
Computational Prediction and Experimental Validation of the Unique Molecular Mode of Action of Scoulerine


 and
and 
Abstract
:1. Introduction
2. Result and Discussion
2.1. Scoulerine in Cancer Cell
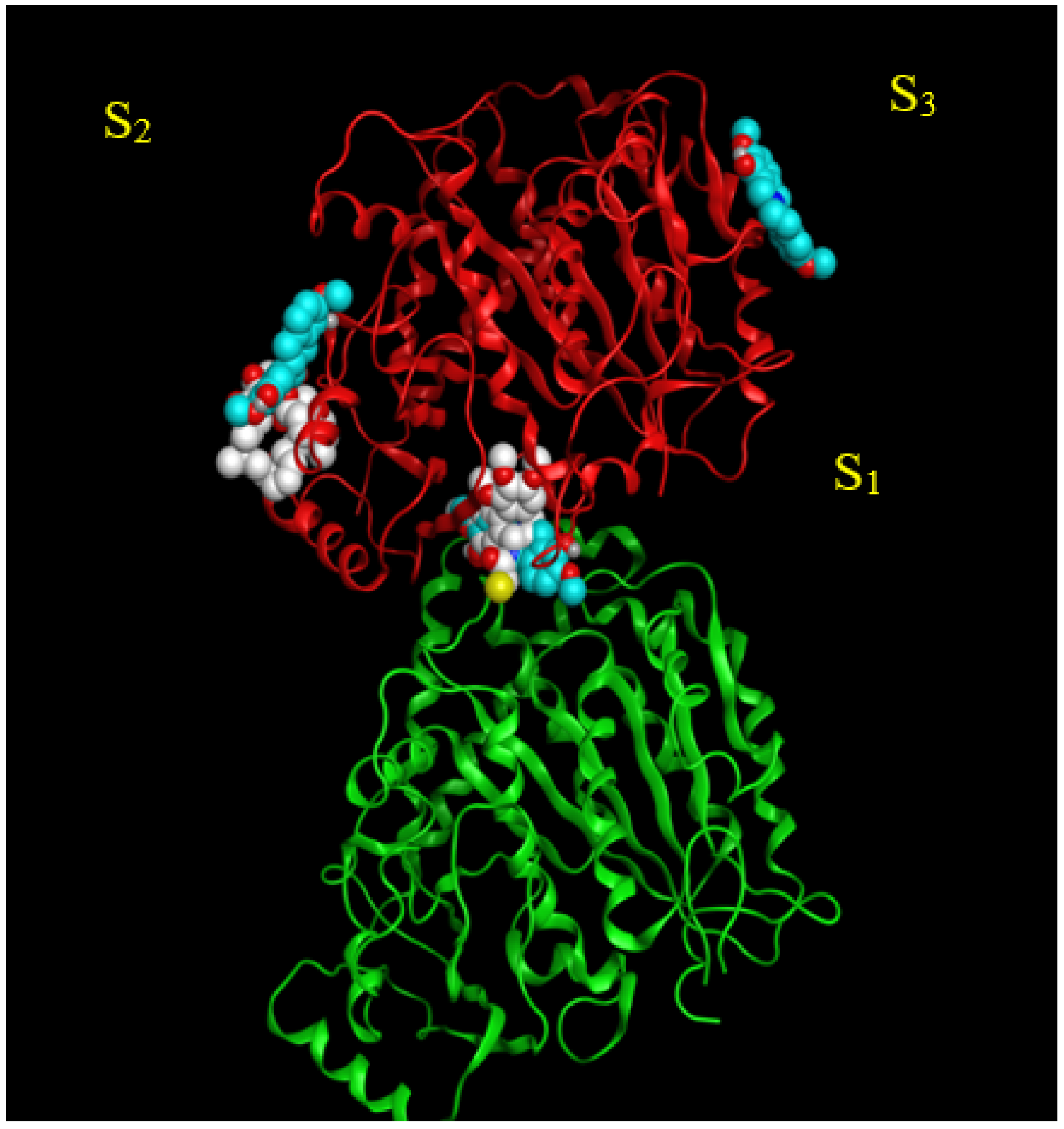
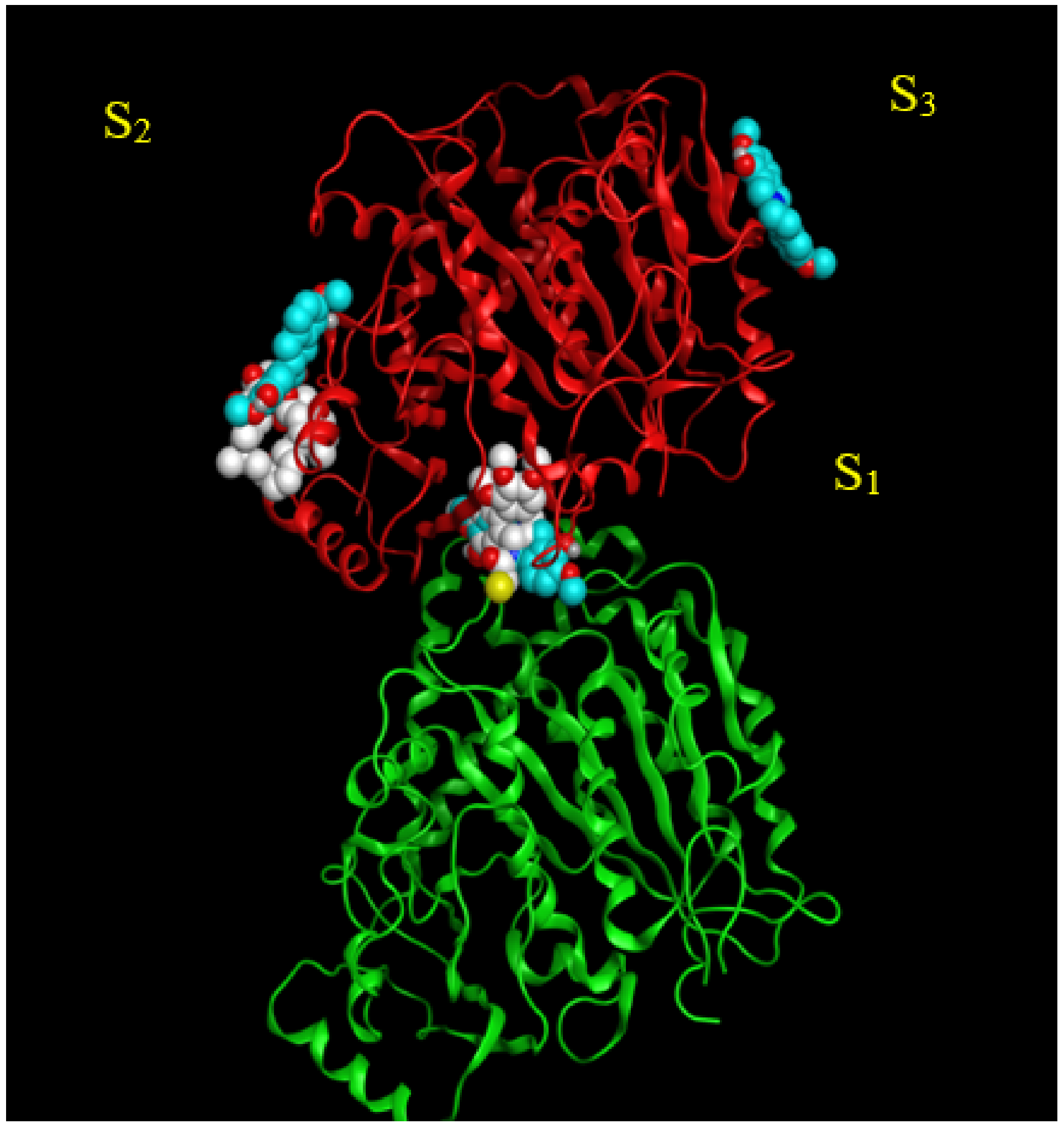
2.2. Analysis of Potential Scoulerine Binding Sites on β Tubulin
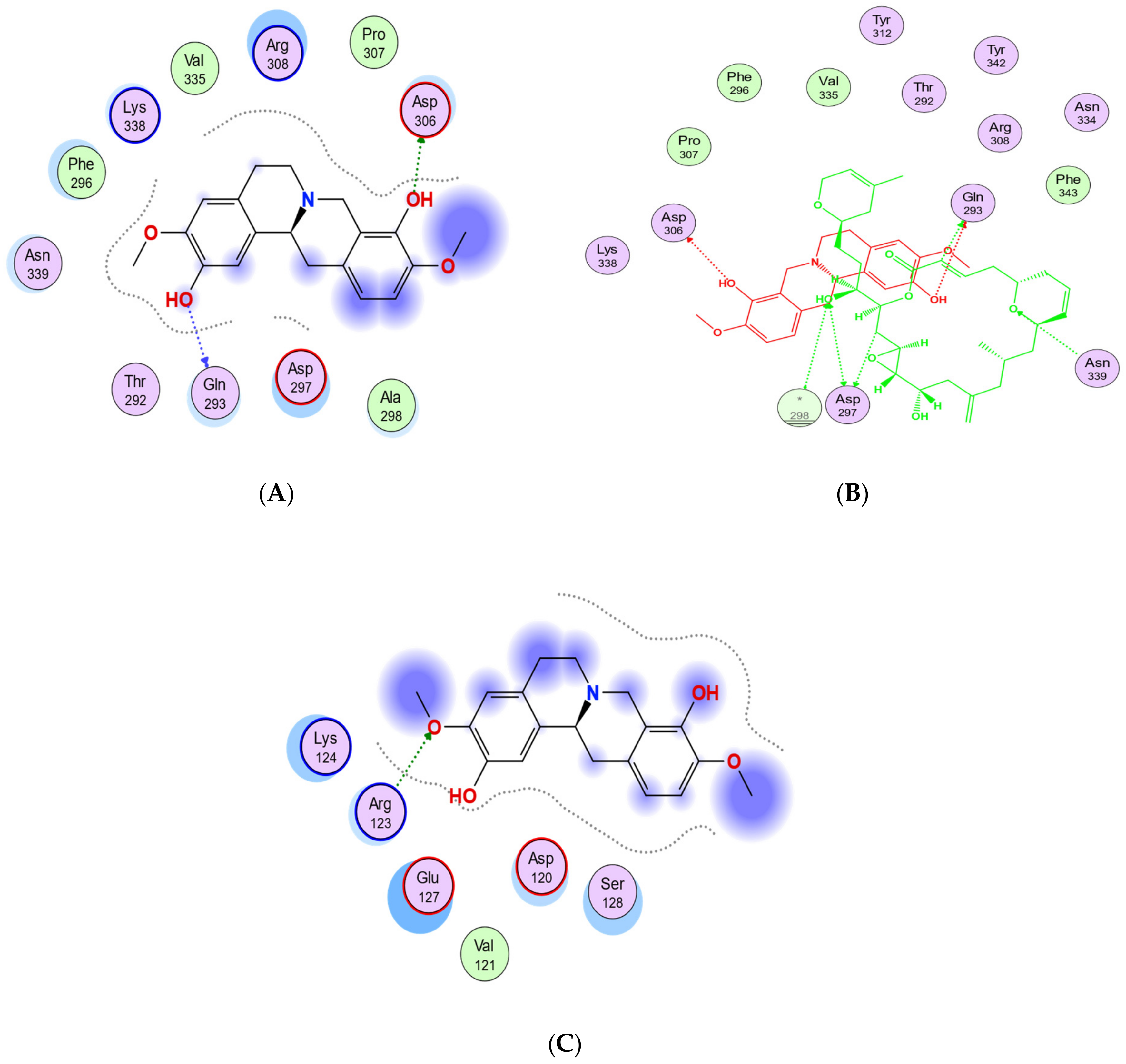
2.3. Binding Affinities and Pose Analysis of Potential Scoulerine Binding Sites
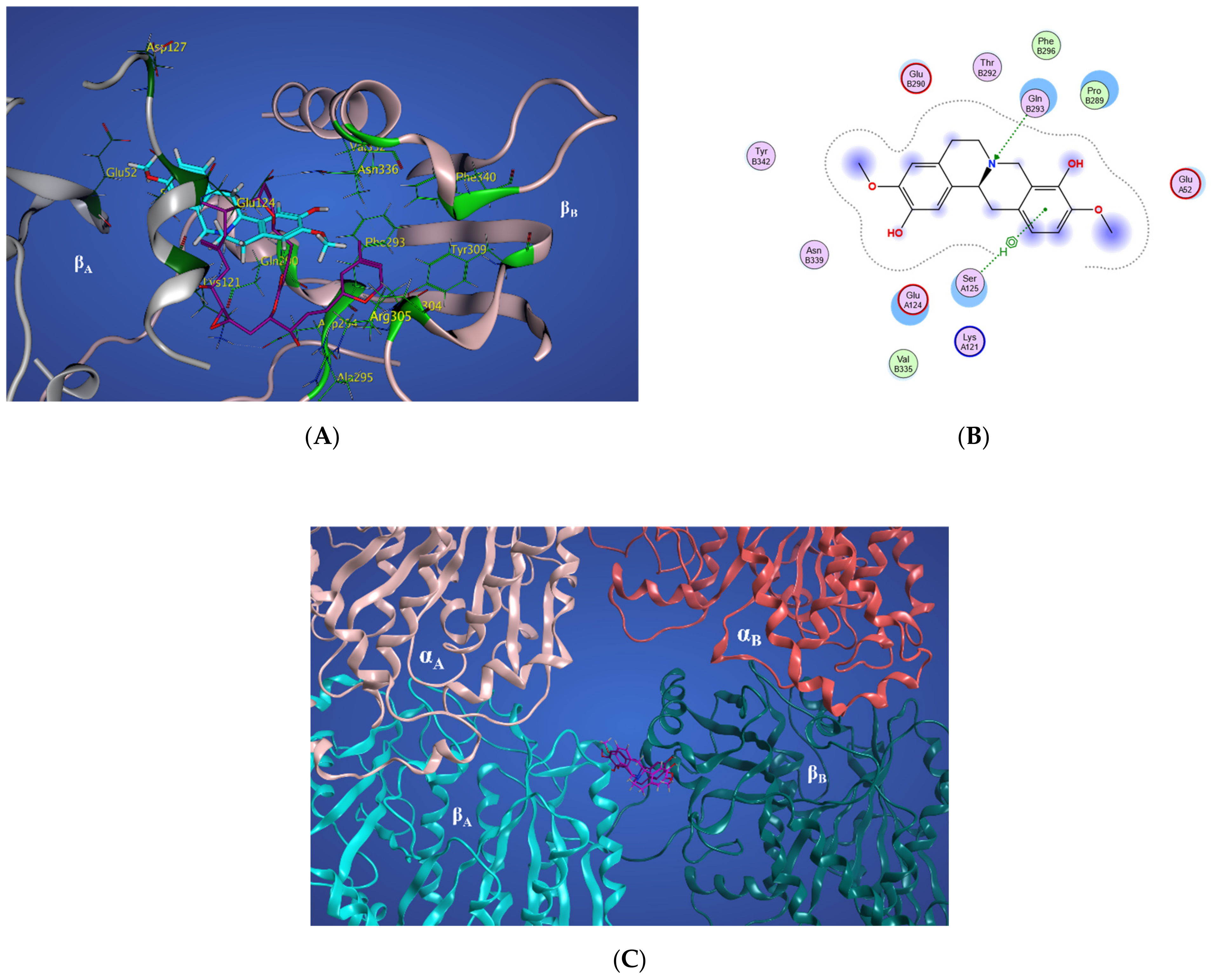
2.4. Colchicine Site
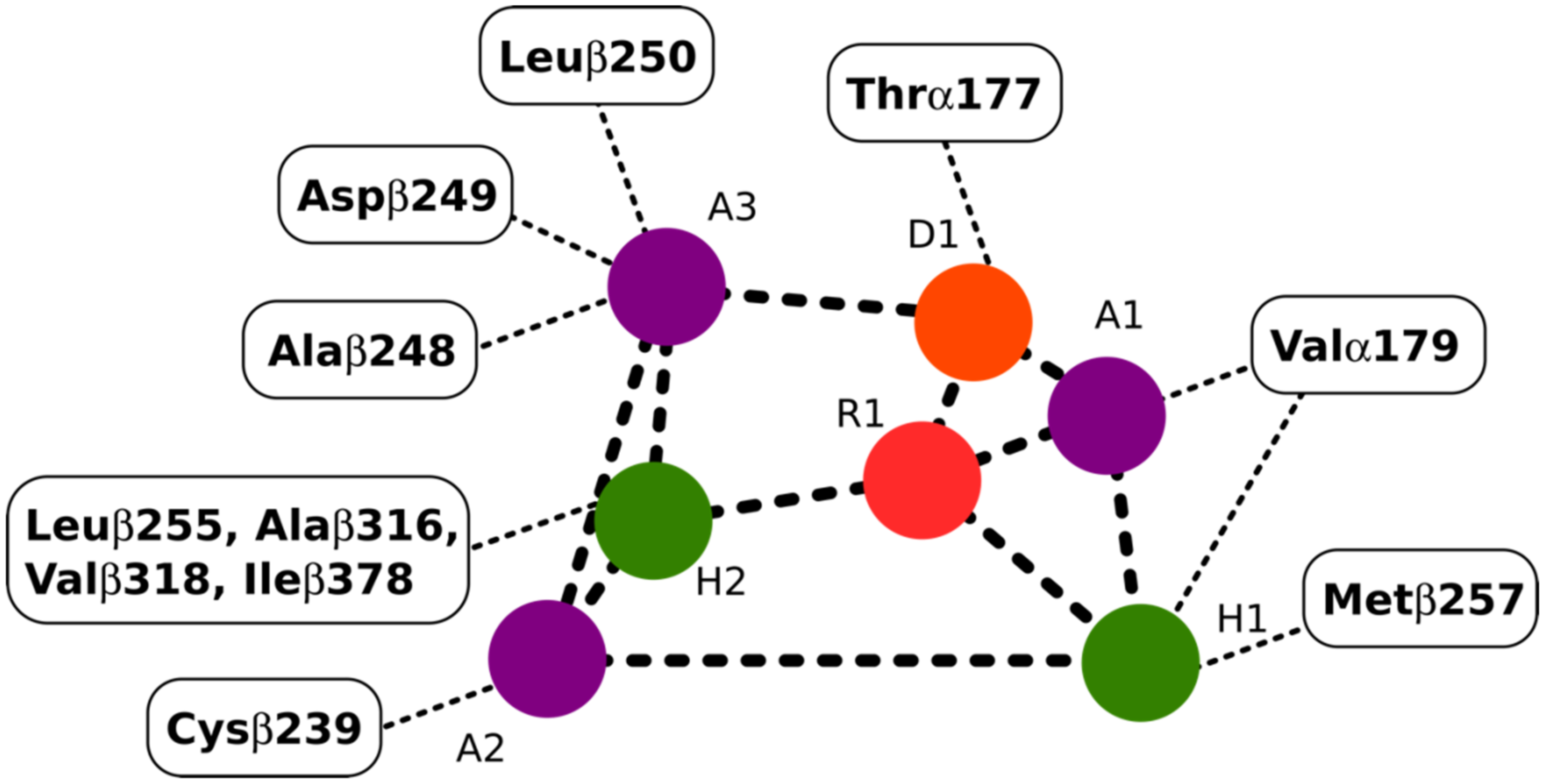
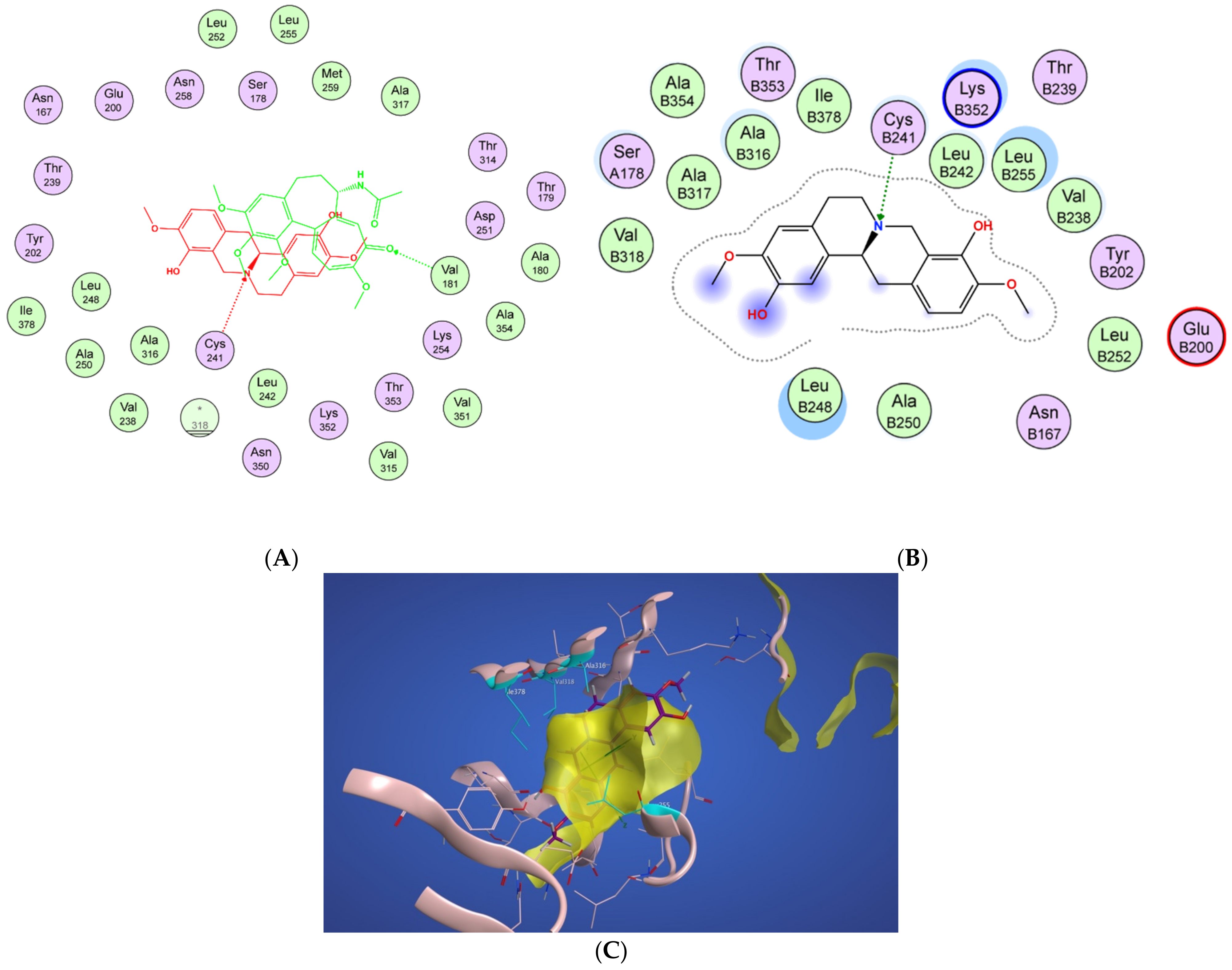
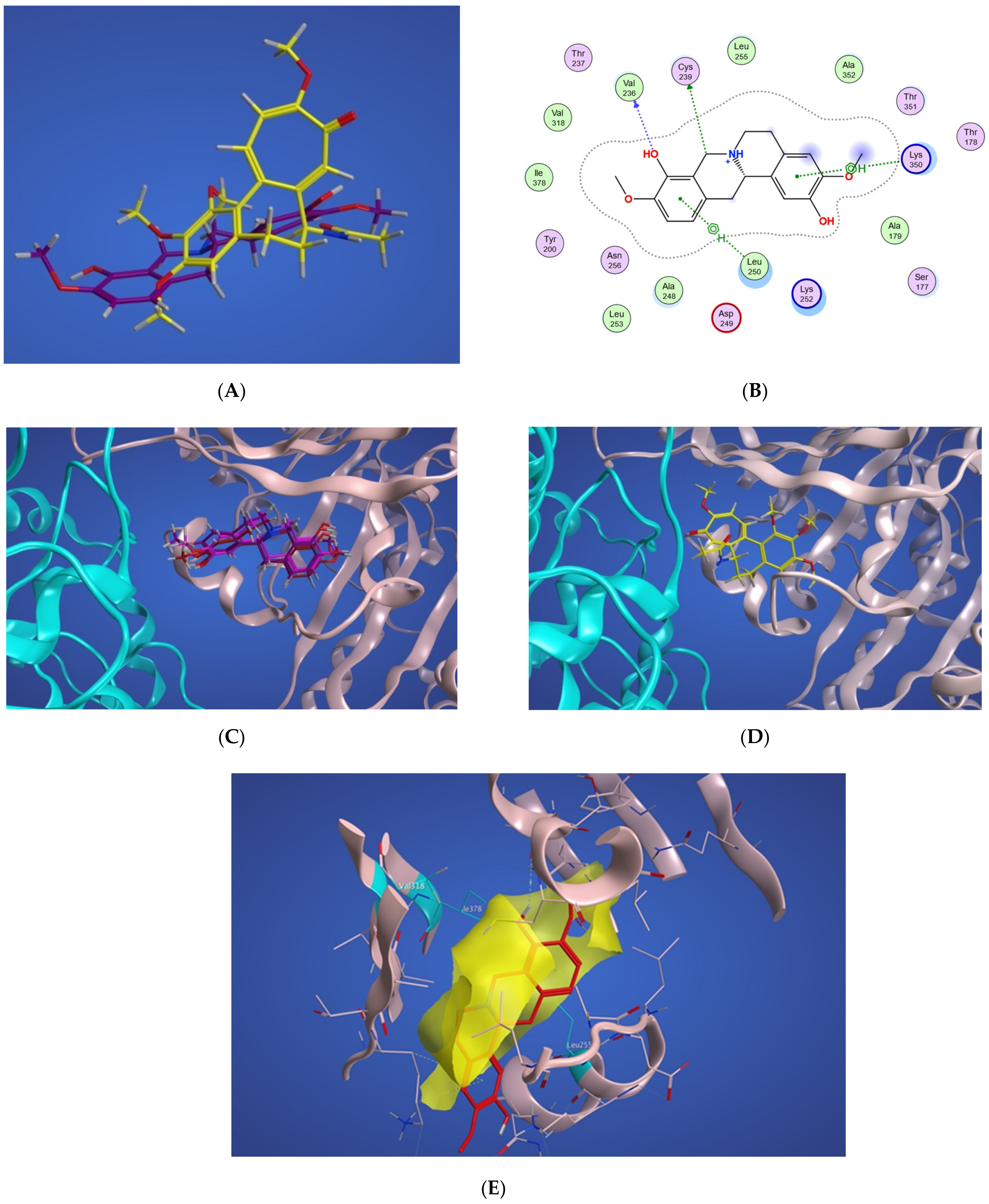
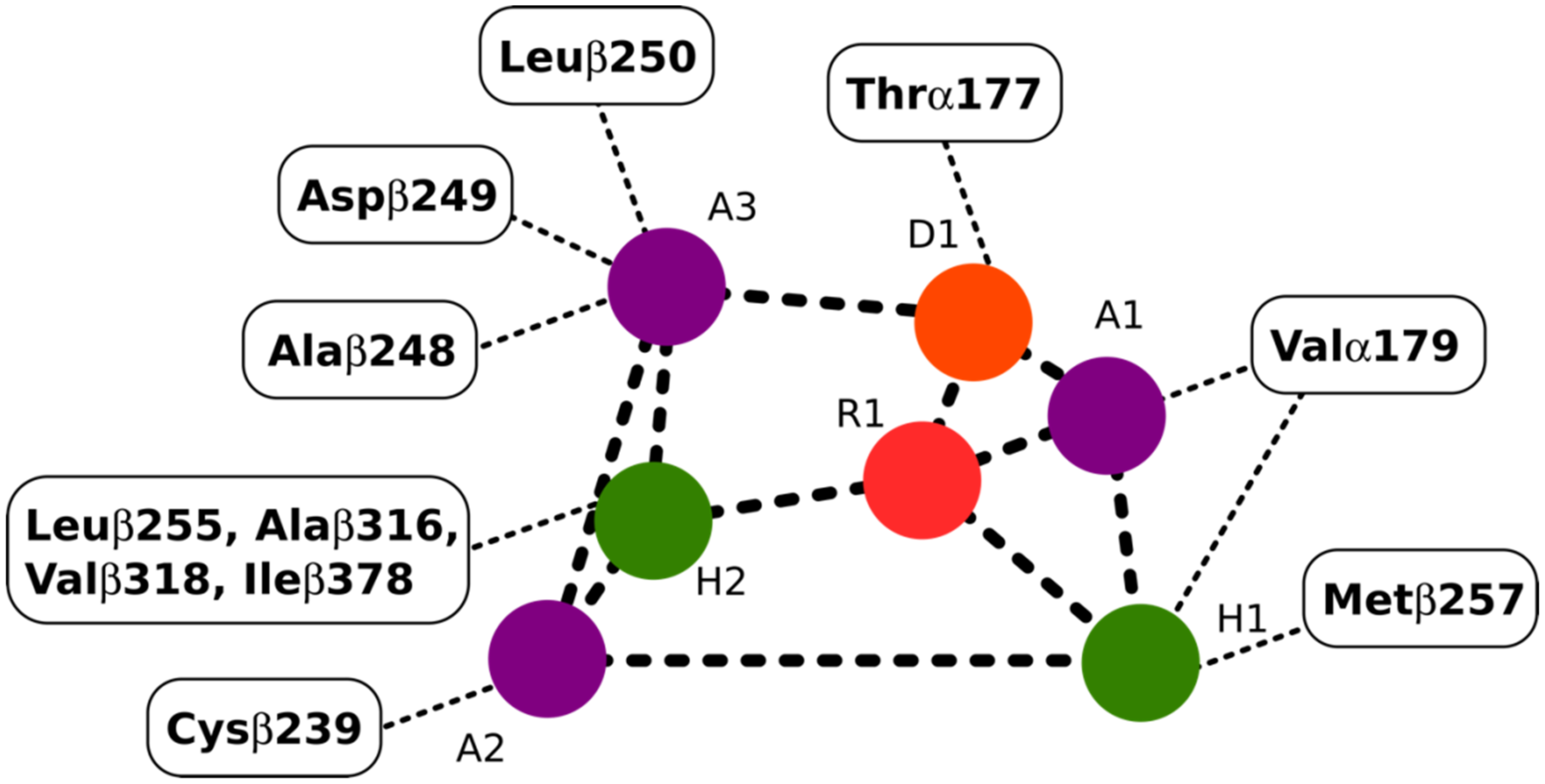
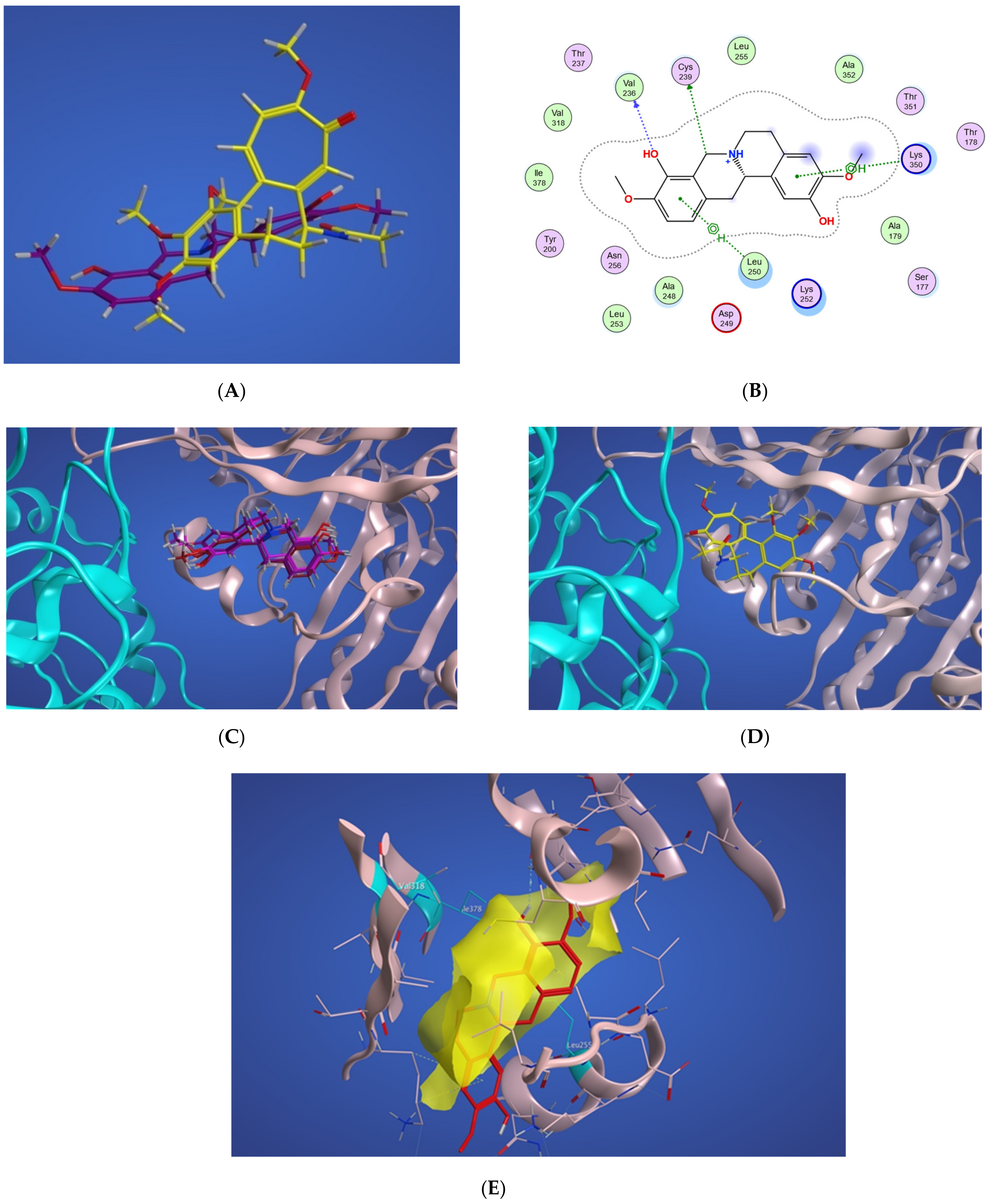
2.4.1. Potential Scoulerine Binding Site (S1)
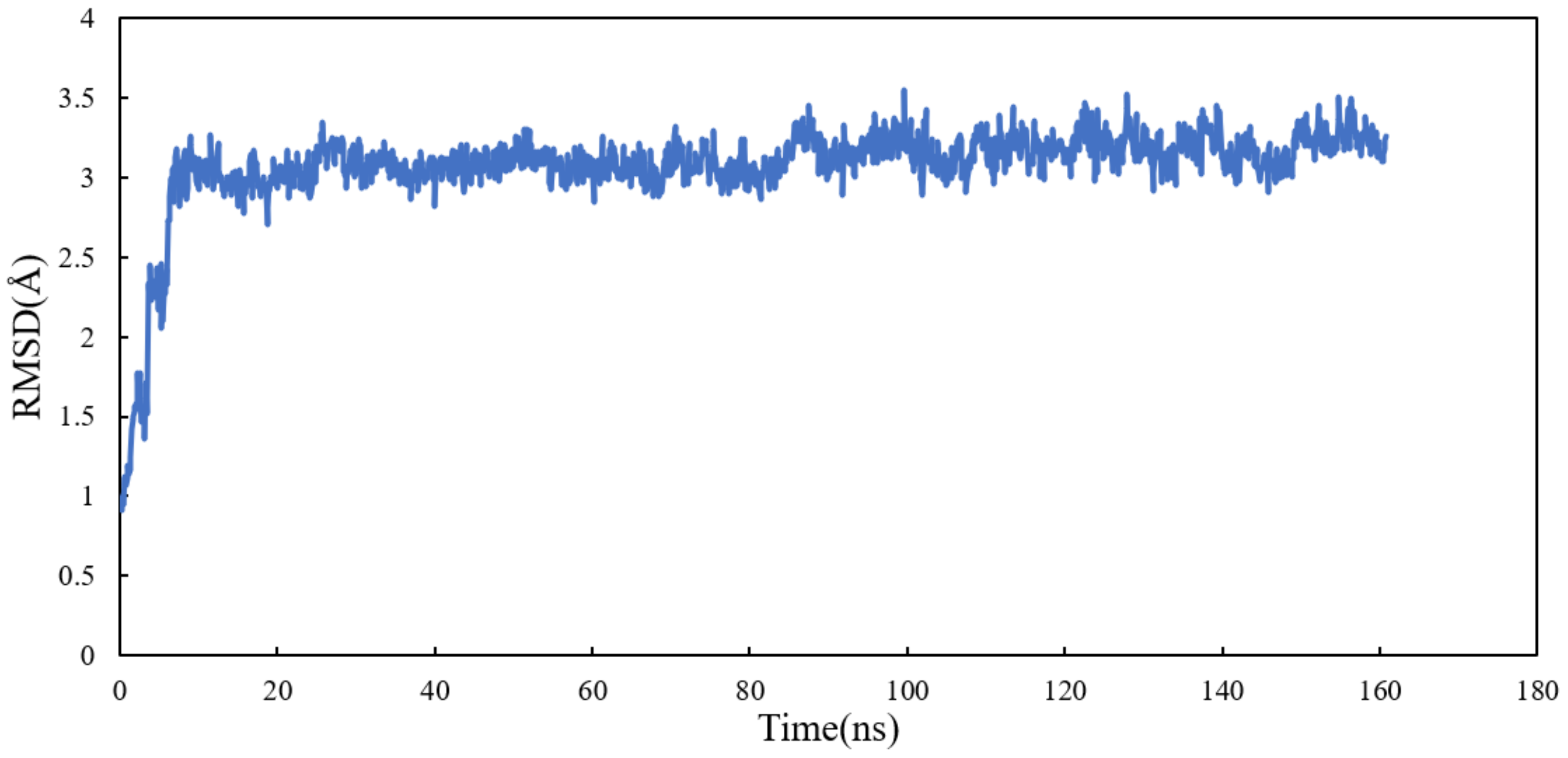
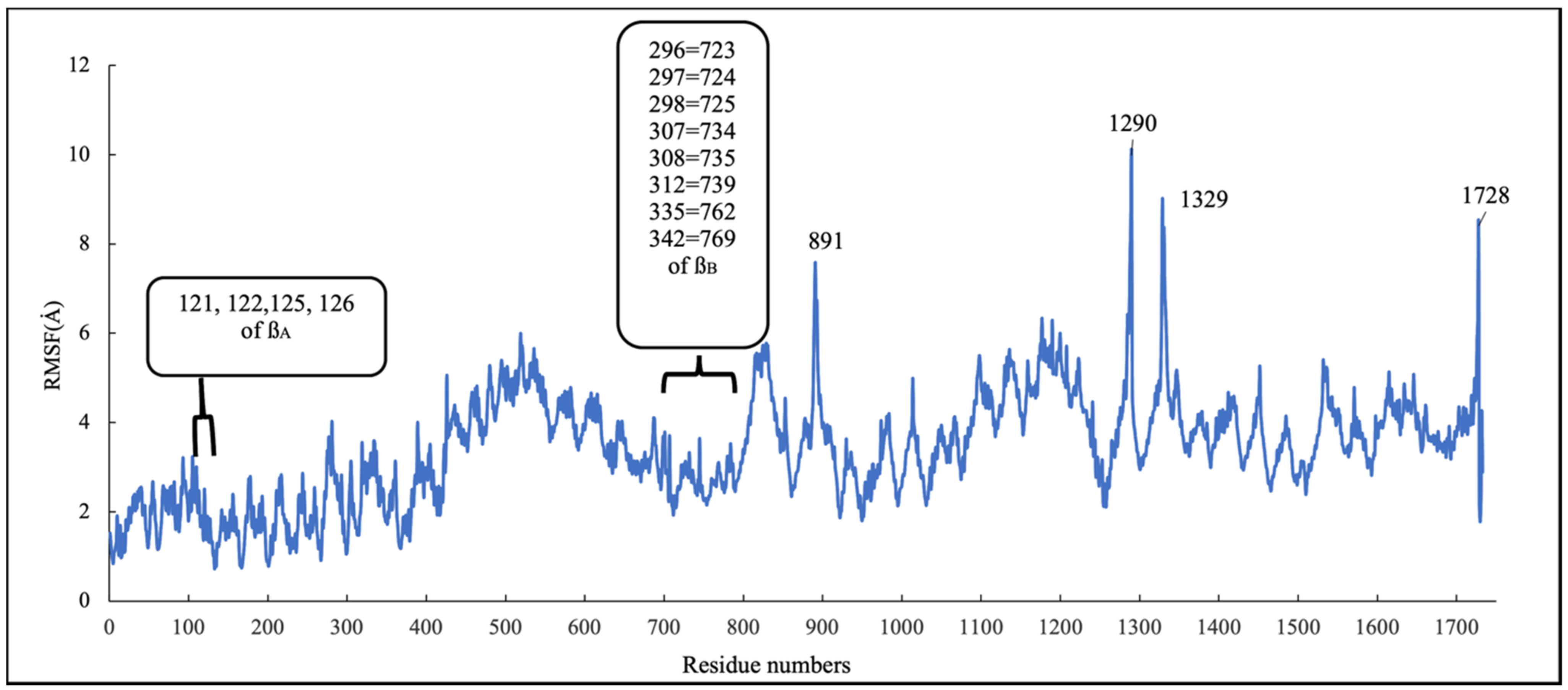
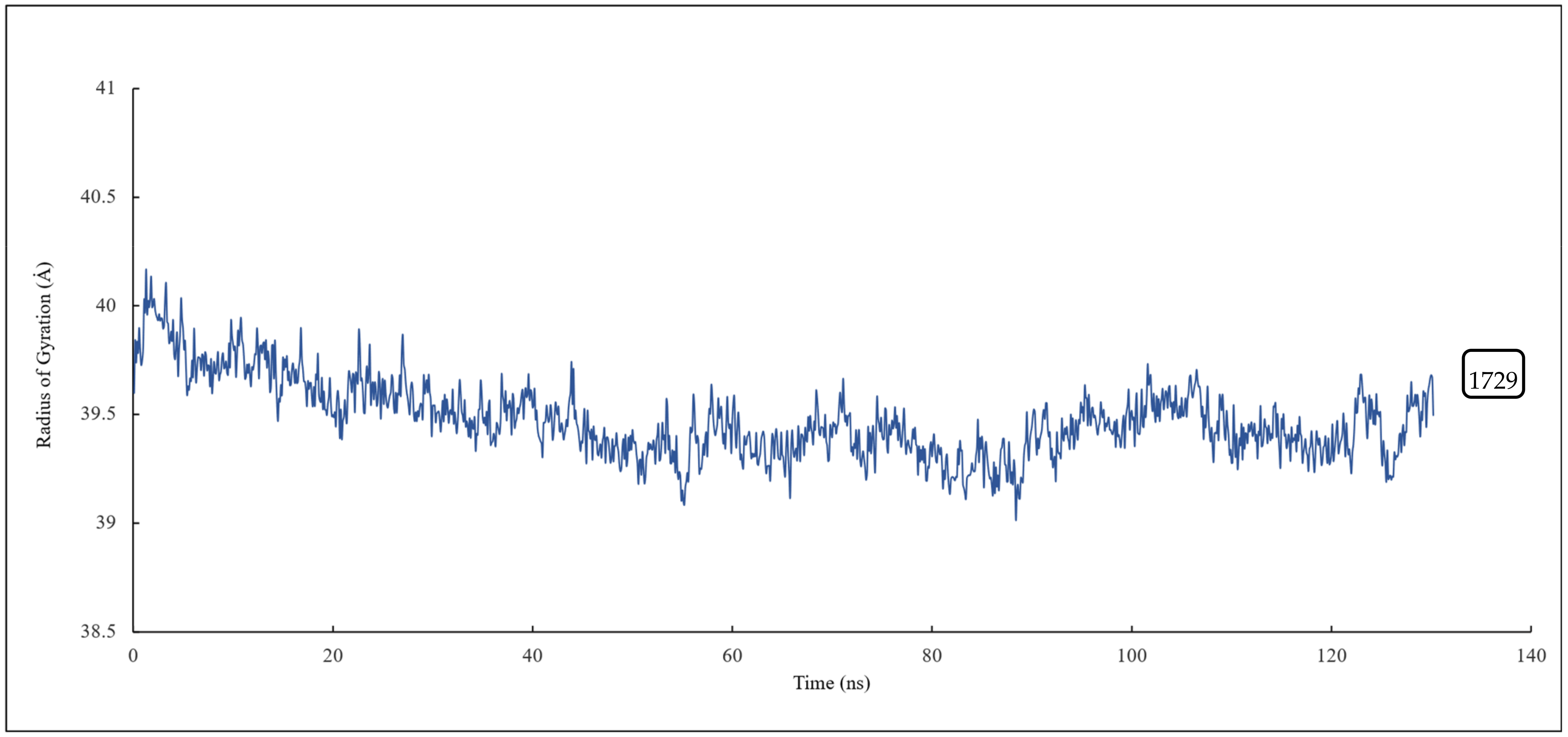
2.4.2. Conformational Analysis
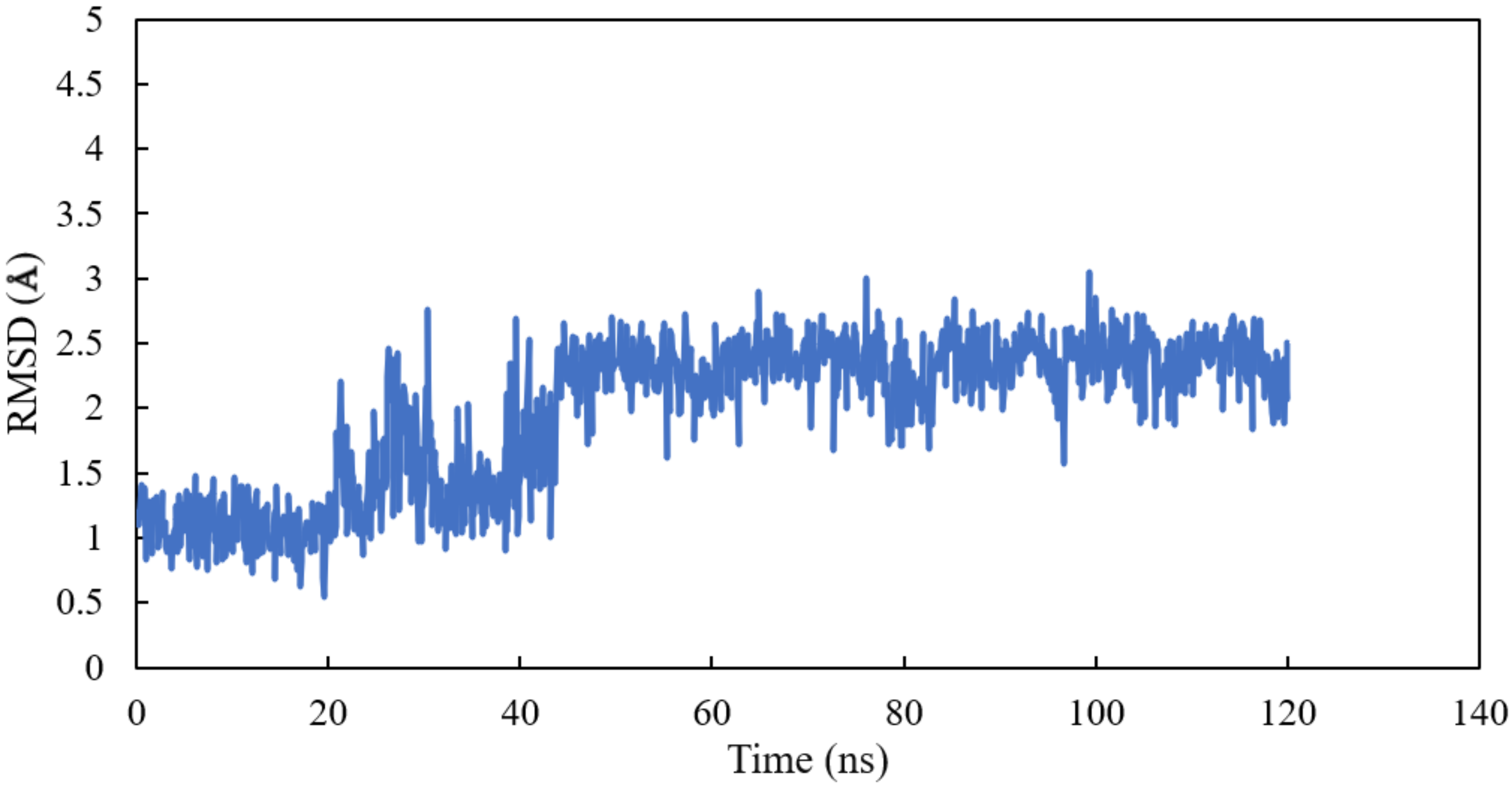
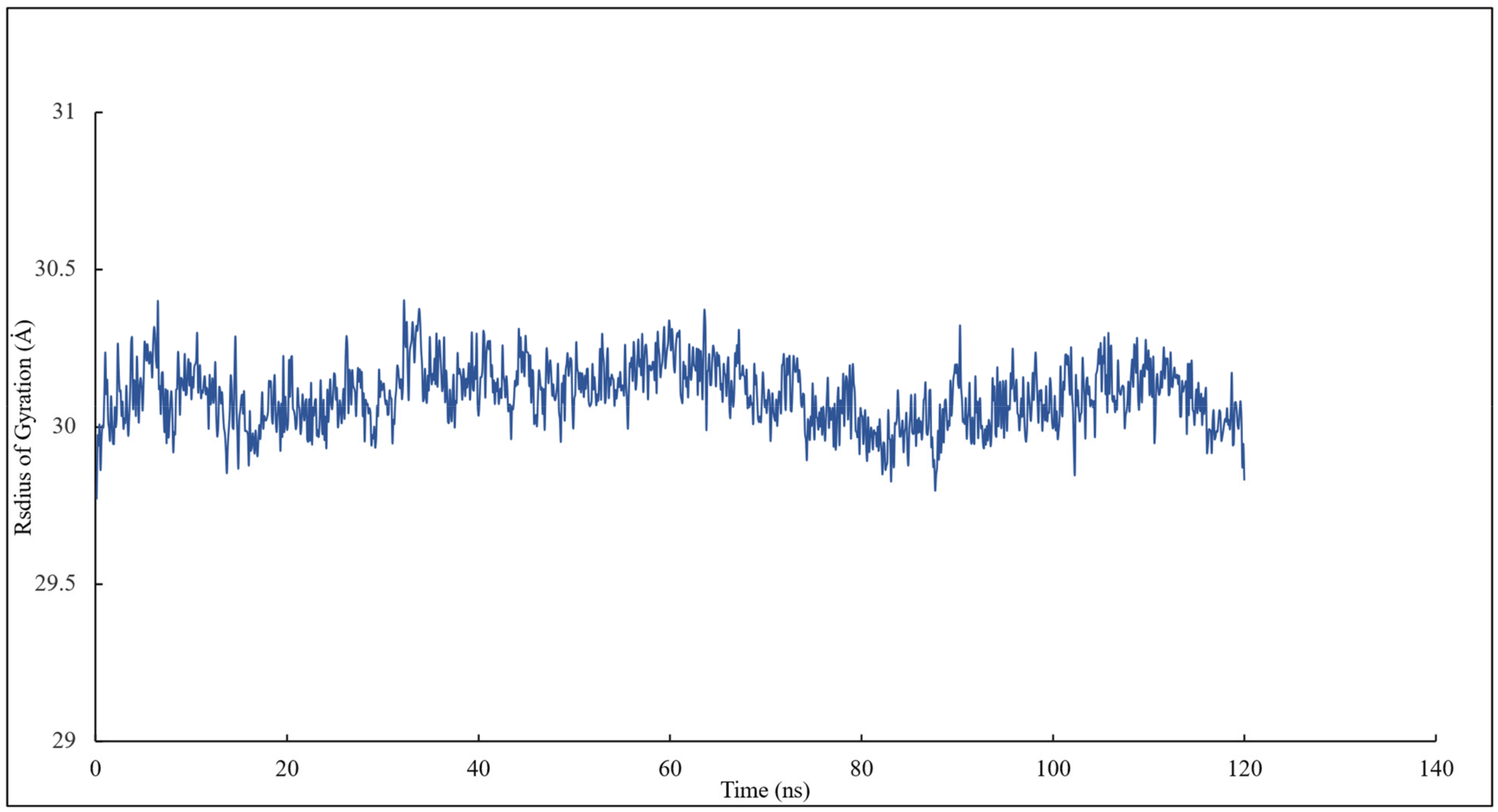
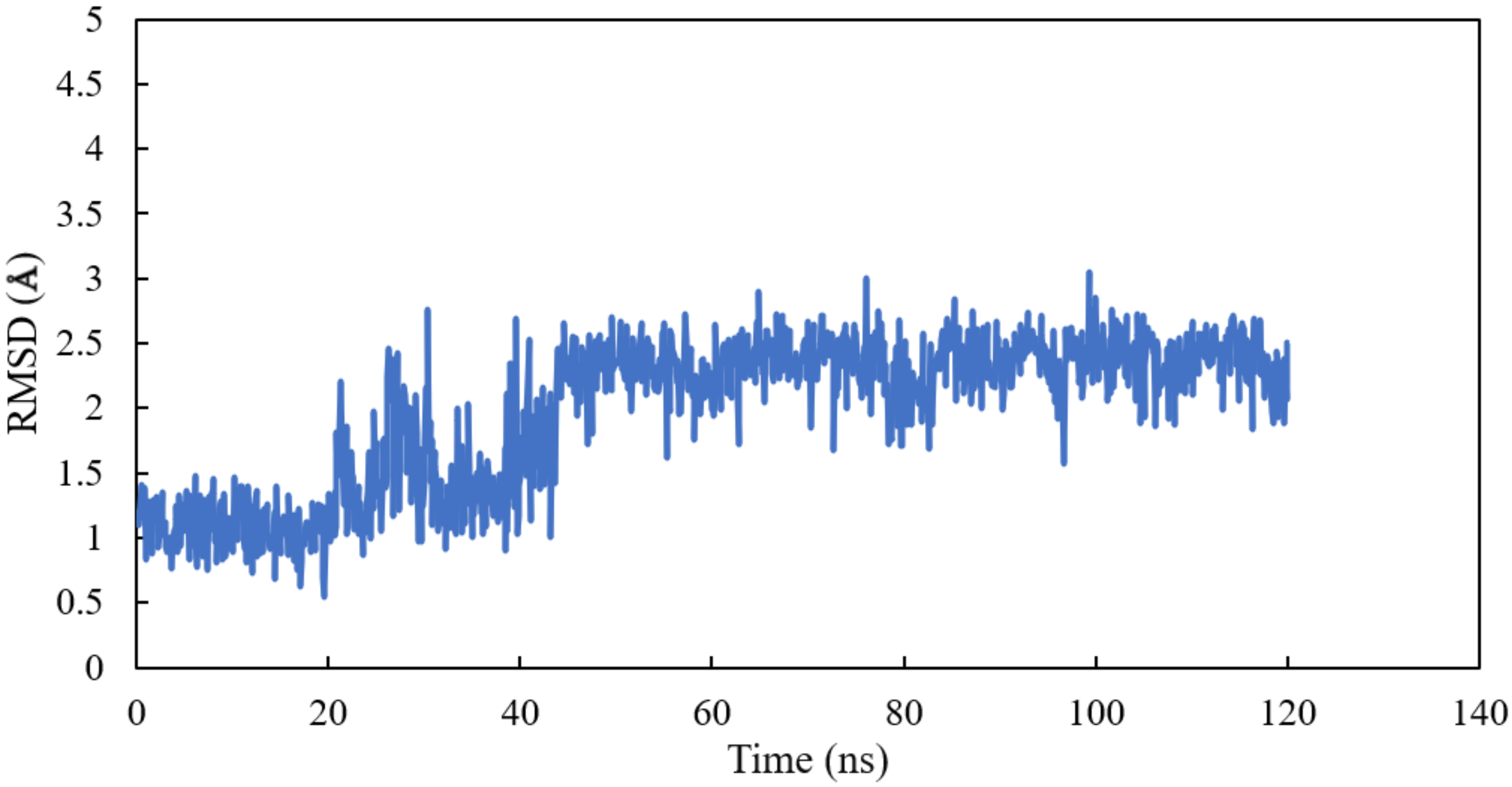
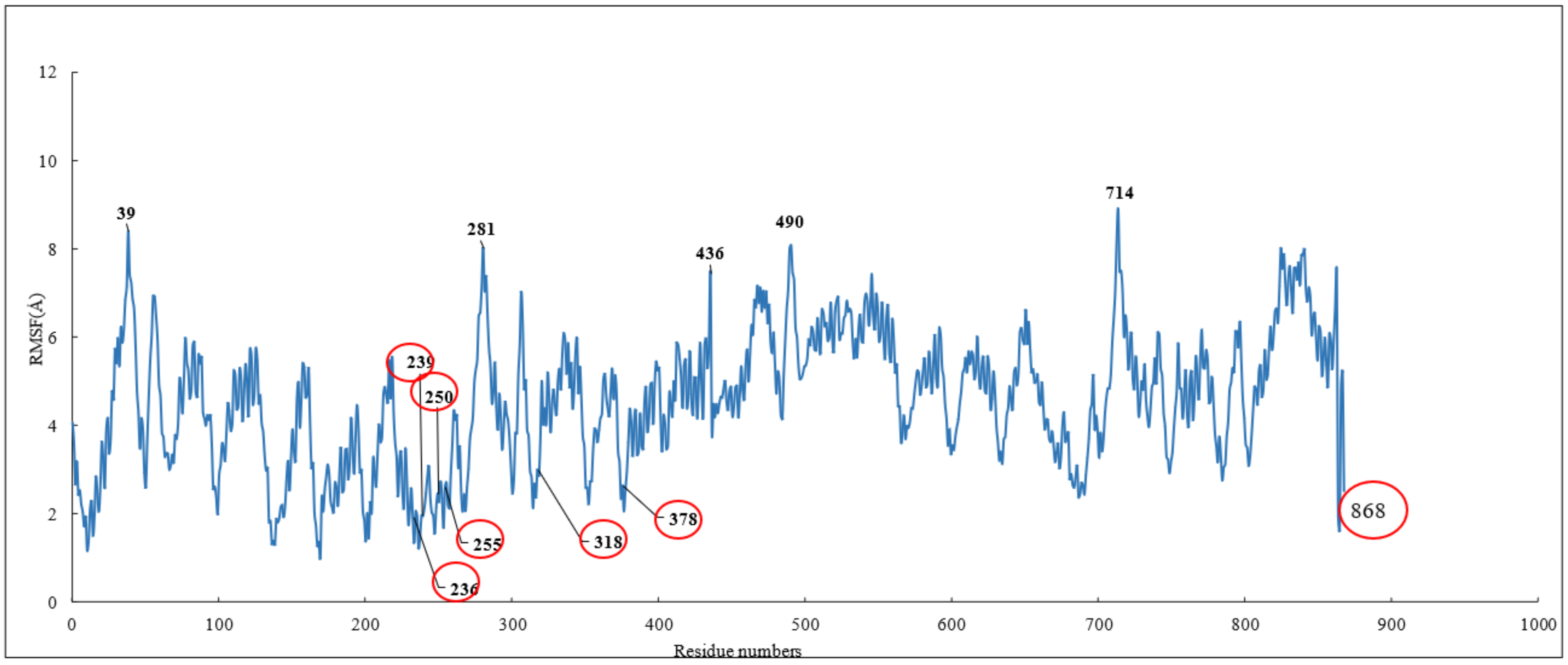
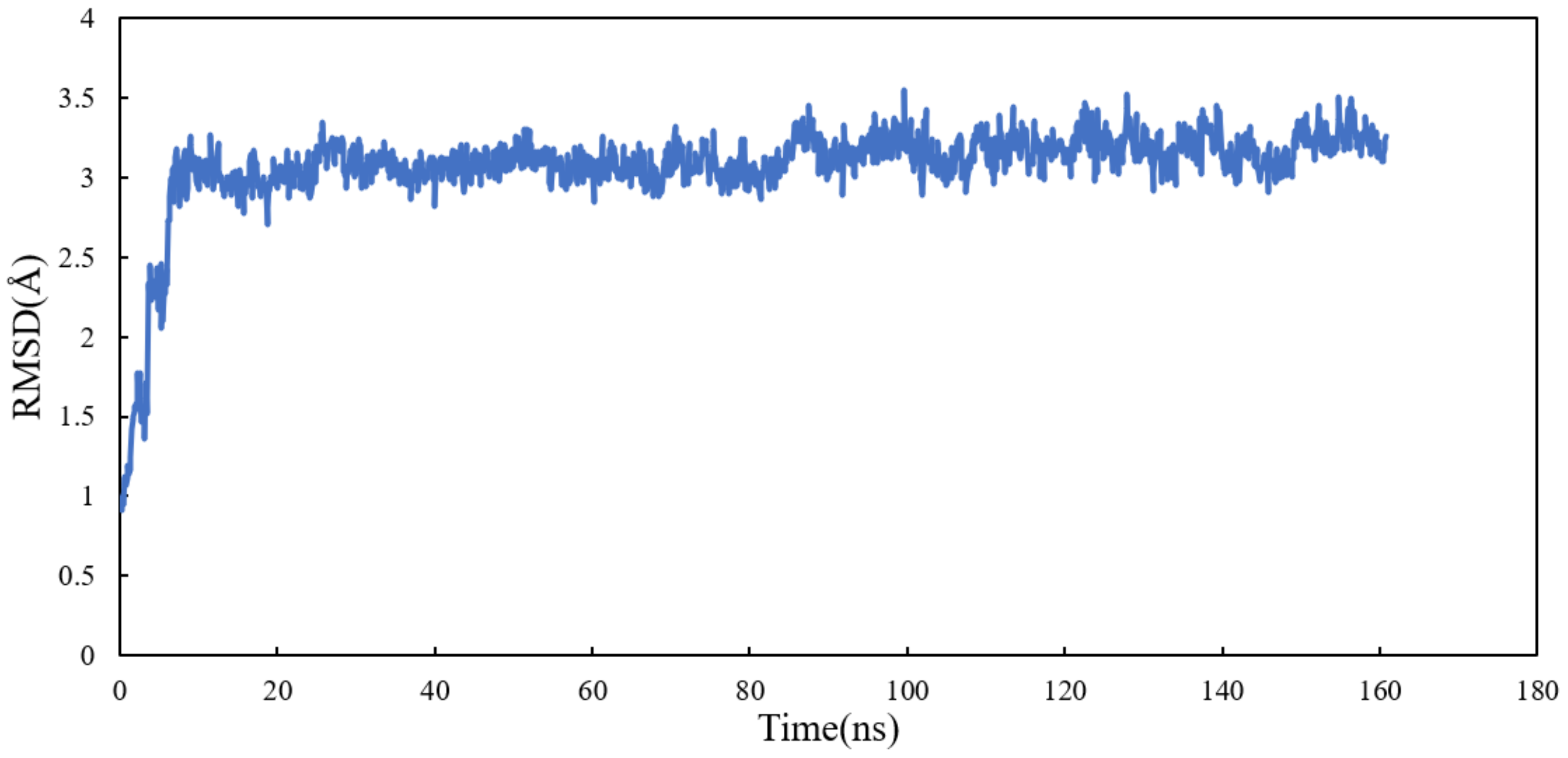
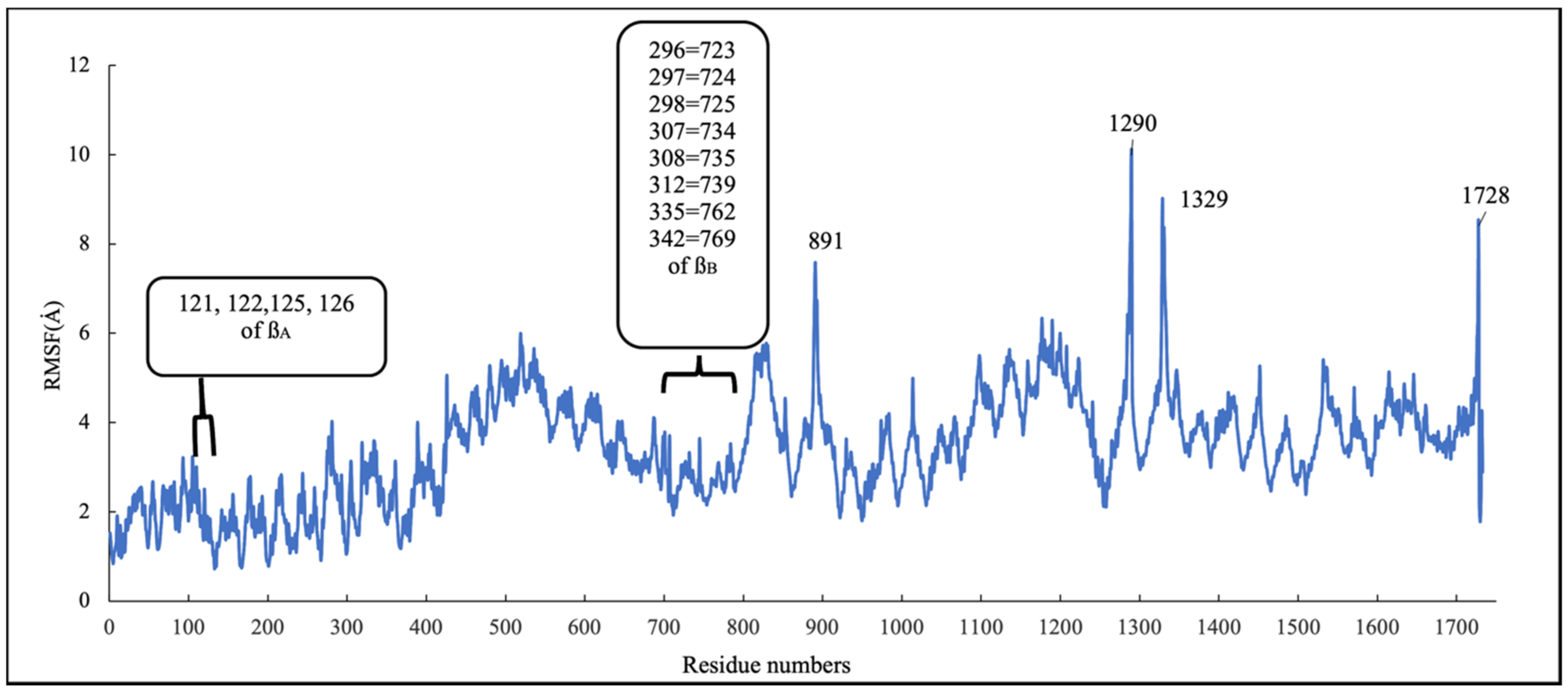
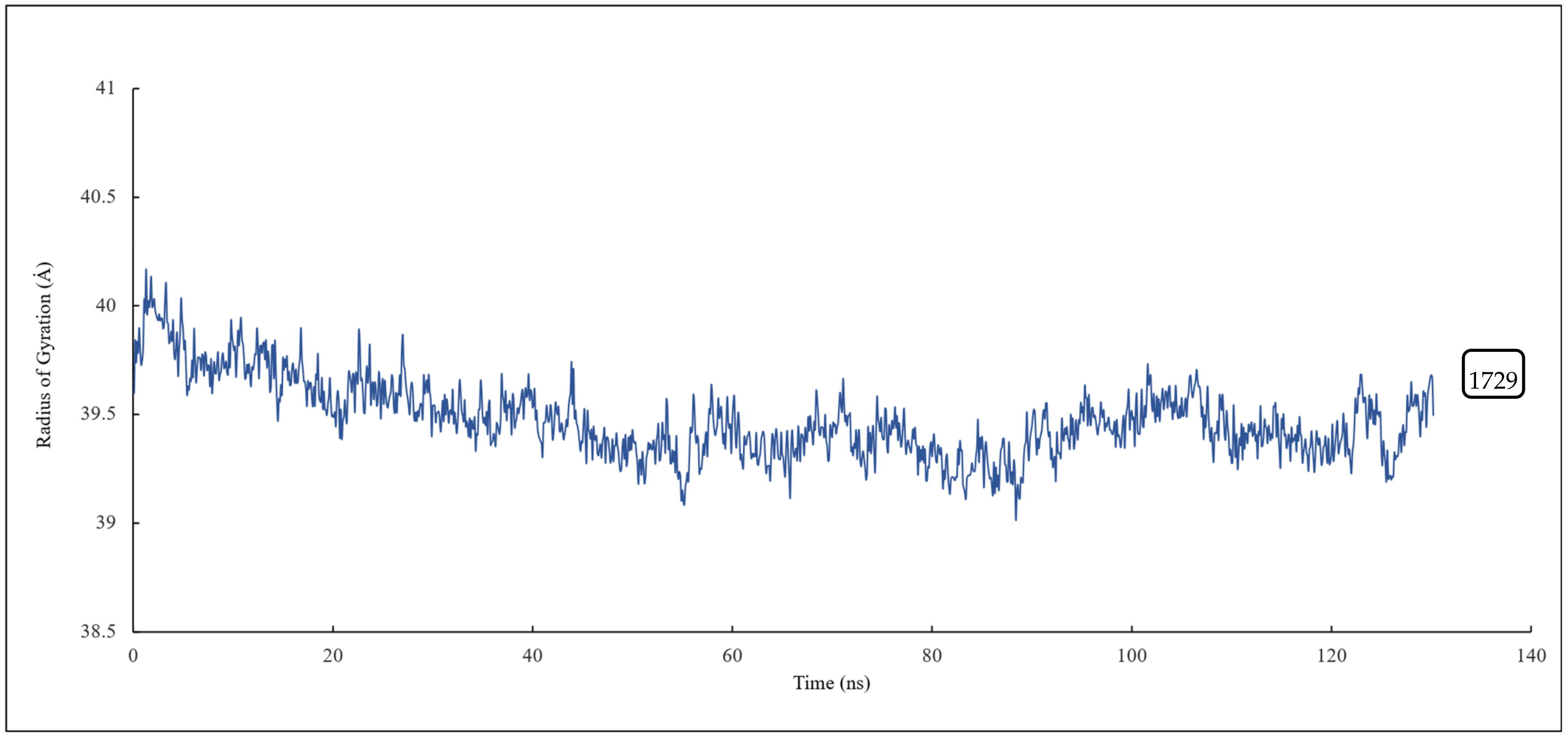
