
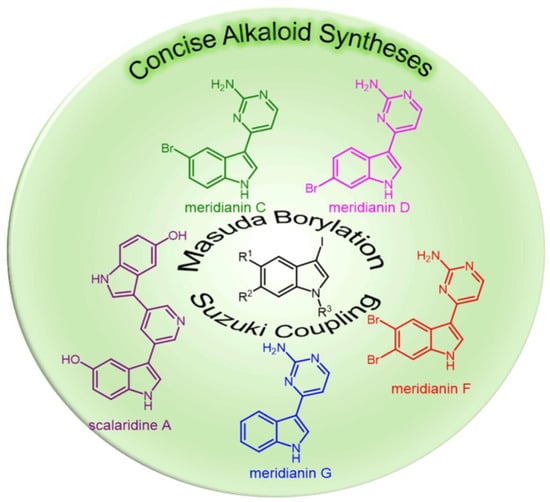
Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence



Abstract
:
{kind=link}
{kind=link}
{kind=link}
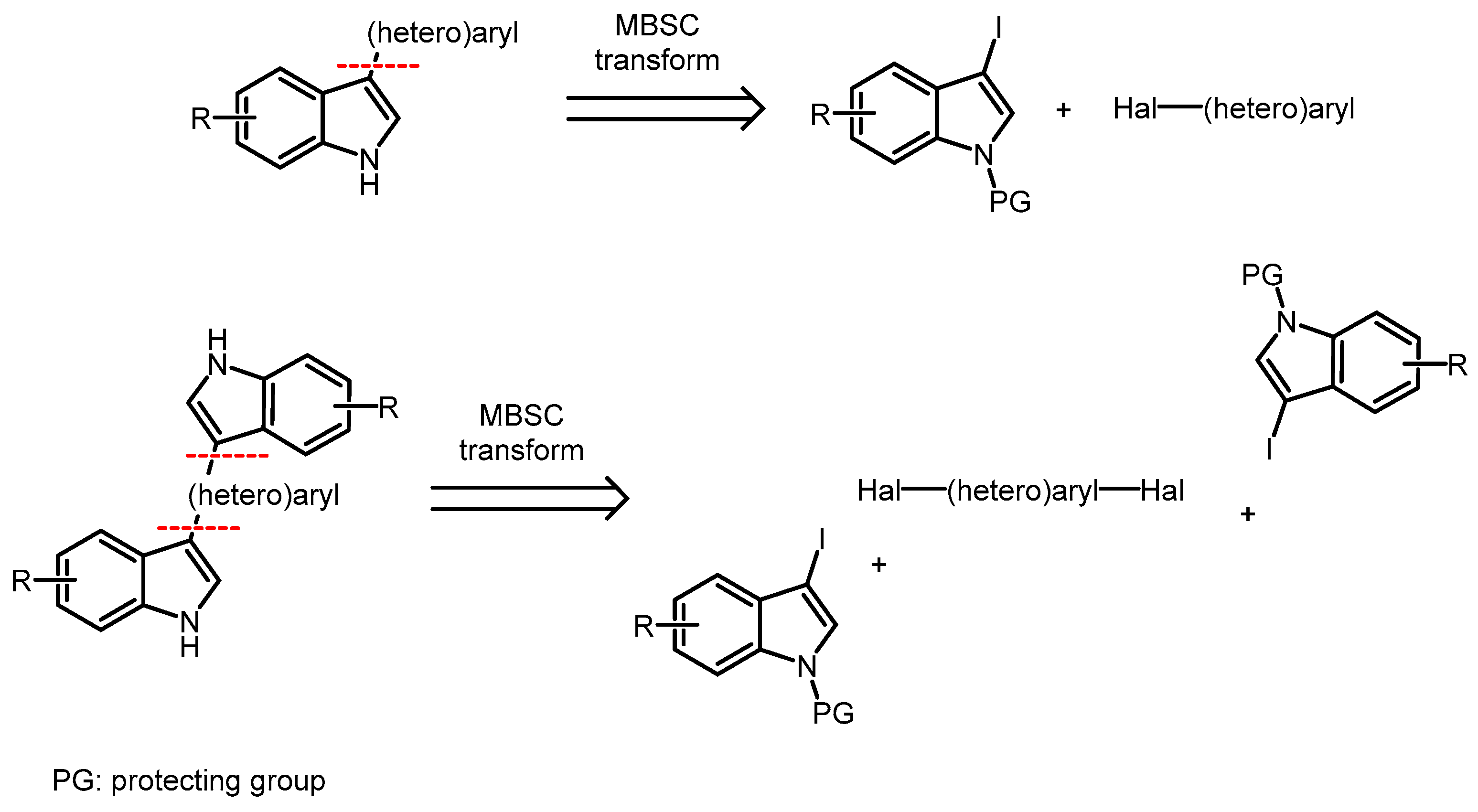{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
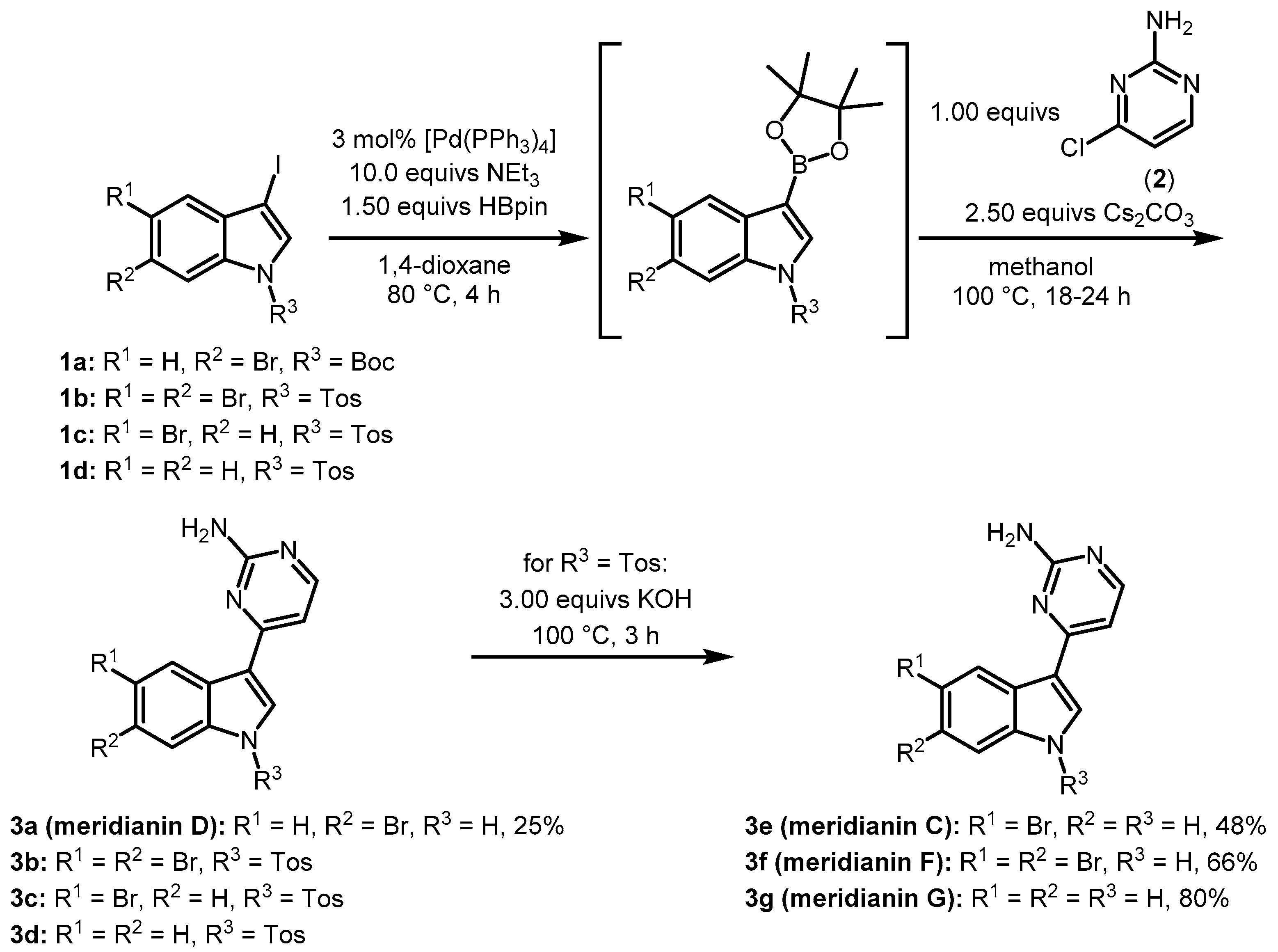
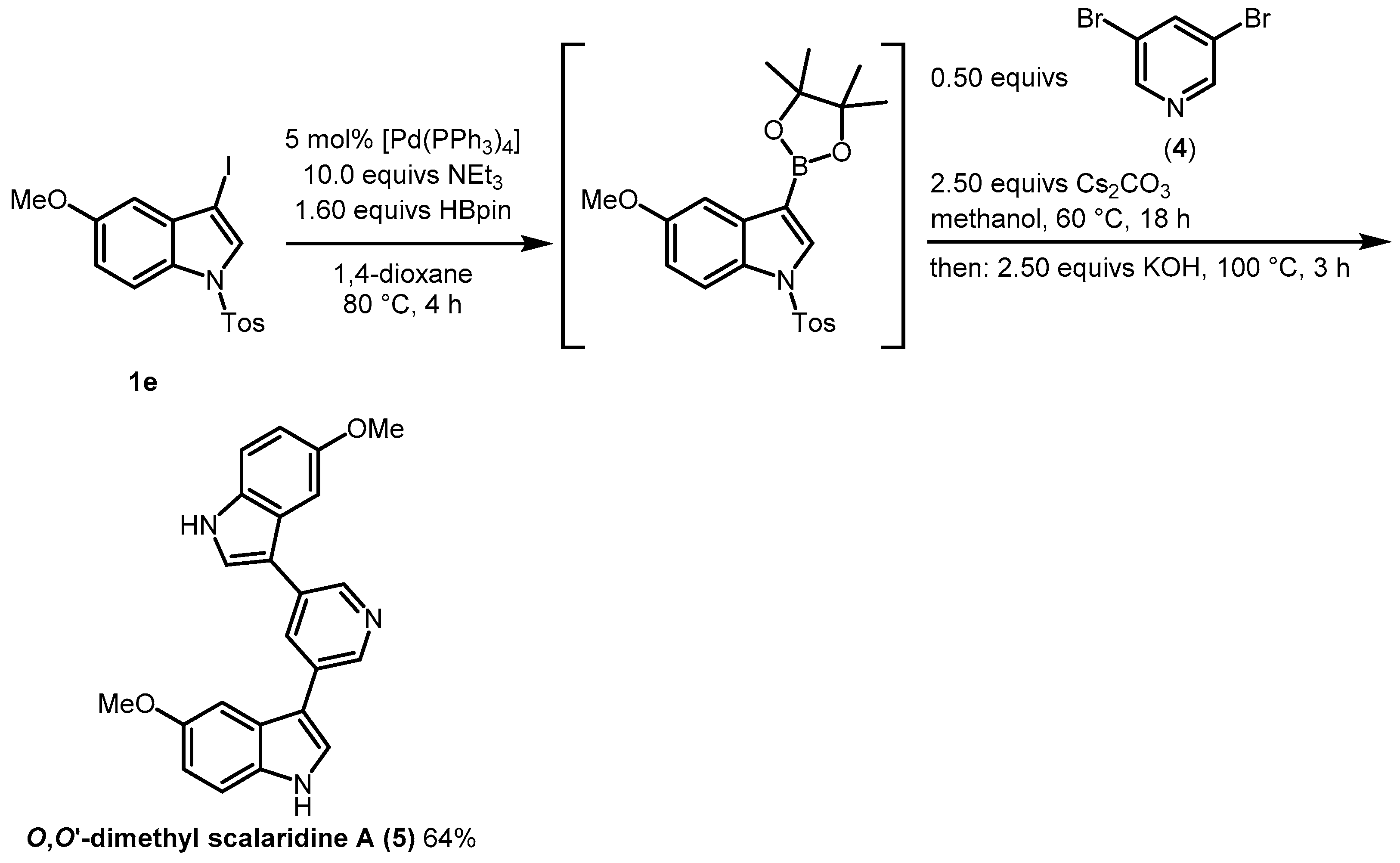
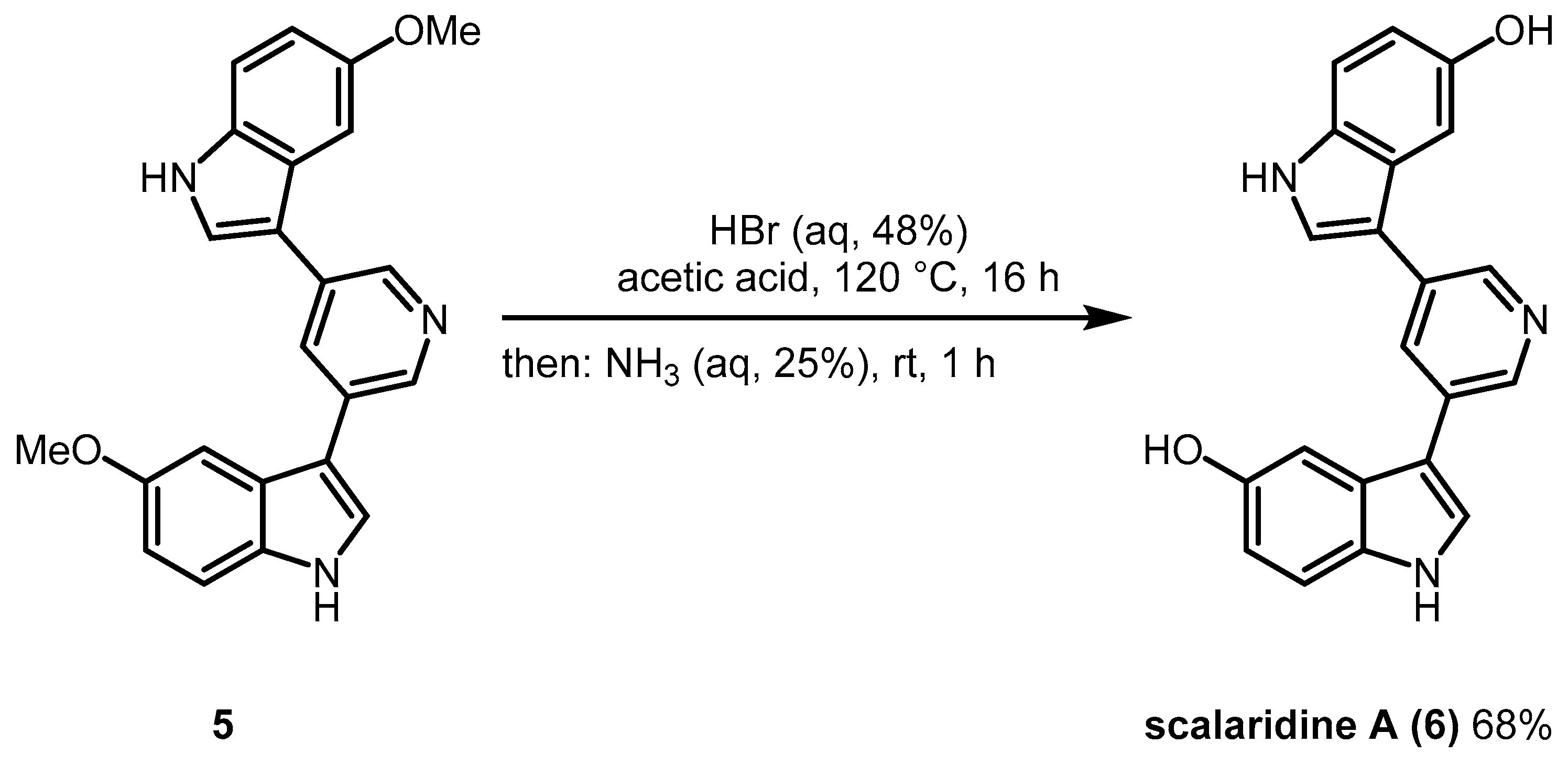
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of Meridianin D (3a)
3.3. Synthesis of Meridianin C (3e), Meridianin F (3f), and Meridianin G (3g)
3.3.1. General Procedure for the Synthesis of Meridianins 3e–g via MBSC Sequence
3.3.2. Synthesis of 4-(5-Bromo-1H-indol-3-yl)pyrimidin-2-amine, Meridianin C (3e)
3.3.3. Synthesis of 4-(5,6-Dibromo-1H-indol-3-yl)pyrimidin-2-amine, Meridianin F (3f)
3.3.4. Synthesis of 4-(1H-indol-3-yl)pyrimidin-2-amine, Meridianin G (3g)
3.4. Synthesis of Scalaridine A (6)
3.4.1. Synthesis of 3,5-Bis(5-methoxy-1H-indol-3-yl)pyridine (5)
3.4.2. Synthesis of 3,3′-(Pyridine-3,5-diyl)bis(1H-indol-5-ol), Scalaridine A (6) by Demethylation of Compound 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 2010, 9, 475–489. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Debbab, A.; Aly, A.H.; Lin, W.H.; Proksch, P. Bioactive Compounds from Marine Bacteria and Fungi. Microb. Biotechnol. 2010, 3, 544–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, J.I.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Takuma, S. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis(indolyl) imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Hughes, R.G.; Mizsak, S.A.; Scahill, T.A. Eudistomins C, E, K, and L, potent antiviral compounds containing a novel oxathiazepine ring from the Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1524–1526. [Google Scholar] [CrossRef]
- Bobzin, S.C.; Faulkner, D.J. Aromatic alkaloids from the marine sponge Chelonaplysilla sp. J. Org. Chem. 1991, 56, 4403–4407. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Jiang, B.; Smallheer, J.M.; Amaral-Ly, C.; Wuonola, M.A. Total Synthesis of (±)-Dragmacidin: A Cytotoxic Bis(indole)alkaloid of Marine Origin. J. Org. Chem. 1994, 59, 6823–6827. [Google Scholar] [CrossRef]
- Bifulco, G.; Bruno, I.; Minale, L.; Riccio, R.; Calignano, A.; Debitus, C. (±)-Gelliusines A and B, Two Diastereomeric Brominated Tris-indole Alkaloids from a Deep Water New Caledonian Marine Sponge (Gellius or Orina sp.). J. Nat. Prod. 1994, 57, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.H.; de Kier Joffé, E.B.; Puricelli, L.; Tatian, M.; Seldes, A.M.; Palermo, J.A. Indole alkaloids from the tunicate Aplidiummeridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef]
- Seldes, A.; Rodriguez Brasco, M.; Hernández Franco, L.; Palermo, J. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007, 21, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Gompel, M.; Leost, M.; de Kier Joffé, E.B.