3.4. Synthesis of Chiral Oxazolines
3.4.1. 2-(Benzyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 26
A solution of 2-(benzyloxy)benzoyl chloride
25 (6.27 g, 25.4 mmol) in CH
2Cl
2 (40 cm
3) was added dropwise to a stirred 0 °C solution of (±)-2-amino-2-phenylethan-1-ol [
22] (3.77 g, 27.5 mmol) and triethylamine (3.9 cm
3, 2.83 g, 28.0 mmol) in CH
2Cl
2 (40 cm
3). Once the addition was complete, the reaction mixture was allowed to warm to rt for 20 h before being poured into water (100 cm
3). The two layers were separated and the aqueous layer was re-extracted with CH
2Cl
2 (2 × 50 cm
3). The combined organic layers were washed with 2 M HCl (100 cm
3), 2 M NaOH (100 cm
3) and water (100 cm
3) before being dried and evaporated to give
26 (6.83 g, 77%) as a pale yellow solid which was used without further purification; mp 143–146 °C; ν
max/cm
−1 3377, 3032, 2943, 1622, 1597, 1549, 1485, 1449, 1302, 1236, 1070, 989, 748 and 694; δ
H (400 MHz) 8.55 (1 H, d,
J 6.8, NH), 8.23 (1 H, dd,
J 7.8, 1.8, ArH), 7.48–7.38 (6 H, m, ArH), 7.21–7.14 (3 H, m, ArH), 7.12–7.07 (2 H, m, ArH), 7.01–6.98 (2 H, m, ArH), 5.24 (1 H, td,
J 6.8, 4.4, NCH), 5.16 and 5.12 (2 H, AB pattern,
JAB 10.2, OCH
2Ph), 3.76–3.67 (2 H, m, C
H2OH) and 2.61 (1 H, br s, OH); δ
C (100 MHz) 165.7 (C=O), 156.9 (C–O), 138.9 (C), 135.2 (C), 133.0 (CH), 132.5 (CH), 129.0 (CH), 128.9 (2CH), 128.72 (2CH), 128.70 (2CH), 127.5 (CH), 126.6 (2CH), 121.6 (CH), 121.3 (C), 112.3 (CH), 71.5 (OCH
2Ph), 67.4 (CH
2OH) and 56.4 (NCH); HRMS (NSI
+): found 348.1595. C
22H
22NO
3 (M + H) requires 348.1594.
3.4.2. 2-(Benzyloxy)-N-(2-chloro-1-phenylethyl)benzamide 27
Thionyl chloride (1.7 cm3, 2.77 g, 23.3 mmol) was added to a solution of 2-(benzyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 26 (6.50 g, 18.7 mmol) in CH2Cl2 (100 cm3) and the reaction mixture was stirred at rt for 18 h. The reaction mixture was washed with 2 M NaOH (100 cm3) and water (100 cm3) before being dried and evaporated to give, after purification of the crude residue by column chromatography (SiO2, Et2O/hexane 3:2), at Rf 0.70, 27 (5.21 g, 76%) as a colourless solid; mp 90–93 °C; νmax/cm−1 3368, 3059, 1645, 1595, 1522, 1479, 1285, 1217, 1157, 991, 752, 694 and 621; δH (500 MHz) 8.63 (1 H, d, J 8.0, NH), 8.25 (1 H, dd, J 8.0, 2.0, ArH), 7.51–7.40 (6 H, m, ArH), 7.35–7.27 (1 H, m, ArH), 7.23–7.18 (2 H, m, ArH), 7.13–7.10 (2 H, m, ArH), 7.03–7.00 (2 H, m, ArH), 5.51 (1 H, dt, J 8.0, 5.5, NCH), 5.21 and 5.17 (2 H, AB pattern, JAB 10.0, OCH2Ph), 3.74 (1 H, half AB pattern of d, JAB 11.0, JAX 5.0, CHHCl), 3.62 (1 H, half AB pattern of d, JAB 11.0, JAX 5.5, CHHCl); δC (125 MHz) 164.5 (C=O), 157.0 (C–O), 138.7 (C), 135.2 (C), 133.1 (CH), 132.6 (CH), 129.1 (CH), 129.0 (2CH), 128.9 (2CH), 128.6 (2CH), 127.7 (CH), 126.7 (2CH), 121.6 (CH), 121.0 (C), 112.3 (CH), 71.5 (OCH2), 54.1 (NCH) and 48.0 (CH2Cl); HRMS (NSI+): found 366.1250. C22H2135ClNO2 (M + H) requires 366.1255.