RMSD and RMSF Analysis on S1 Site
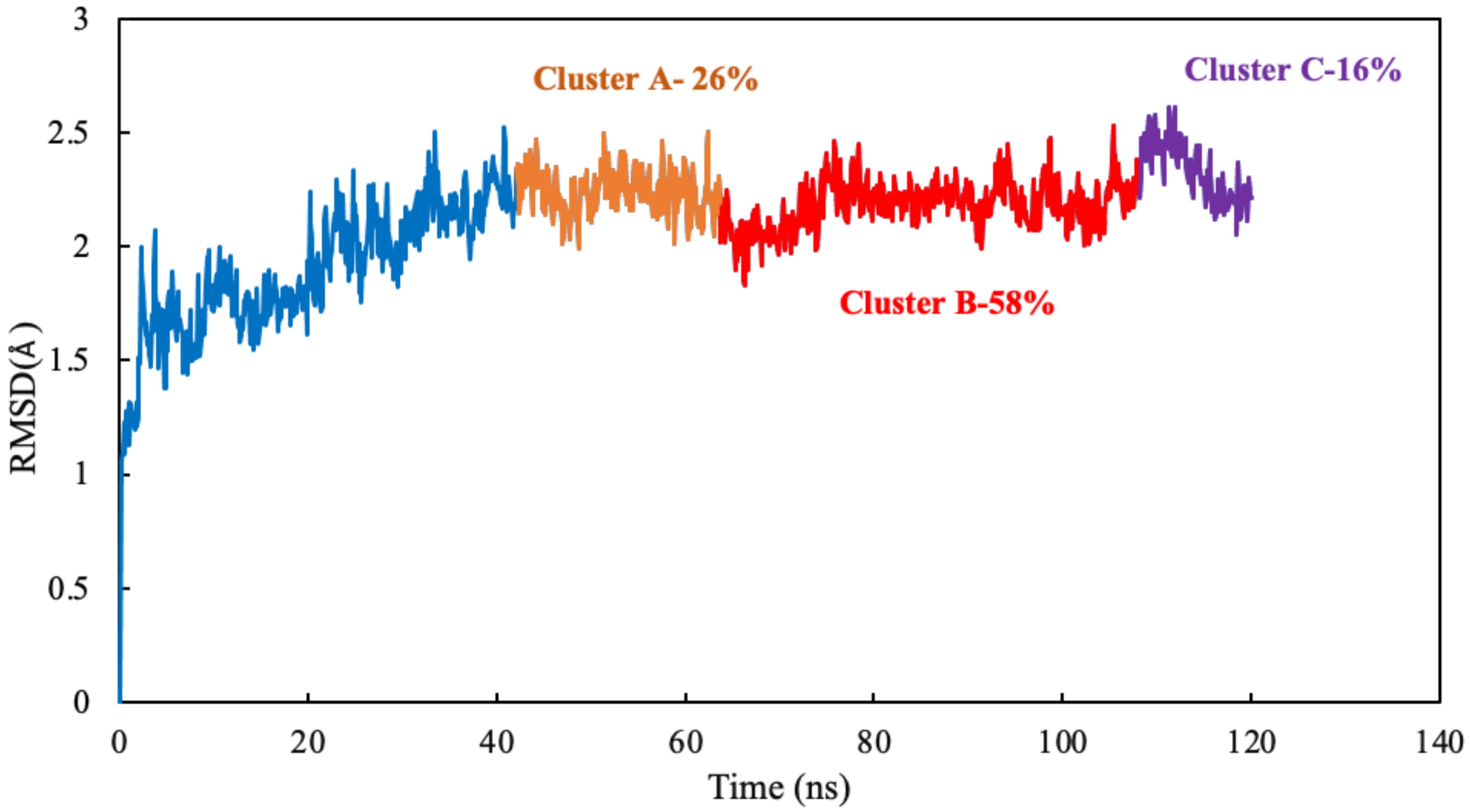
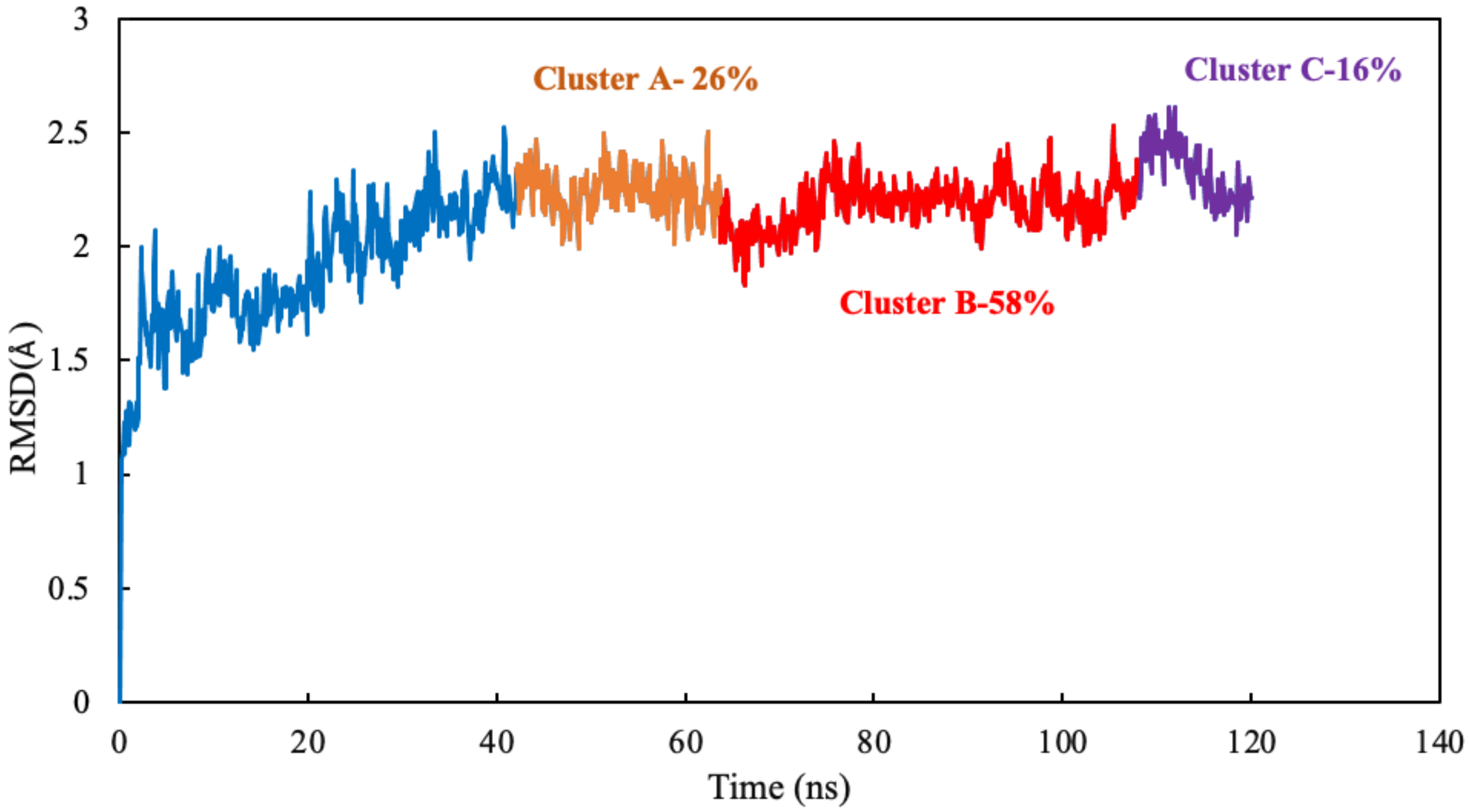
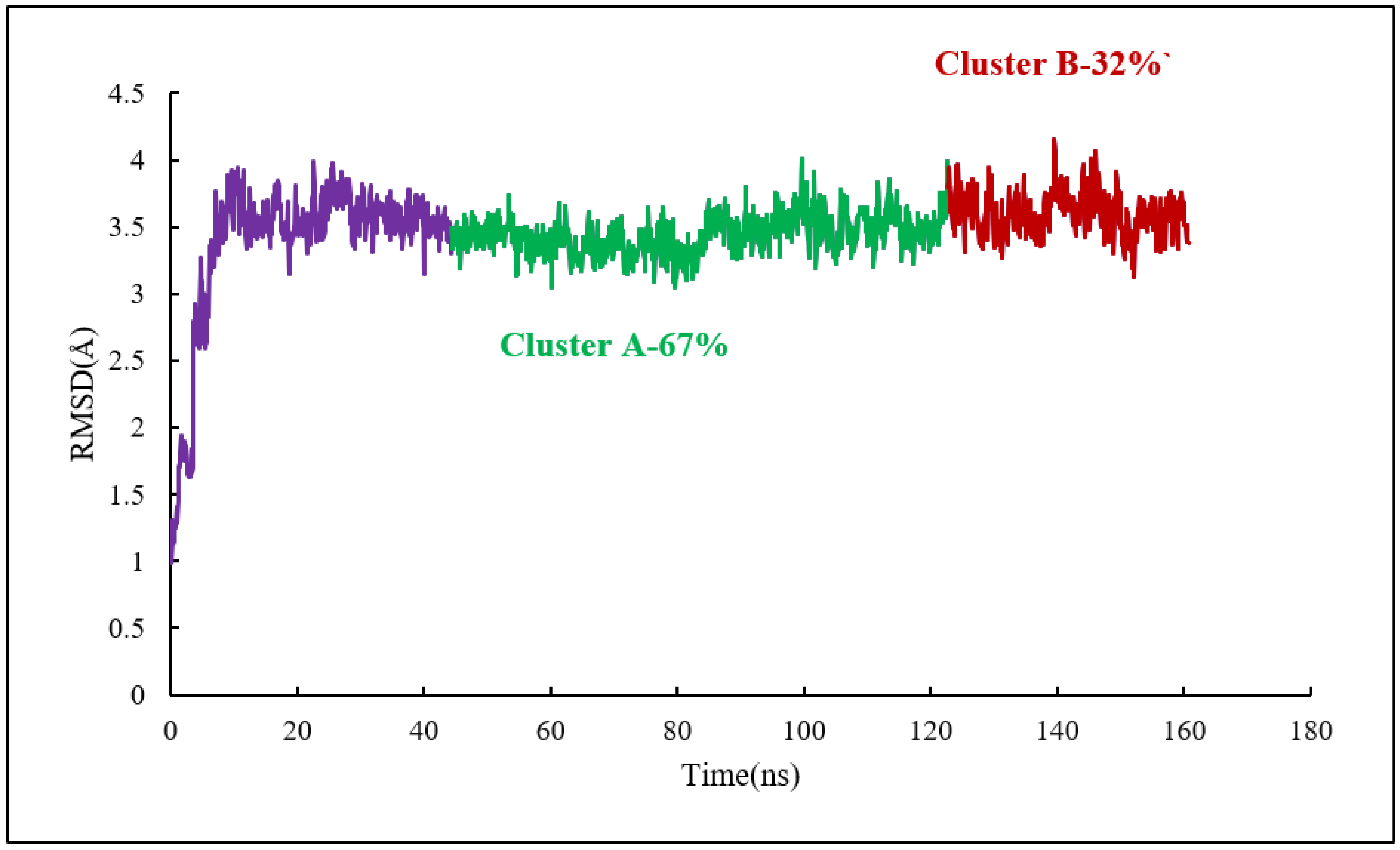
Clustering Analysis
2.5. Laulimalide Binding Sites on β Tubulin
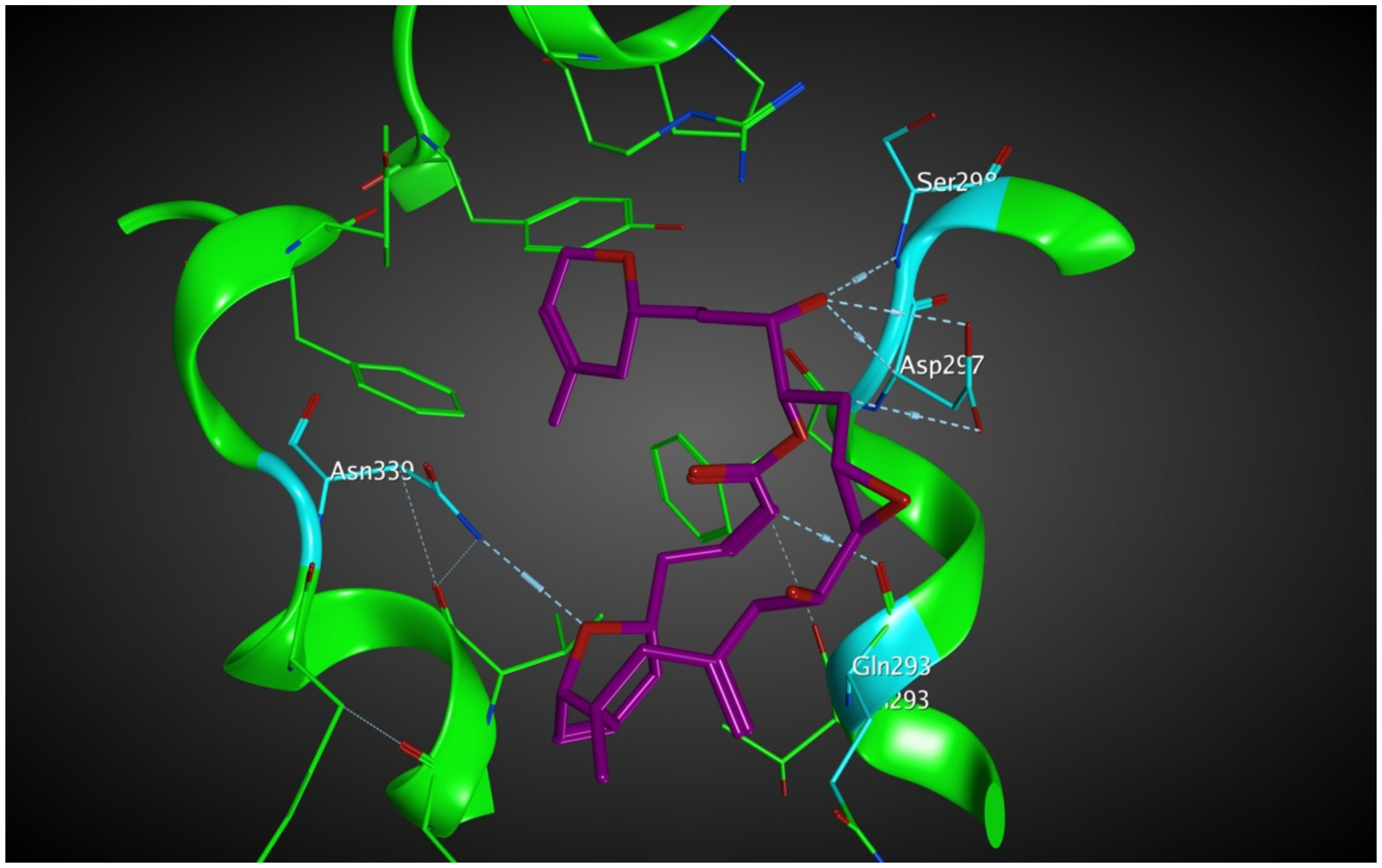
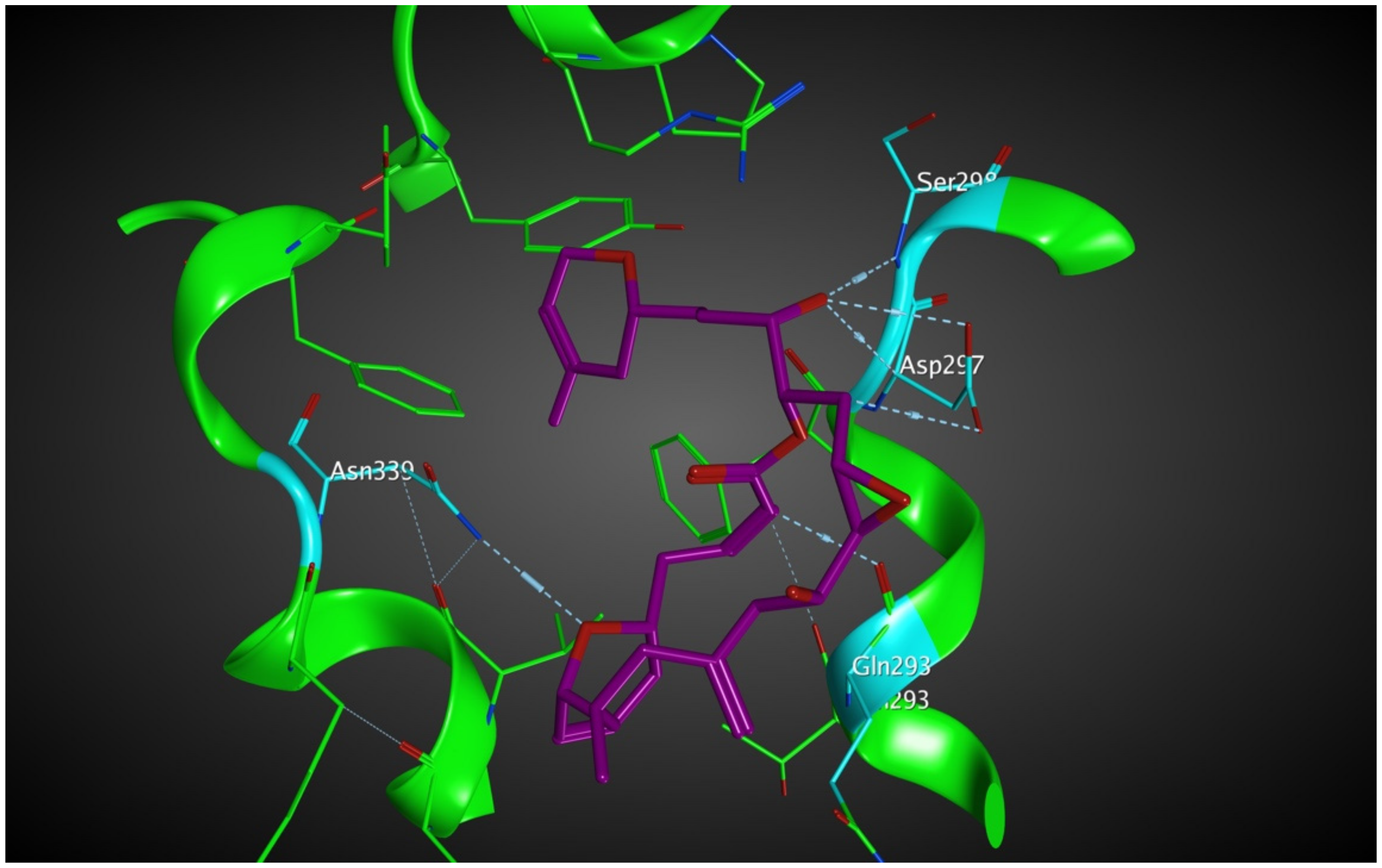
2.5.1. Potential Scoulerine Binding Site (S2)
2.5.2. Conformational Analysis
Clustering Analysis
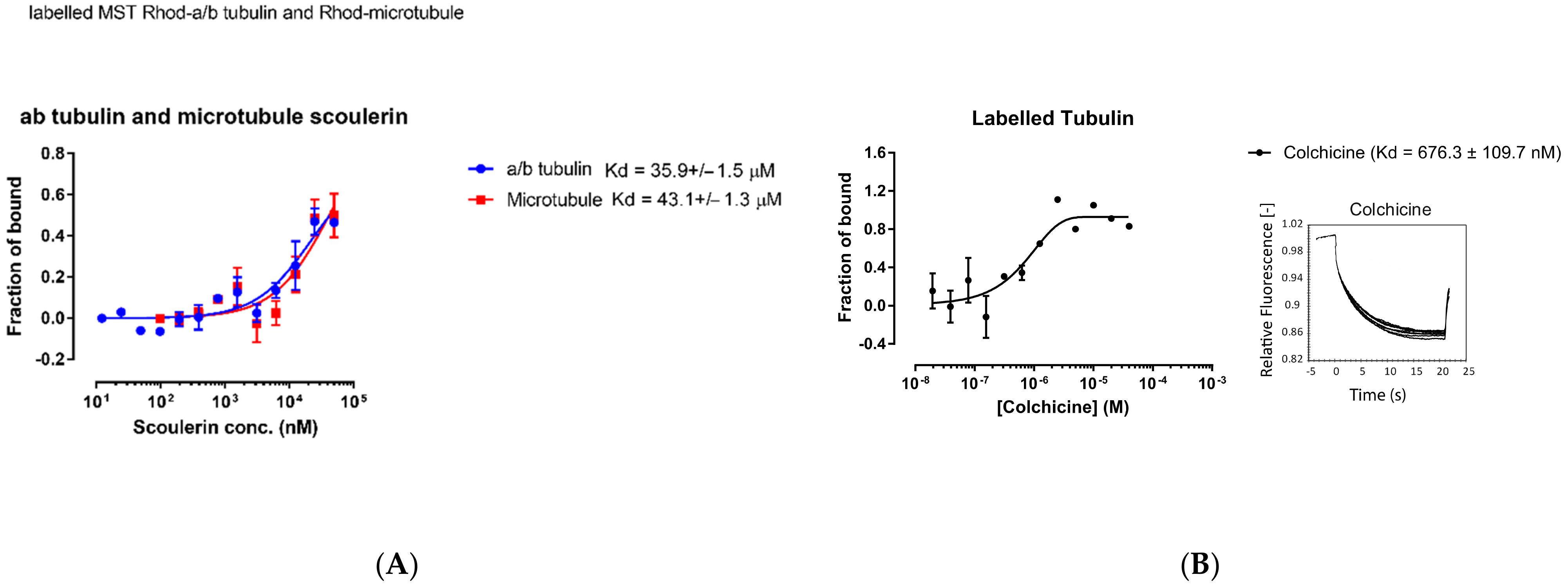
2.6. Experimental Validation
3. Materials and Methods
3.1. 3D Structure Preparation of the Ligand
3.2. Blind Docking
3.3. 3D Structure Preparation of Complexes for MD Simulation
3.3.1. Scoulerine in the Colchicine Binding Site
3.3.2. Scoulerine in the Laulimalide Binding Sites of Microtubule
3.3.3. Molecular Dynamic Simulations
3.3.4. Clustering Analysis
3.3.5. Microscale Thermophoresis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cragg, G.M.; Pezzuto, J.M. Natural products as a vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Meng, X.; Wu, D.; Qiu, Z.; Luo, H. A Natural isoquinoline alkaloid with antitumor activity: Studies of the biological activities of berberine. Front. Pharmacol. 