Weigel, L.F.; Schilz, J.; Graf, D.; Bartenschlager, R.; Hilgenfeld, R.; Klein, C.D. Peptide–Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. J. Med. Chem. 2017, 60, 511–516. [Google Scholar] [CrossRef]
- Ramharack, P.; Soliman, M.E. Zika virus NS5 protein potential inhibitors: An enhanced in silico approach in drug discovery. J. Biomol. Struct. Dyn. 2018, 36, 1118–1133. [Google Scholar] [CrossRef]
- Gharbi-Ayachi, A.; Santhanakrishnan, S.; Wong, Y.H.; Chan, K.W.K.; Tan, S.T.; Bates, R.W.; Vasudevan, S.G.; El Sahili, A.; Lescar, J. Non-nucleoside Inhibitors of Zika Virus RNA-Dependent RNA Polymerase. J. Virol. 2020, 94, 2287–2300. [Google Scholar] [CrossRef]
- Noreen; Ali, R.; Badshah, S.L.; Faheem, M.; Abbasi, S.W.; Ullah, R.; Bari, A.; Jamal, S.B.; Mahmood, H.M.; Haider, A.; et al. Identification of potential inhibitors of Zika virus NS5 RNA-dependent RNA polymerase through virtual screening and molecular dynamic simulations. Saudi Pharm. J. 2020, 28, 1580–1591. [Google Scholar] [CrossRef]
- Song, W.; Zhang, H.; Zhang, Y.; Chen, Y.; Lin, Y.; Han, Y.; Jiang, J. Identification and Characterization of Zika Virus NS5 Methyltransferase Inhibitors. Front. Cell. Infect. Microbiol. 2021, 11, 665379. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Bespalov, M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.W.; Sam, I.-C.; Chong, W.L.; Lee, V.S.; Chan, Y.F. Polysulfonate suramin inhibits Zika virus infection. Antivir. Res. 2017, 143, 186–194. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, J.F.-W.; Den-Haan, H.; Chik, K.K.-H.; Zhang, A.J.; Chan, C.C.-S.; Poon, V.K.-M.; Yip, C.C.-Y.; Mak, W.W.-N.; Zhu, Z.; et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antivir. Res. 2017, 145, 33–43. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.; Pooe, O.J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des. Dev. Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef]
- Li, Y.H.; Wang, P.; Li, X.X.; Yu, C.Y.; Yang, H.; Zhou, J.; Xue, W.; Tan, J.; Zhu, F. The Human Kinome Targeted by FDA Approved Multi-Target Drugs and Combination Products: A Comparative Study from the Drug-Target Interaction Network Perspective. PLoS ONE 2016, 11, e0165737. [Google Scholar] [CrossRef] [Green Version]
- Subapriya, R.; Nagini, S. Medicinal Properties of Neem Leaves: A Review. Curr. Med. Chem. Agents 2005, 5, 149–156. [Google Scholar] [CrossRef]
- Gupta, S.C.; Prasad, S.; Tyagi, A.K.; Kunnumakkara, A.B.; Aggarwal, B.B. Neem (Azadirachta indica): An indian traditional panacea with modern molecular basis. Phytomedicine 2017, 34, 14–20. [Google Scholar] [CrossRef]
- Yadav, D.K.; Bharitkar, Y.P.; Chatterjee, K.; Ghosh, M.; Mondal, N.B.; Swarnaka, S. Importance of Neem Leaf: An insight into its role in combating diseases. Indian J. Exp. Biol. 2016, 54, 708–718. [Google Scholar] [PubMed]
- Braun, N.J.; Quek, J.P.; Huber, S.; Kouretova, J.; Rogge, D.; Lang-Henkel, H.; Cheong, E.Z.K.; Chew, B.L.A.; Heine, A.; Luo, D.; et al. Structure-Based Macrocyclization of Substrate Analogue NS2B-NS3 Protease Inhibitors of Zika, West Nile and Dengue viruses. ChemMedChem 2020, 15, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2018-4: Protein Preparation Wizard;Epik; Schrödinger, LLC.: New York, NY, USA, 2018.
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Sacramento, C.Q.; de Melo, G.R.; de Freitas, C.S.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, H.; Song, W.; Si, S.; Han, Y.; Jiang, J. Identification and characterization of Zika virus NS5 RNA-dependent RNA polymerase inhibitors. Int. J. Antimicrob. Agents 2019, 54, 502–506. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sac-erdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Association for Computing Machinery, New York, NY, USA, 11 November 2006. [Google Scholar]
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System, D.E. Shaw Research: New York, NY, 2020; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020.