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, D.G.; Rho, H.S.; Krasokhin, V.B.; Shin, H.J.; Lee, J.S.; Lee, H.S. Cytotoxic 5-Hydroxyindole Alkaloids from the Marine Sponge Scalarispongia sp. J. Heterocycl. Chem. 2013, 50, 1400–1404. [Google Scholar] [CrossRef]
- Murata, M.; Watanabe, S.; Masuda, Y. Novel Palladium(0)-Catalyzed Coupling Reaction of Dialkoxyborane with Aryl Halides: Convenient Synthetic Route to Arylboronates. J. Org. Chem. 1997, 62, 6458–6459. [Google Scholar] [CrossRef]
- Murata, M.; Oyama, T.; Watanabe, S.; Masuda, Y. Palladium-Catalyzed Borylation of Aryl Halides or Triflates with Dialkoxyborane: A Novel and Facile Synthetic Route to Arylboronates. J. Org. Chem. 2000, 65, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O.; Guénard, D.; Guéritte, F. Palladium-Catalyzed Borylation of Ortho-Substituted Phenyl Halides and Application to the One-Pot Synthesis of 2,2′-Disubstituted Biphenyls. J. Org. Chem. 2000, 65, 9268–9271. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, O.; Cesario, M.; Guénard, D.; Guéritte, F. Application of the Palladium-Catalyzed Borylation/Suzuki Coupling (BSC) Reaction to the Synthesis of Biologically Active Biaryl Lactams. J. Org. Chem. 2002, 67, 1199–1207. [Google Scholar] [CrossRef]
- Penhoat, M.; Levacher, V.; Dupas, G. Novel Extension of Meyers’ Methodology: Stereoselective Construction of Axially Chiral 7,5-Fused Bicyclic Lactams. J. Org. Chem. 2003, 68, 9517–9520. [Google Scholar] [CrossRef]