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Mo, J.; Xu, L.; Zhang, R.; Qiao, Y.; Liu, B.; Jiang, L.; Ma, S.; Shi, G. Scoulerine promotes cell viability reduction and apoptosis by activating ROS-dependent endoplasmic reticulum stress in colorectal cancer cells. Chem. Biol. Interact. 2020, 327, 109184. [Google Scholar] [CrossRef] [PubMed]
- Kukula-Koch, W.A.; Widelski, J. Alkaloids. In Pharmacognosy; Badal, S., Delgoda, R., Eds.; Elsevier: Boston, MA, USA, 2017; pp. 163–198. ISBN 978-0-12-802104-0. [Google Scholar]
- Habartova, K.; Havelek, R.; Seifrtova, M.; Kralovec, K.; Cahlikova, L.; Chlebek, J.; Cermakova, E.; Mazankova, N.; Marikova, J.; Kunes, J.; et al. Scoulerine affects microtubule structure, inhibits proliferation, arrests cell cycle and thus culminates in the apoptotic death of cancer cells. Sci. Rep. 2018, 8, 4829. [Google Scholar] [CrossRef] [Green Version]
- Hagel, J.M.; Morris, J.S.; Lee, E.-J.; Desgagné-Penix, I.; Bross, C.D.; Chang, L.; Chen, X.; Farrow, S.C.; Zhang, Y.; Soh, J.; et al. Transcriptome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol. 2015, 15, 227. [Google Scholar] [CrossRef] [Green Version]
- Alisaraie, L.; Tuszynski, J.A. Determination of noscapine’s localization and interaction with the tubulin-α/β heterodimer. Chem. Biol. Drug Des. 2011, 78, 535–546. [Google Scholar] [CrossRef]