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef]
- Negahdari, R.; Bohlouli, S.; Sharifi, S.; Dizaj, S.M.; Saadat, Y.R.; Khezri, K.; Jafari, S.; Ahmadian, E.; Jahandizi, N.G.; Raeesi, S. Therapeutic benefits of rutin and its nanoformulations. Phytotherapy Res. 2021, 35, 1719–1738. [Google Scholar] [CrossRef]
- Caparica, R.; Júlio, A.; Araújo, M.E.M.; Baby, A.R.; Fonte, P.; Costa, J.G.; Santos de Almeida, T. Anticancer Activity of Rutin and Its Combination with Ionic Liquids on Renal Cells. Biomolecules 2020, 10, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budzynska, B.; Faggio, C.; Kruk-Slomka, M.; Samec, D.; Nabavi, S.F.; Sureda, A.; Devi, K.P.; Nabavi, S.M. Rutin as neuroprotective agent: From bench to bedside. Curr. Med. Chem. 2019, 26, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Rutin as a Natural Therapy for Alzheimer’s Disease: Insights into its Mechanisms of Action. Curr. Med. Chem. 2016, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Shaik, Y.B.; Castellani, M.L.; Perrella, A.; Conti, F.; Salini, V.; Tete, S.; Madhappan, B.; Vecchiet, J.; A De Lutiis, M.; Caraffa, A.; et al. Role of quercetin (a natural herbal compound) in allergy and inflammation. J. Boil. Regul. Homeost. Agents 2006, 20, 47–52. [Google Scholar]
- Thapa, M.; Kim, Y.; Desper, J.; Chang, K.-O.; Hua, D.H. Synthesis and antiviral activity of substituted quercetins. Bioorganic Med. Chem. Lett. 2012, 22, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savov, V.M.; Galabov, A.S.; Tantcheva, L.P.; Mileva, M.M.; Pavlova, E.L.; Stoeva, E.S.; Braykova, A.A. Effects of rutin and quercetin on monooxygenase activities in experimental influenza virus infection. Exp. Toxicol. Pathol. Off. J. Ges. Toxikol. Pathol. 2006, 58, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S.; Lentini, G. The therapeutic potential of rutin for diabetes: An update. Mini-Rev. Med. Chem. 2015, 15, 524–528. [Google Scholar] [CrossRef]
- Dwivedi, V.D.; Bharadwaj, S.; Afroz, S.; Khan, N.; Ansari, M.A.; Yadava, U.; Tripathi, R.C.; Tripathi, I.P.; Mishra, S.K.; Kang, S.G. Anti-dengue infectivity evaluation of bioflavonoid from Azadirachta indica by dengue virus serine protease inhibition. J. Biomol. Struct. Dyn. 2021, 39, 1417–1430. [Google Scholar] [CrossRef]
- Dubey, R.; Dubey, K. Molecular Docking Studies of Bioactive Nicotiflorin against 6W63 Novel Coronavirus 2019 (COVID-19). Comb. Chem. High Throughput Screen. 2021, 24, 874–878. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, P.; Xu, H. Hyperoside exhibits anticancer activity in non-small cell lung cancer cells with T790M mutations by upregulating FoxO1 via CCAT1. Oncol. Rep. 2020, 43, 617–624. [Google Scholar] [CrossRef]
- Wu, P.; Kong, Y.; Sun, W. Hyperoside exerts potent anticancer activity in skin cancer. Front. Biosci. 2020, 25, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.D.; Singh, A.; El-Kafraway, S.A.; Alandijany, T.A.; Faizo, A.A.; Bajrai, L.H.; Kamal, M.A.; Azhar, E.I. Mechanistic insights into the Japanese encephalitis virus RNA dependent RNA polymerase protein inhibition by bioflavonoids from Azadirachta indica. Sci. Rep. 2021, 11, 18125. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R. Critical review of the role of HTS in drug discovery. Drug Discov. Today 2006, 11, 277–279. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Cataneo, A.H.D.; Ávila, E.P.; Mendes, L.A.D.O.; de Oliveira, V.G.; Ferraz, C.R.; de Almeida, M.V.; Frabasile, S.; dos Santos, C.N.D.; Verri, W.A.J.; Bordignon, J.; et al. Flavonoids as Molecules with Anti-Zika virus Activity. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compounds | Redocking Score (kcal/mol) | H-Bond | * π-Cation Stacking/† π-π Stacking/‡ Salt Bridge | Hydrophobic | ||||
