
Stereoselective Synthesis of β-Glycinamide Ribonucleotide

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
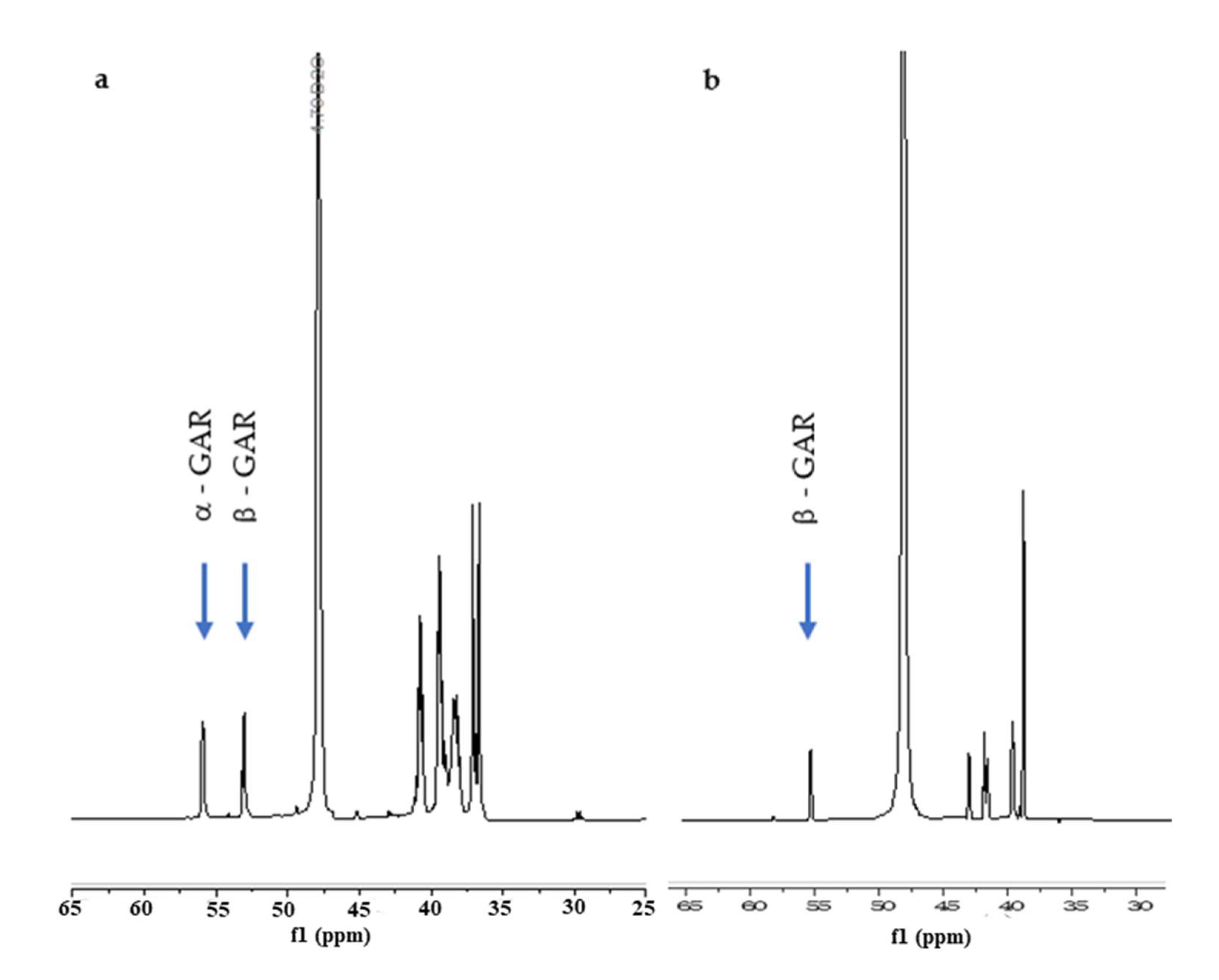
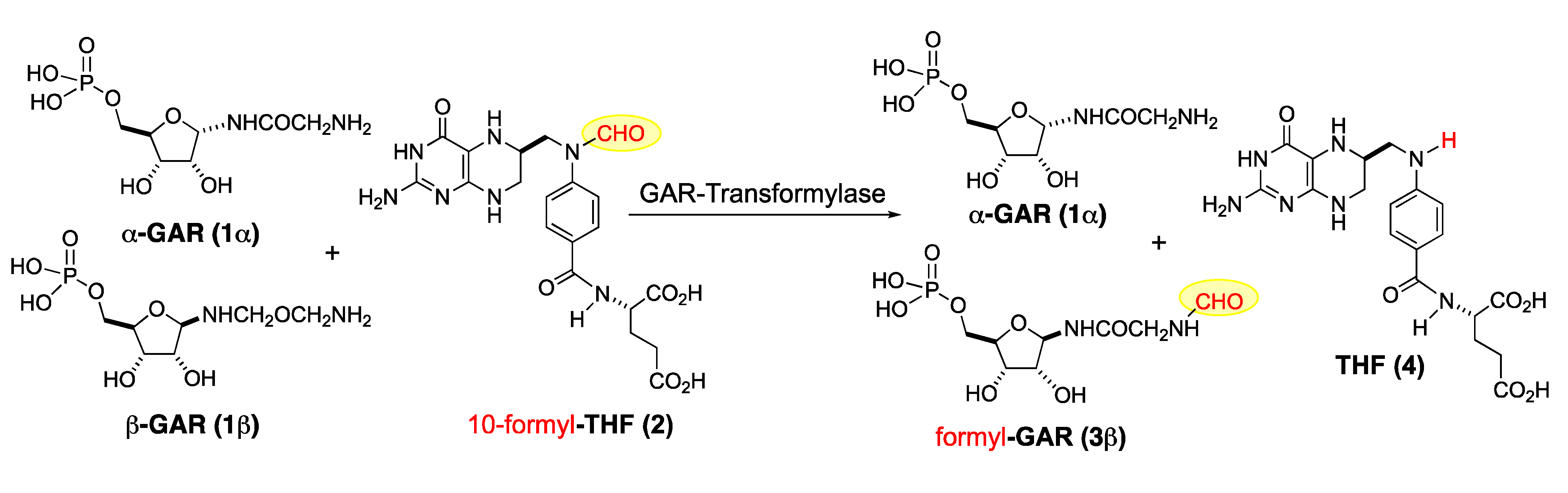
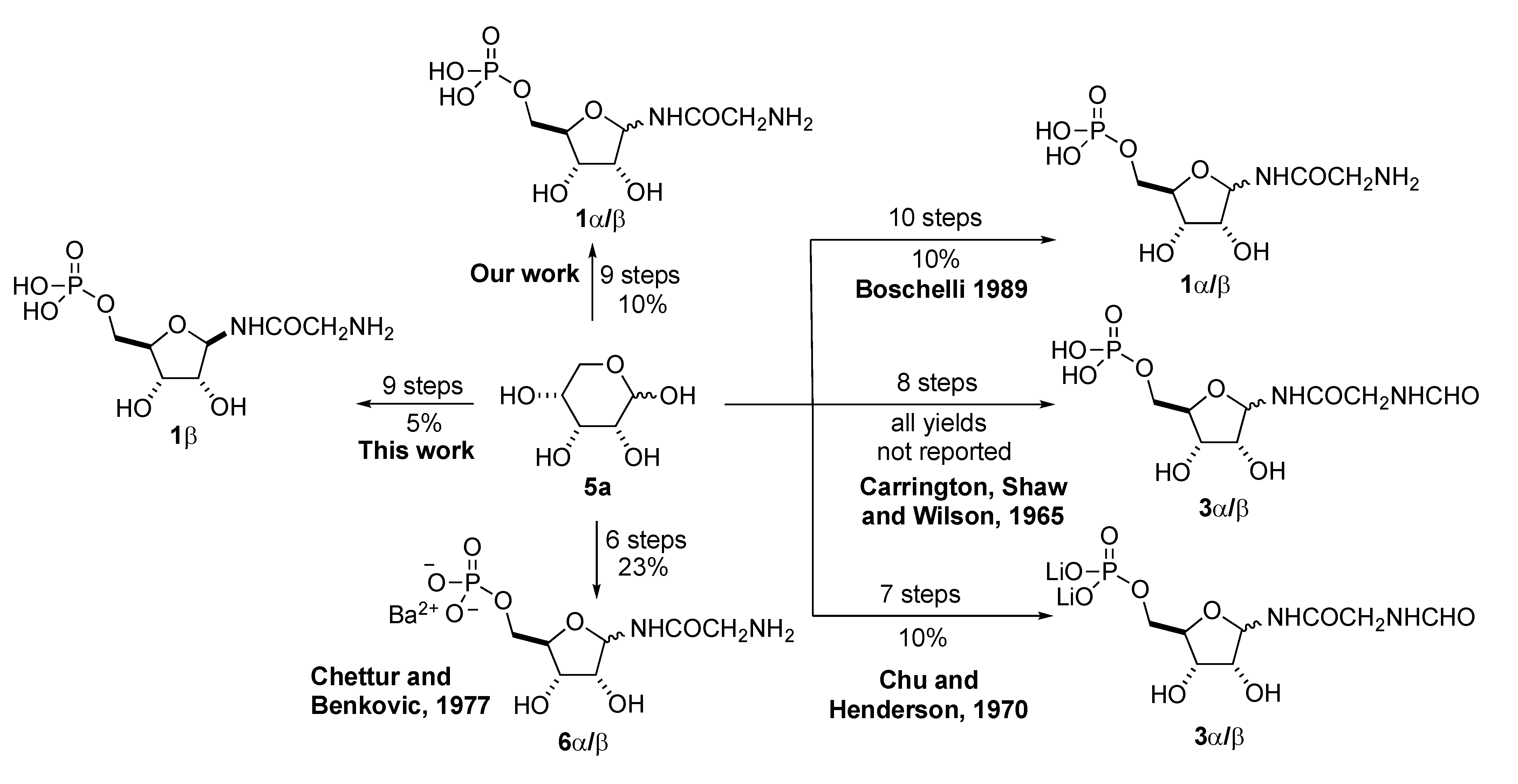
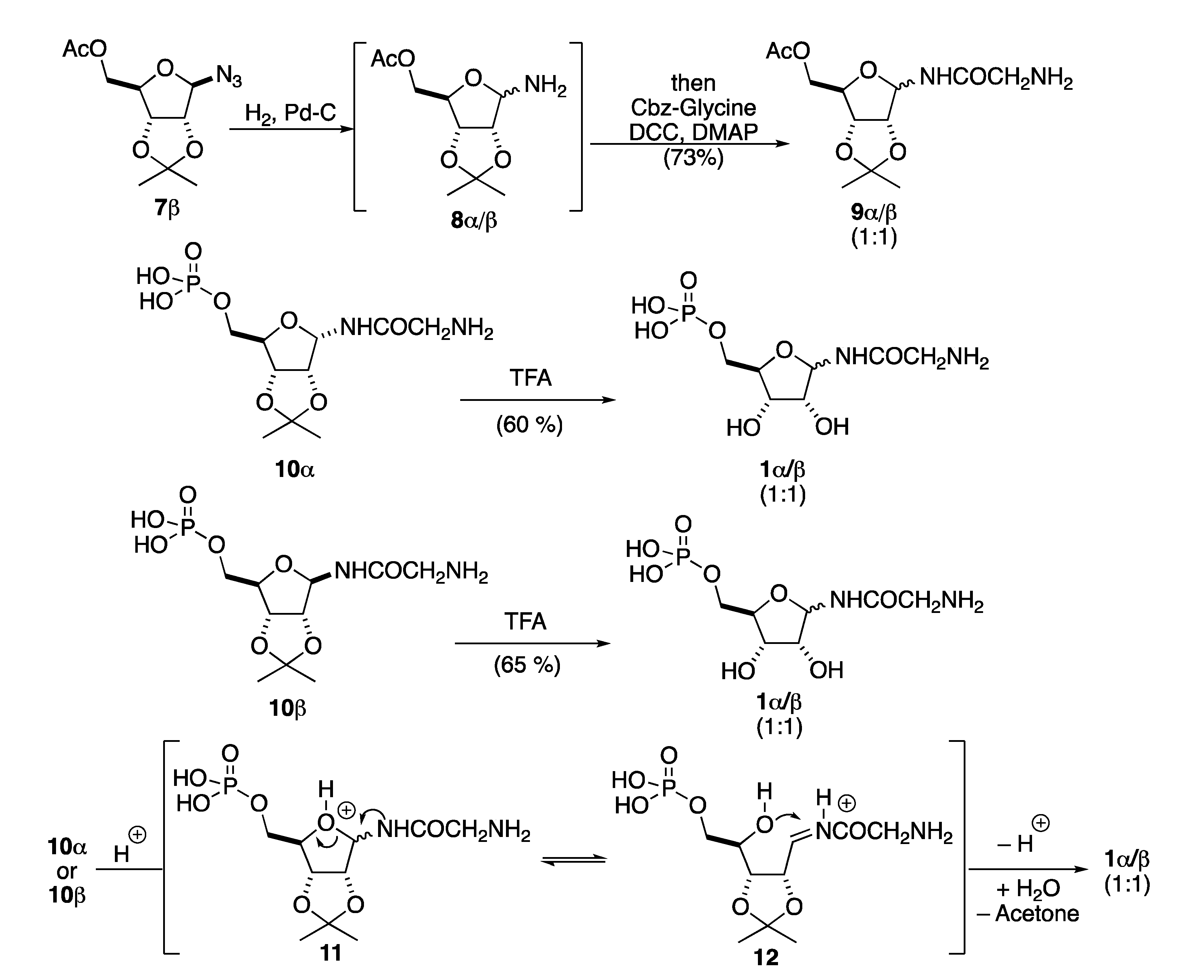
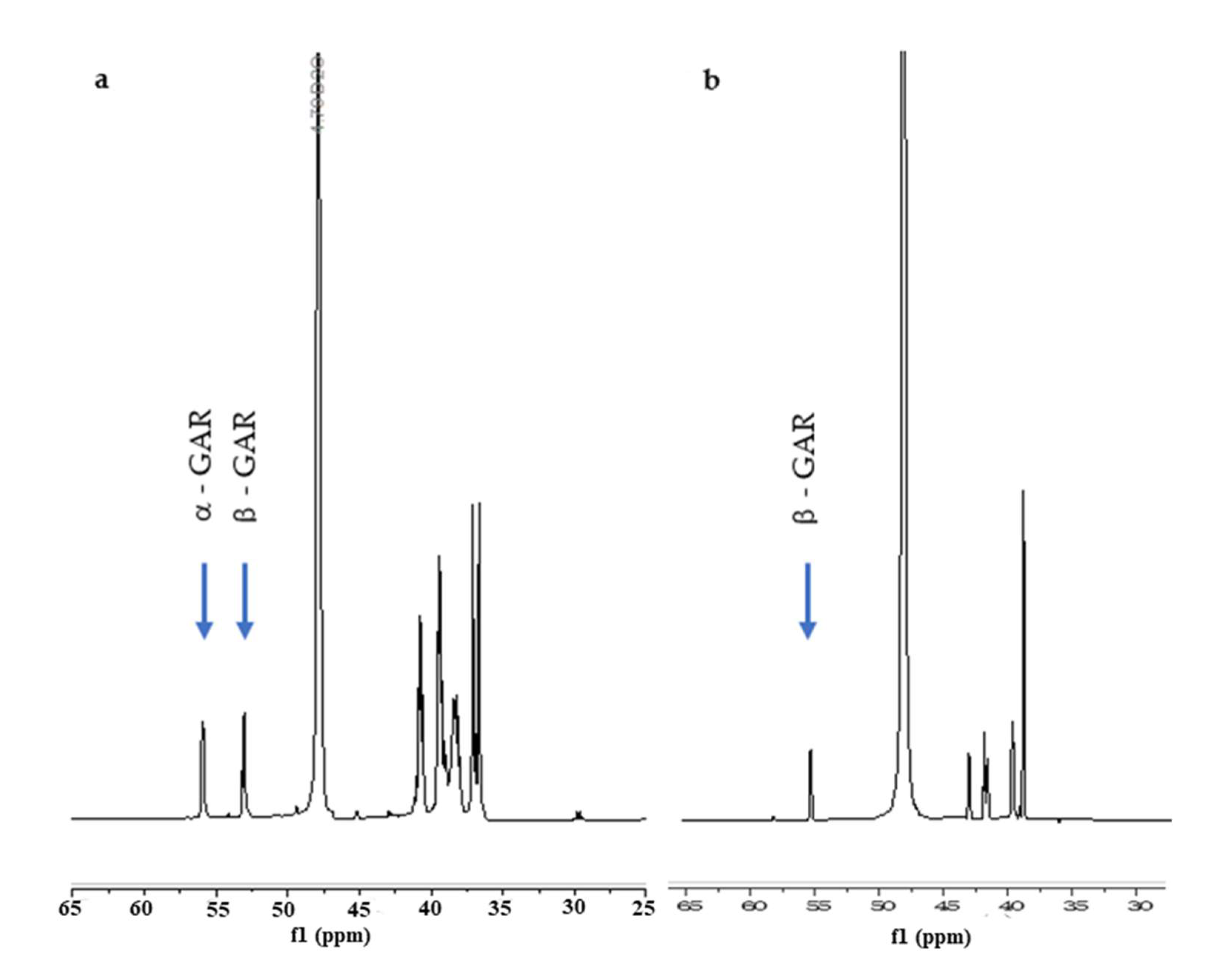
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Methods
4.2. Experimental Procedures:
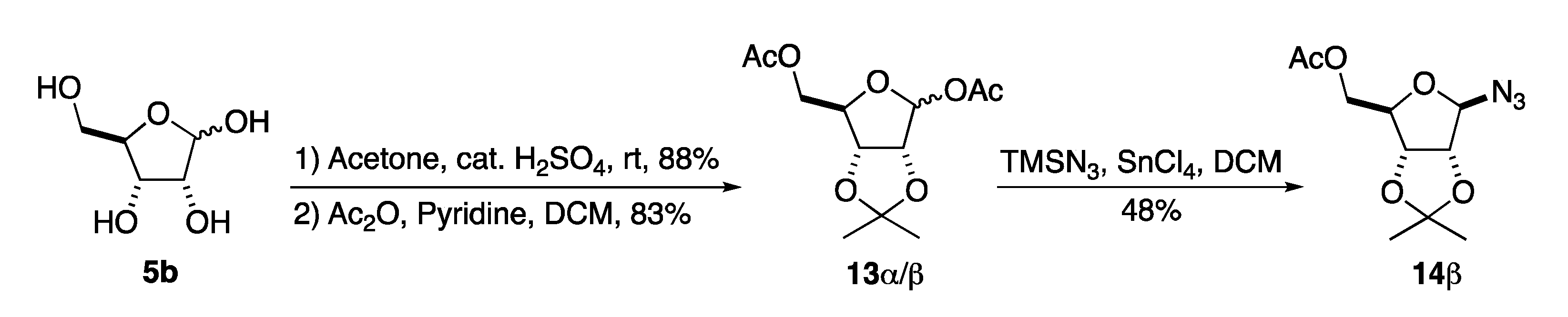
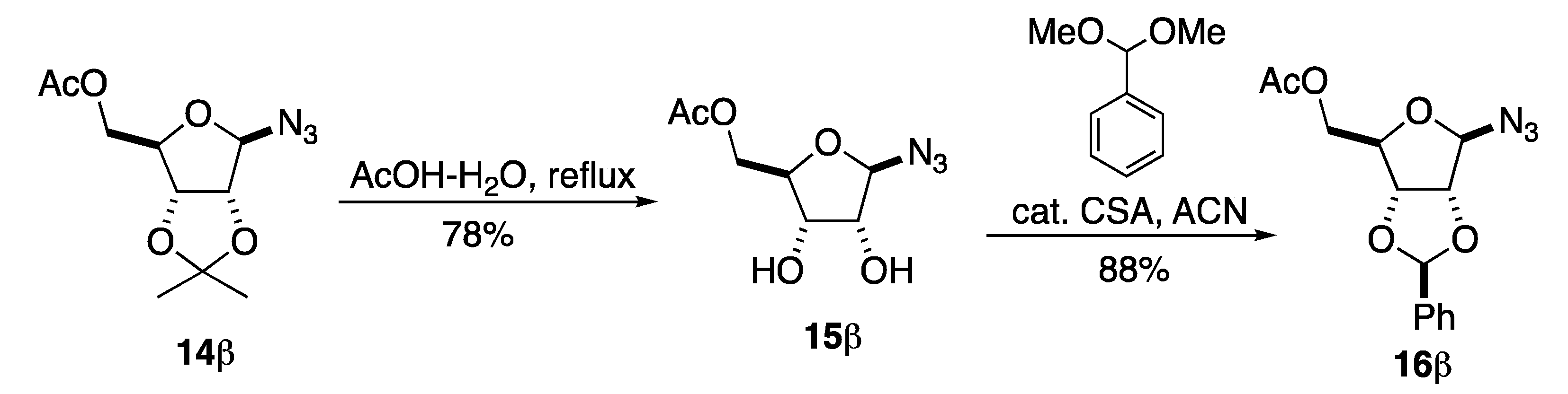
4.2.1. 1-Azido-β-d-ribofuranosyl-5-acetate (15β)
4.2.2. 2,3-O-Benzyliden-5-O-acetyl-β-d-ribofuranosyl Azide (16β)
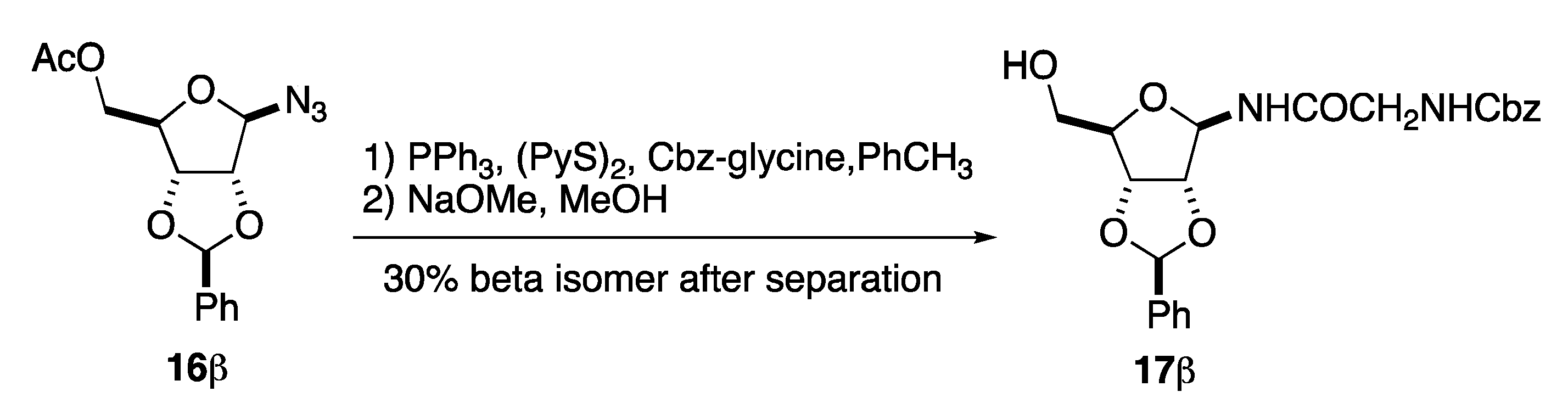
4.2.3. Benzyl(2-(2,3-O-benzyliden-5-O-acetyl-β-d-ribofuranosylamino)-2-oxoethyl)carbamate (17β)
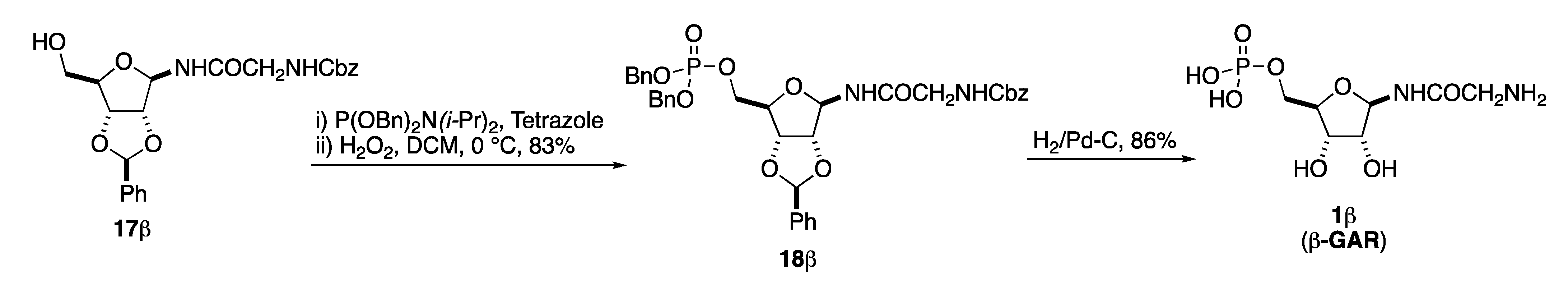
4.2.4. N-Cbz-Dibenzyl-β-glycinamide Ribonucleotide (18β)
4.2.5. β-Glycinamide Ribonucleotide (1β)