3.4.3. 2-(2-(Benzyloxy)phenyl)-4-phenyl-4,5-dihydrooxazole 28
A literature procedure [
15] was adapted as follows: A mixture of 2-(benzyloxy)-
N-(2-chloro-1-phenylethyl)benzamide
27 (4.74 g, 13.0 mmol) and sodium hydroxide (0.82 g, 20.5 mmol) in methanol (100 cm
3) was heated at reflux for 3 h. After cooling to rt, the reaction mixture was diluted with Et
2O (250 cm
3) and washed with brine (3 × 100 cm
3). The organic layer was dried and evaporated to give
28 (4.24 g, 99%) as a colourless oil which solidified on standing; mp 47–51 °C; ν
max/cm
−1 3061, 3034, 1665, 1493, 1447, 1250, 1034, 1001, 750 and 696; δ
H (500 MHz) 7.84 (1 H, dd,
J 7.8, 1.8, ArH), 7.49 (2 H, d,
J 7.0, Ph), 7.44–7.40 (1 H, m, ArH), 7.35–7.27 (8 H, m, ArH), 7.05–7.00 (2 H, m, ArH), 5.42 (1 H, dd,
J 10.0, 8.0, NCH), 5.22 and 5.20 (2 H, AB pattern,
JAB 12.0, OCH
2Ph), 4.78 (1 H, dd,
J 10.0, 8.0, oxazoline C
HH) and 4.26 (1 H, t,
J 8.0, oxazoline CH
H); δ
C (125 MHz) 163.8 (C=N), 157.5 (C–O), 142.6 (C), 136.8 (C), 132.3 (CH), 131.4 (CH), 128.6 (2CH), 128.4 (2CH), 127.6 (CH), 127.4 (CH), 127.0 (2CH), 126.8 (2CH), 120.7 (CH), 118.0 (C), 113.5 (CH), 74.4 (CH
2), 70.6 (CH
2) and 70.2 (NCH); HRMS (NSI
+): found 330.1488. C
22H
20NO
2 (M + H) requires 330.1489.
3.4.4. 2-(Allyloxy)benzoyl Chloride 29
Thionyl chloride (6.51 cm
3, 10.68 g, 89.8 mmol) was added dropwise to a stirred solution of 2-(allyloxy)benzoic acid [
23]
2 (8.00 g, 44.9 mmol) in toluene (70 cm
3) at rt and the mixture heated to reflux for 3 h then cooled to rt and concentrated. The crude residue was purified via Kugelrohr distillation to give
29 (5.79 g, 66%) as a colourless oil, bp 182 °C/20 Torr (lit. [
24] bp 80−82 °C/0.2 Torr); δ
H (400 MHz) 8.12 (1H, dd,
J 8.1, 1.8, ArH), 7.58 (1H, ddd,
J 8.4, 7.4, 1.8, ArH), 7.08 (1H, ddd,
J 8.1, 7.4 1.0, ArH), 7.01 (1H, dd,
J 8.4, 1.0, ArH), 6.07 (1H, ddt,
J 17.2, 10.6, 4.8, C
H=CH
2), 5.56 (1H, dq,
J 17.3, 1.7, CH=C
HH), 5.36 (1H, dq,
J 10.6, 1.5, CH=CH
H) and 4.68 (2H, dt,
J 4.8, 1.7, OCH
2); δ
C (100 MHz) 163.7 (C=O), 158.5 (C-O), 136.0 (CH), 134.5 (CH), 131.8 (CH), 122.4 (C) 120.4 (CH), 117.8 (=CH
2), 113.2 (CH) and 69.3 (OCH
2). The
1H spectral data were in accordance with those previously reported [
25]. The
13C spectral are reported for the first time.
3.4.5. (S)-2-(Allyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 30
A solution of 2-(allyloxy)benzoyl chloride
29 (5.00 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3) was stirred at 0 °C while triethylamine (3.53 cm
3, 2.57 g, 25.4 mmol) was added dropwise, followed by dropwise addition of a solution of (
S)-2-amino-2-phenylethan-1-ol [
26] (3.48 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3). After stirring at RT for 18 h, the mixture was poured into water (50 cm
3) and the organic layer was separated. Extraction of the aqueous layer with CH
2Cl
2 (2 × 20 cm
3) followed by drying and evaporation of the combined organic solutions gave, after recrystallisation from EtOAc/hexane,
30 (5.86 g, 78%), colourless crystals; mp 65−67 °C; ν
max/cm
−1 3300, 2947, 1632, 1601, 1539, 1233, 993, 934, 752, 719, 694 and 529; δ
H (400 MHz) 8.72 (1H, d,
J 6.7 NH), 8.18 (1H, dd,
J 7.8, 1.9, ArH), 7.44 (1H, ddd,
J 8.3, 7.4, 1.9 ArH), 7.39−7.33 (4H, m, ArH), 7.33−7.28 (1H, m, ArH), 7.05 (1H, ddd,
J 8.1, 7.4, 1.0, ArH), 6.93 (1H, dd,
J 8.4, 1.0, ArH), 5.98 (1H, ddt,
J 17.2, 10.4, 5.9, C
H=CH
2), 5.38 (1H, dq,
J 17.2, 1.4, CH=C
HH), 5.34−5.26 (2H, m, CH=CH
H and CHN), 4.63−4.57 (2H, m, OCH
2), 3.92−3.88 (2H, m, CHC
H2OH) and 3.49 (1H, s, OH); δ
C (100 MHz) 165.8 (C=O), 156.6 (C-O), 139.2 (C), 132.9 (
CH=CH
2), 132.2 (CH), 131.8 (CH), 128.7 (2CH), 127.6 (CH), 126.8 (2CH), 121.4 (CH), 121.1 (C), 119.7 (=CH
2), 112.4 (CH), 70.0 (OCH
2), 67.2 (CH
2OH) and 56.7 (
CHNH); [α]
D −32.4 (
c 1.002, CH
2Cl
2); HRMS (NSI
+): found 298.1438. C
18H
20NO
3 (M + H) requires 298.1443.