- Broutin, P.-E.; Čerňa, I.; Campaniello, M.; Leroux, F.; Colobert, F. Palladium-Catalyzed Borylation of Phenyl Bromides and Application in One-Pot Suzuki−Miyaura Biphenyl Synthesis. Org. Lett. 2004, 6, 4419–4422. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.S.; Ferreira, P.M.T.; Queiroz, M.-J.R.P.; Ferreira, I.C.F.R.; Calhelha, R.C.; Estevinho, L.M. Synthesis of β-Benzo[b]thienyldehydrophenylalanine Derivatives by One-Pot Palladium-Catalyzed Borylation and Suzuki Coupling (BSC) and Metal-Assisted Intramolecular Cyclization–Studies of Fluorescence and Antimicrobial Activity. Eur. J. Org. Chem. 2005, 2005, 2951–2957. [Google Scholar] [CrossRef] [Green Version]
- Merkul, E.; Schäfer, E.; Müller, T.J.J. Rapid synthesis of bis(hetero)aryls by one-pot Masuda borylation-Suzuki coupling sequence and its application to concise total syntheses of meridianins A and G. Org. Biomol. Chem. 2011, 9, 3139–3141. [Google Scholar] [CrossRef]
- Tasch, B.O.A.; Merkul, E.; Müller, T.J.J. One-Pot Synthesis of Diazine-Bridged Bisindoles and Concise Synthesis of the Marine Alkaloid Hyrtinadine A. Eur. J. Org. Chem. 2011, 2011, 4532–4535. [Google Scholar] [CrossRef]
- Tasch, B.O.A.; Antovic, D.; Merkul, E.; Müller, T.J.J. One-Pot Synthesis of Camalexins and 3,3′-Biindoles by the Masuda Borylation–Suzuki Arylation (MBSA) Sequence. Eur. J. Org. Chem. 2013, 2013, 4564–4569. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P.; Delgado, S.; Bleda, J.A. Synthetic studies towards the 2-aminopyrimidine alkaloids variolins and meridianins from marine origin. Tetrahedron Lett. 2000, 41, 4777–4780. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P.; Bleda, J.A. Synthesis of the indole alkaloids meridianins from the tunicate Aplidium meridianum. Tetrahedron 2001, 57, 2355–2363. [Google Scholar] [CrossRef]
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clément, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.C.; Endicott, J.A.; et al. Meriolins (3-(Pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/Cyclin A/Meriolin Complex. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef]
- Sperry, J. A concise synthesis of meridianin F. Tetrahedron Lett. 2011, 52, 4537–4538. [Google Scholar] [CrossRef]
- Tibiletti, F.; Simonetti, M.; Nicholas, K.M.; Palmisano, G.; Parravicini, M.; Imbesi, F.; Tollari, S.; Penoni, A. One-pot synthesis of meridianins and meridianin analogues via indolization of nitrosoarenes. Tetrahedron 2010, 66, 1280–1288. [Google Scholar] [CrossRef]
- Parsons, T.B.; Ghellamallah, C.; Male, L.; Spencer, N.; Grainger, R.S. Regioselective dibromination of methyl indole-3-carboxylate and application in the synthesis of 5,6-dibromoindoles. Org. Biomol. Chem. 2011, 9, 5021–5023. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Czyz, M.L.; Morris, J.C. Concise Syntheses of Meridianins and Meriolins Using a Catalytic Domino Amino-Palladation Reaction. Org. Lett. 2014, 16, 708–711. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.-G. Synthesis of Indolylpyrimidines via Cross-Coupling of Indolylboronic Acid with Choropyrimidines: Facile Synthesis of Meridianin D. Heterocycles 2000, 53, 1489–1498. [Google Scholar] [CrossRef]
- Dorsch, D.; Sirrenberg, C.; Müller, T.J.J. Preparation of 4-(Pyrrolopyridinyl)pyrimidinyl-2-amines as Antitumor Agents. Patent WO2007107221 A1, 27 September 2007. [Google Scholar]
- Dorsch, D.; Wuchrer, M.; Burgdorf, L.T.; Sirrenberg, C.; Esdar, C.; Müller, T.J.J.; Merkul, E. Preparation of Pyrrolopyridinylpyrimidinylamines as Inhibitors of Cell Proliferation. Patent WO2008155000 A1, 24 December 2008. [Google Scholar]
- Dorsch, D.; Sirrenberg, C.; Müller, T.J.J.; Merkul, E. Preparation of 3-(4-pyridinyl)-1H-pyrrolo[2,3-b]pyridines as Anti-Tumor Agents. Patent DE102008025751 A1, 3 December 2009. [Google Scholar]
- Dorsch, D.; Sirrenberg, C.; Müller, T.J.J.; Merkul, E.; Karapetyan, G. 7-Azaindole Derivatives as Kinase Inhibitors and Their Preparation and Use in the Treatment of Tumors. Patent WO2012104007 A2, 9 August 2012. [Google Scholar]
- Drießen, D.; Stuhldreier, F.; Frank, A.; Stark, H.; Wesselborg, S.; Stork, B.; Müller, T.J.J. Novel meriolin derivatives as rapid apoptosis inducers. Bioorg. Med. Chem. 2019, 27, 3463–3468. [Google Scholar] [CrossRef]
- Müller, T.J.J. Sequentially Palladium-Catalyzed Processes. Top. Organomet. Chem. 2006, 19, 149–205. [Google Scholar] [CrossRef]