- Ghaly, P.E.; Abou El-Magd, R.M.; Churchill, C.D.M.; Tuszynski, J.A.; West, F.G. A new antiproliferative noscapine analogue: Chemical synthesis and biological evaluation. Oncotarget 2016, 7, 40518–40530. [Google Scholar] [CrossRef]
- Ghaly, P.E.; Churchill, C.D.M.; Abou El-Magd, R.M.; Hájková, Z.; Dráber, P.; West, F.G.; Tuszynski, J.A. Synthesis and biological evaluation of structurally simplified noscapine analogues as microtubule binding agents. Can. J. Chem. 2017, 95, 649–655. [Google Scholar] [CrossRef]
- Chlebek, J.; De Simone, A.; Hošťálková, A.; Opletal, L.; Pérez, C.; Pérez, D.I.; Havlíková, L.; Cahlíková, L.; Andrisano, V. Application of BACE1 immobilized enzyme reactor for the characterization of multifunctional alkaloids from Corydalis cava (Fumariaceae) as Alzheimer’s disease targets. Fitoterapia 2016, 109, 241–247. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of tubulin binding ligands to target cancer cells: Updates on their therapeutic potential and clinical trials. Curr. Cancer Drug Targets 2017, 17, 357–375. [Google Scholar] [CrossRef]