4.2.6. Isomerization of β-GAR
4.2.7. GART Activity
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Warren, L.; Flaks, J.G. Single-carbon transfer reactions and purine biosynthesis. Fed. Proc. 1956, 15, 379. [Google Scholar]
- Warren, L. Transformylation and purine biosynthesis. Fed. Proc. 1957, 16, 267. [Google Scholar]
- Warren, L.; Buchanan, J.M. Biosynthesis of the purines. J. Biol. Chem. 1957, 229, 613–626. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A new view into the regulation of purine metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic screening in cancer drug discovery—Past, present and future. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Almassy, R.J.; Janson, C.A.; Kan, C.C.; Hostomska, Z. Structures of apo and complexed Escherichia coli glycinamide ribonucleotide transformylase. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoll, J.; Johnson, A.M. Quinazoline analogues of folic acid. J. Chem. Soc. 1970, 8, 997–1002. [Google Scholar] [CrossRef]
- Caperelli, C.A.; Conigliaro, J. Synthesis of 10-Acetyl-5,8-dideazafolic acid: A potent inhibitor of glycinamide ribonucleotide transformylase. J. Med. Chem. 1986, 29, 2117–2119. [Google Scholar] [CrossRef]
- Greasley, S.E.; Yamashita, M.M.; Cai, H.; Benkovic, S.J.; Boger, D.L.; Wilson, I.A. New insights into inhibitor design from the crystal structure and NMR studies of Escherichia coli GAR transfomylase. Biochemistry 1999, 38, 16783–16793. [Google Scholar] [CrossRef]
- Smith, G.K.; Mueller, W.T.; Benkovic, P.A.; Benkovic, S.J. On the cofactor specificity of glycinamide ribonucleotide and 5-Aminoimidazole-4-carboxamide ribonucleotide transformylase from chicken liver. Biochemistry 1981, 20, 1241–1245. [Google Scholar] [CrossRef]