- Lessing, T.; Müller, T.J.J. Sequentially Palladium-Catalyzed Processes in One-Pot Syntheses of Heterocycles. Appl. Sci. 2015, 5, 1803–1836. [Google Scholar] [CrossRef] [Green Version]
- Rehberg, N.; Sommer, G.A.; Drießen, D.; Kruppa, M.; Adeniyi, E.T.; Chen, S.; Wang, L.; Wolf, K.; Tasch, B.O.A.; Ioerger, T.R.; et al. Nature-Inspired (di)Azine-Bridged Bisindole Alkaloids with Potent Antibacterial In Vitro and In Vivo Efficacy against Methicillin-Resistant Staphylococcus aureus. J. Med. Chem. 2020, 63, 12623–12641. [Google Scholar] [CrossRef]
- Karpov, A.S.; Merkul, E.; Rominger, F.; Müller, T.J.J. Concise Syntheses of Meridianins by Carbonylative Alkynylation and a Four-Component Pyrimidine Synthesis. Angew. Chem. Int. Ed. 2005, 44, 6951–6956. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sperry, J. Synthesis of scalaridine A. Tetrahedron Lett. 2015, 56, 5914–5915. [Google Scholar] [CrossRef]
- Ansari, N.H.; Söderberg, B.C.G. Short syntheses of the indole alkaloids alocasin A, scalaridine A, and hyrtinadine A-B. Tetrahedron 2016, 72, 4214–4221. [Google Scholar] [CrossRef] [Green Version]
- Ranjani, G.; Nagarajan, R. Insight into Copper Catalysis: In Situ Formed Nano Cu2O in Suzuki–Miyaura Cross-Coupling of Aryl/Indolyl Boronates. Org. Lett. 2017, 19, 3974–3977. [Google Scholar] [CrossRef] [PubMed]