- Dustin, P. The role of MT in mitosis. In Microtubules; Springer: Berlin/Heidelberg, Germany, 1978; pp. 340–397. ISBN 978-3-642-96436-7. [Google Scholar]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltai, T. Cancer: Fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther. 2016, 9, 6343–6360. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density functional with spherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Morris, P.G.; Fornier, M.N. Microtubule active agents: Beyond the taxane frontier. Clin. Cancer Res. 2008, 14, 7167–7172. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and evaluation of azetidinone analogues of combretastatin A-4 as tubulin targeting agents. J. Med. Chem. 2010, 53, 8569–8584. [Google Scholar] [CrossRef] [PubMed]
- Churchill, C.D.M.; Klobukowski, M.; Tuszynski, J.A. The unique binding mode of laulimalide to two tubulin protofilaments. Chem. Biol. Drug Des. 2015, 86, 190–199. [Google Scholar] [CrossRef]
- Demir, Ö.; Baronio, R.; Salehi, F.; Wassman, C.D.; Hall, L.; Hatfield, G.W.; Chamberlin, R.; Kaiser, P.; Lathrop, R.H.; Amaro, R.E. Ensemble-based computational approach discriminates functional activity of p53 cancer and rescue mutants. PLoS Comput. Biol. 2011, 7, e1002238. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Churchill, C.D.M.; Klobukowski, M.; Tuszynski, J.A. Analysis of the binding mode of laulimalide to microtubules: Establishing a laulimalide-tubulin pharmacophore. J. Biomol. Struct. Dyn. 2016, 34, 1455–1469. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.-H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Pryor, D.E.; O’Brate, A.; Bilcer, G.; Díaz, J.F.; Wang, Y.; Wang, Y.; Kabaki, M.; Jung, M.K.; Andreu, J.M.; Ghosh, A.K.; et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002, 41, 9109–9115. [Google Scholar] [CrossRef]
- Autodock; Chemical Computing Group ULC: Montreal, Canada, 2018.
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Fourniol, F.J.; Sindelar, C.V.; Amigues, B.; Clare, D.K.; Thomas, G.; Perderiset, M.; Francis, F.; Houdusse, A.; Moores, C.A. Template-free 13-protofilament microtubule-MAP assembly visualized at 8 A resolution. J. Cell Biol. 2010, 191, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; Gohlke, H.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the effect of the g245s single point mutation on the structure of p53 and the binding of the protein to DNA. Molecules 2017, 22, 1358. [Google Scholar] [CrossRef] [Green Version]
- Eisenreich, W.J.; Höfner, G.; Bracher, F. Alkaloids from Croton flavens L. and their affinities to GABA-receptors. Nat. Prod. Res. 2003, 17, 437–440. [Google Scholar] [CrossRef]
- Wangchuk, P.; Keller, P.A.; Pyne, S.G.; Willis, A.C.; Kamchonwongpaisan, S. Antimalarial alkaloids from a Bhutanese traditional medicinal plant Corydalis dubia. J. Ethnopharmacol. 2012, 143, 310–313. [Google Scholar] [CrossRef] [Green Version]
- Chlebek, J.; Macáková, K.; Cahlíková, L.; Kurfürst, M.; Kuneš, J.; Opletal, L. Acetylcholinesterase and butyrylcholinesterase inhibitory compounds from corydalis cava (fumariaceae). Nat. Prod. Commun. 2011, 6, 607–610. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of Theory | Sco | ScoH+ | H2O | H3O+ | OH− | ΔG(1) | ΔG(2) |
|---|---|---|---|---|---|---|---|
| APFD/aug-cc-pVDZ | −1091.278 | −1091.716 | −76.385 | −76.764 | −75.885 | −37.0 | +38.9 |
| APFD/6-311++(2d,p) | −1091.443 | −1091.882 | −76.400 | −76.779 | −75.899 | −37.7 | +38.9 |
| Crystal Structure (Reference) | Docked Scoulerine | RMSD (Å) |
|---|---|---|
| 1SA0 (CN2) | S1 | 3.4 |
| 5NM5 (colchicine) | S1 | 3.5 |
| 4O4H (laulimalide) | S2 | 1.6 |
| Colchicine Binding Site A | Laulimalide Binding Site B | |||