- Cook, R.J. Use of 10-Formyl-5,8-dideazafolate as substrate from rat 10-formyltetrahydrofolate dehydrogenase. Methods Enzymol. 1997, 281, 129–134. [Google Scholar] [PubMed]
- Antle, V.D.; Liu, D.; Mckeller, B.R.; Caperelli, C.A.; Hua, M.; Vince, R. Substrate specificity of glycinamide ribonucleotide transformylase from chicken liver. J. Biol. Chem. 1996, 271, 6045–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schendel, F.J.; Stubbe, J. Substrate Specificity of Formylglycinamide Synthetase. Biochemistry 1986, 25, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Beuning, P.J.; Ondrechen, M.J.; O’Doherty, G.A. A practical synthesis of glycinamide ribonucleotide. Heterocycles 2018, 97, 776–784. [Google Scholar]
- Chettur, G.; Benkovic, S.J. A new chemical synthesis of 2-amino-(N-D-ribofuranosyl)acetamide 5’-phosphate. Carbohydr. Res. 1977, 56, 75–86. [Google Scholar] [CrossRef]
- Carrington, R.; Shaw, G.; Wilson, J. Purines, pyrimidines, and imidazole. Part XXIII. The use of 5-phospho-β-d-ribosyl azide in a new direct synthesis of nucleotides. J. Chem. Soc. 1965, 6864–6870. [Google Scholar] [CrossRef]
- Boschelli, D.H.; Powell, D.; Sharky, V.; Semmelhack, M.F. An improved synthesis of glycinamide ribonucleotide. Tetrahedron Lett. 1989, 30, 1599–1600. [Google Scholar] [CrossRef]
- Chu, S.Y.; Henderson, J.F. New chemical synthesis of 5’-phosphoribosyl-N-formylglycineamide. Can. J. Chem. 1970, 48, 2306–2309. [Google Scholar] [CrossRef]
- Coral, J.A.; Guo, H.; Shan, M.; O’Doherty, G.A. A De Novo Asymmetric Approach To 8a-epi-Swainsonine. Heterocycles 2009, 79, 521–529. [Google Scholar]
- Abrams, J.N.; Babu, R.S.; Guo, H.; Le, D.; Le, J.; Osbourn, J.M.; O’Doherty, G.A. De Novo Asymmetric Synthesis of 8a-epi-Swainsonine. J. Org. Chem. 2008, 73, 1935–1940. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De Novo Asymmetric Syntheses of D-, L- and 8-epi-Swainsonine. Tetrahedron 2008, 64, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of D- and L-Swainsonine. Org. Lett. 2006, 8, 1609–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haukaas, M.H.; O’Doherty, G.A. Enantioselective Synthesis of N-Cbz-Protected 6-Amino-6-deoxy-Mannose, Gulose and Talose. Org. Lett. 2001, 3, 3899–3992. [Google Scholar] [CrossRef] [PubMed]
- Haukaas, M.H.; O’Doherty, G.A. Synthesis of D- and L-Deoxymannojirimycin via an Asymmetric Aminohydroxylation of Vinylfuran. Org. Lett. 2001, 3, 401–404. [Google Scholar] [CrossRef]
- Wang, H.-Y.L.; Guo, H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Rhamno Di- and Tri-saccharides related to the Anthrax Tetrasaccharide. Tetrahedron 2013, 69, 3432–3436. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Anthrax Tetrasaccharide and Analogue. J. Org. Chem. 2008, 73, 5211–5220. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of the Anthrax Tetrasaccharide via a Palladium Catalyzed Glycosylation Reaction. Angew. Chem. Int. Ed. 2007, 46, 5206–5208. [Google Scholar] [CrossRef]
- Olimpieri, F.; Volonterio, A.; Zanda, M. Three-component, one-pot sequential synthesis of 1,3-disubstituted 5-arylhydantoins. Synlett 2008, 19, 3016–3020. [Google Scholar]
- Kitagawa, M.; Ara, T.; Arifuzzaman, M.; Ioka-Nakamichi, T.; Inamoto, E.; Toyonaga, H.; Mori, H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005, 12, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Warren, M.S.; Marolewski, A.E.; Benkovic, S.J. A rapid screen of active site mutants in glycinamide ribonucleotide transformylase. Biochemistry 1996, 35, 8855–8862. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngu, L.; Ray, D.; Watson, S.S.; Beuning, P.J.; Ondrechen, M.J.; O’Doherty, G.A. Stereoselective Synthesis of β-Glycinamide Ribonucleotide. Molecules 2022, 27, 2528. https://doi.org/10.3390/molecules27082528
Ngu L, Ray D, Watson SS, Beuning PJ, Ondrechen MJ, O’Doherty GA. Stereoselective Synthesis of β-Glycinamide Ribonucleotide. Molecules. 2022; 27(8):2528. https://doi.org/10.3390/molecules27082528
Chicago/Turabian StyleNgu, Lisa, Debarpita Ray, Samantha S. Watson, Penny J. Beuning, Mary Jo Ondrechen, and George A. O’Doherty. 2022. "Stereoselective Synthesis of β-Glycinamide Ribonucleotide" Molecules 27, no. 8: 2528. https://doi.org/10.3390/molecules27082528
APA StyleNgu, L., Ray, D., Watson, S. S., Beuning, P. J., Ondrechen, M. J., & O’Doherty, G. A. (2022). Stereoselective Synthesis of β-Glycinamide Ribonucleotide. Molecules, 27(8), 2528. https://doi.org/10.3390/molecules27082528