- Witulski, B.; Buschmann, N.; Bergsträßer, U. Hydroboration and Suzuki–Miyaura Coupling Reactions with the Electronically Modulated Variant of an Ynamine: The Synthesis of (E)-β-Arylenamides. Tetrahedron 2000, 56, 8473–8480. [Google Scholar] [CrossRef]
- Kim, S.H.; Sperry, J. Synthesis of Alocasin A. J. Nat. Prod. 2015, 78, 3080–3082. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, G.E.; Johnson, T.B. Researches on pyrimidines. CXIII. An improved method for the synthesis of cytosine1. J. Am. Chem. Soc. 1930, 52, 1152–1157. [Google Scholar] [CrossRef]
- Kofler, A.; Kolšek, J. Beitrag zur mikroskopischen Identifizierung organischer Stoffe nach L. Kofler. IV. Microchim. Acta 1970, 58, 1063–1088. [Google Scholar] [CrossRef]
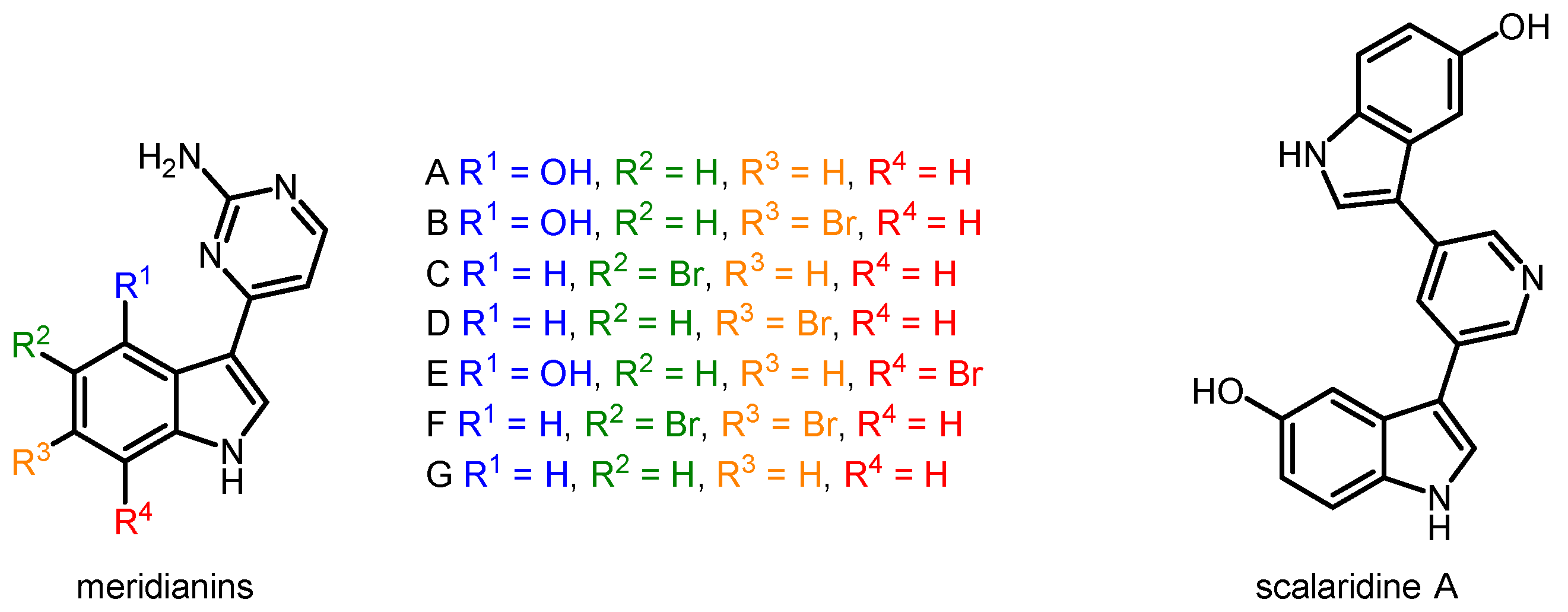

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruppa, M.; Sommer, G.A.; Müller, T.J.J. Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence. Molecules 2022, 27, 2233. https://doi.org/10.3390/molecules27072233
Kruppa M, Sommer GA, Müller TJJ. Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence. Molecules. 2022; 27(7):2233. https://doi.org/10.3390/molecules27072233
Chicago/Turabian StyleKruppa, Marco, Gereon A. Sommer, and Thomas J. J. Müller. 2022. "Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence" Molecules 27, no. 7: 2233. https://doi.org/10.3390/molecules27072233
APA StyleKruppa, M., Sommer, G. A., & Müller, T. J. J. (2022). Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence. Molecules, 27(7), 2233. https://doi.org/10.3390/molecules27072233