|---|---|---|---|---|
| Name | colchicine | scoulerine | Laulimalide | scoulerine |
| B.A (kcal/mol) | −9.23 | −7.96 | −7.50 | −6.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshari, M.; Wang, Q.; Michalak, M.; Klobukowski, M.; Tuszynski, J.A. Computational Prediction and Experimental Validation of the Unique Molecular Mode of Action of Scoulerine. Molecules 2022, 27, 3991. https://doi.org/10.3390/molecules27133991
Moshari M, Wang Q, Michalak M, Klobukowski M, Tuszynski JA. Computational Prediction and Experimental Validation of the Unique Molecular Mode of Action of Scoulerine. Molecules. 2022; 27(13):3991. https://doi.org/10.3390/molecules27133991
Chicago/Turabian StyleMoshari, Mahshad, Qian Wang, Marek Michalak, Mariusz Klobukowski, and Jack Adam Tuszynski. 2022. "Computational Prediction and Experimental Validation of the Unique Molecular Mode of Action of Scoulerine" Molecules 27, no. 13: 3991. https://doi.org/10.3390/molecules27133991
APA StyleMoshari, M., Wang, Q., Michalak, M., Klobukowski, M., & Tuszynski, J. A. (2022). Computational Prediction and Experimental Validation of the Unique Molecular Mode of Action of Scoulerine. Molecules, 27(13), 3991. https://doi.org/10.3390/molecules27133991