|---|---|---|---|---|---|---|---|---|---|
| ZIKVpro | ZIKVRdRp | ZIKVpro | ZIKVRdRp | ZIKVpro | ZIKVRdRp | ZIKVpro | ZIKVRdRp | ||
| 1. | Rutin | −10.645 | −11.038 | A:Ser81, B:Val36, B:Asn152, B:Gly153 | Glu419, Gly604, Asp666, Ser798, Ilu799 | * B:His51 | † Trp797 | A:Phe84, B:Val36, B:Trp50, B:Val52, B:Tyr130, B:Ala132, B:Tyr150, B:Val154, B:Tyr161 | Ile475, Tyr477, Val606, Tyr609, Cys711, Trp797, Ile799 |
| 2. | Nicotiflorin | −9.986 | −10.593 | A:Ser81, B:Val36, B:Asn152, B:Gly153, B:Tyr161 | Trp539, Asp540, Asp665, Asp666, Cys711 | * B:His51 | -- | A:Phe84, B:Val36, B:Trp50, B:Val52, B:Tyr130, B:Ala132, B:Tyr150, B:Val154, B:Tyr161 | Ala474, Trp539, Tyr609, Cys711, Trp797, Ile799 |
| 3. | Isoquercitrin | −8.666 | −8.877 | A:Asp83, A:Phe84, B:Asn152, B:Gly153 | Asp540, Asp665, Asp666, Cys711, Ilu799 | † B:Tyr161 | -- | A:Phe84, B:Tyr130, B:Pro131, B:Ala132, B:Tyr150, B:Val154, B:Val155, B:Tyr161 | Trp539, Tyr609, Cys711, Trp797, Ile799 |
| 4. | Hyperoside | −8.4 | −7.907 | A:Asp83, B:Val36, B:Asp75, B:Tyr150, B:Gly153 | Asp540, Asp666 | * B:His51, † B:Tyr161 | -- | A:Phe84, B:Val36, B:Val52, B:Tyr130, B:Ala132, B:Tyr150, B:Val154, B:Val155, B:Tyr161 | Ile475, Val606, Tyr609, Cys711, Trp797, Ile799 |
| 5. | O7N (ZIKVpro reference inhibitor) | −6.629 | -- | A:Asp83, B:Gly153, B:Tyr161 | -- | ‡ A:Asp83, ‡ B:Asp75 | -- | A:Phe84, B:Trp50, B:Ala132, B:Tyr150, B:Val154, B:Val155, B:Tyr161 | -- |
| 6. | Sofosbuvir (ZIKVRdRp reference inhibitor) | -- | −6.033 | -- | Asn612, Asp665, Arg731, Arg739, Thr796, Trp797, Ser798 | -- | -- | -- | Leu513, Tyr609, Cys711, Leu736, Tyr760, Met763, Tyr768, Trp797, Ile799 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; El-Kafrawy, S.A.; Bharadwaj, S.; Maitra, S.S.; Alandijany, T.A.; Faizo, A.A.; Khateb, A.M.; Dwivedi, V.D.; Azhar, E.I. Discovery of Bispecific Lead Compounds from Azadirachta indica against ZIKA NS2B-NS3 Protease and NS5 RNA Dependent RNA Polymerase Using Molecular Simulations. Molecules 2022, 27, 2562. https://doi.org/10.3390/molecules27082562
Kumar S, El-Kafrawy SA, Bharadwaj S, Maitra SS, Alandijany TA, Faizo AA, Khateb AM, Dwivedi VD, Azhar EI. Discovery of Bispecific Lead Compounds from Azadirachta indica against ZIKA NS2B-NS3 Protease and NS5 RNA Dependent RNA Polymerase Using Molecular Simulations. Molecules. 2022; 27(8):2562. https://doi.org/10.3390/molecules27082562
Chicago/Turabian StyleKumar, Sanjay, Sherif A. El-Kafrawy, Shiv Bharadwaj, S. S. Maitra, Thamir A. Alandijany, Arwa A. Faizo, Aiah M. Khateb, Vivek Dhar Dwivedi, and Esam I. Azhar. 2022. "Discovery of Bispecific Lead Compounds from Azadirachta indica against ZIKA NS2B-NS3 Protease and NS5 RNA Dependent RNA Polymerase Using Molecular Simulations" Molecules 27, no. 8: 2562. https://doi.org/10.3390/molecules27082562
APA StyleKumar, S., El-Kafrawy, S. A., Bharadwaj, S., Maitra, S. S., Alandijany, T. A., Faizo, A. A., Khateb, A. M., Dwivedi, V. D., & Azhar, E. I. (2022). Discovery of Bispecific Lead Compounds from Azadirachta indica against ZIKA NS2B-NS3 Protease and NS5 RNA Dependent RNA Polymerase Using Molecular Simulations. Molecules, 27(8), 2562. https://doi.org/10.3390/molecules27082562