3.4.6. (R)-2-(Allyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 31
Following the procedure of 3.4.5 using 2-(allyloxy)benzoyl chloride
29 (5.00 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3) and (
R)-2-amino-2-phenylethan-1-ol [
27] (3.48 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3) gave
31 (6.59 g, 87%) as colourless crystals; mp 71−73 °C; ν
max/cm
−1 3348, 1624, 1522, 1234, 1034, 1026, 989, 920, 754, 700, 530 and 521; δ
H (400 MHz) 8.72 (1H, d,
J 6.6, NH), 8.20 (1H, dd,
J 7.8, 1.9, ArH), 7.44 (1H, ddd,
J 8.4, 7.3, 1.9, ArH), 7.39−7.34 (4H, m), 7.33−7.28 (1H, m, ArH), 7.07 (1H, ddd,
J 8.1, 7.3, 1.0 ArH), 6.95 (1H, dd,
J 8.4, 1.0, ArH), 5.99 (1H, ddt,
J 17.2, 10.4, 5.9, C
H=CH
2), 5.39 (1H, dq,
J 17.2, 1.4, CH=C
HH), 5.34−5.27 (2H, m, CH=CH
H and CHN), 4.70−4.56 (2H, m, OCH
2), 4.02−3.91 (2H, m, CHC
H2OH) and 3.33 (1H, s, OH); δ
C (100 MHz) 165.9 (C=O), 156.7 (C-O), 139.2 (C), 133.0 (
CH=CH
2), 132.3 (CH), 131.8 (CH), 128.8 (2CH), 127.7 (CH), 126.8 (2CH), 121.5 (CH), 121.2 (C), 119.8 (=CH
2), 112.4 (CH), 70.1 (OCH
2), 67.5 (CH
2OH) and 56.9 (
CHNH); [α]
D +30.9 (
c 1.00, CH
2Cl
2); HRMS (NSI
+): found 298.1439. C
18H
20NO
3 (M + H) requires 298.1443.
3.4.7. (S)-2-Allyloxy-N-(1-hydroxy-3-phenylpropan-2-yl)benzamide 32
Following the procedure of 3.4.5 using 2-(allyloxy)benzoyl chloride
29 (5.06 g, 25.7 mmol) in CH
2Cl
2 (15 cm
3) and (
S)-2-amino-3-phenylpropan-1-ol [
26] (3.88 g, 25.7 mmol) in CH
2Cl
2 (15 cm
3) gave
32 (7.05 g, 88%) as colourless crystals; mp 98−101 °C; ν
max/cm
−1 3391, 3354, 1616, 1541, 1314, 1229, 1045, 760, 752, 706 and 500; δ
H (400 MHz) 8.24 (1H, d,
J 7.1, NH), 8.19 (1H, dd,
J 7.8, 1.9, ArH), 7.42 (1H, ddd,
J 8.3, 7.3, 1.9, ArH), 7.30−7.19 (5H, m), 7.07 (1H, ddd,
J 7.8, 7.3, 1.0, ArH), 6.93 (1H, dd,
J 8.4, 1.0 ArH), 5.95 (1H, ddt,
J 17.3, 10.5, 5.6, C
H=CH
2), 5.38 (1H, dq,
J 17.3, 1.5, CH=C
HH), 5.33 (1H, dq,
J 10.5, 1.2. CH=H
H), 4.59 (2H, dt,
J 5.6, 1.4, OCH
2), 4.42 (1H, qdd,
J 7.1, 5.5, 3.4, C
HCH
2Ph), 3.80 (1H, dd,
J 11.1, 3.5, CH
HOH), 3.69 (1H, dd,
J 11.1, 5.6, C
HHOH) and 2.96 (2H, dd,
J 7.2, 1.6, CH
2Ph); δ
C (100 MHz) 166.0 (C=O), 156.6 (C-O), 137.8 (C), 132.8 (
CH=CH
2), 132.2 (CH), 132.0 (CH), 129.2 (2CH), 128.5 (2CH), 126.5 (CH), 121.5 (CH), 121.4 (C), 119.2 (=CH
2), 112.7 (CH), 69.9 (OCH
2), 64.9 (CH
2OH), 53.6 (CHN) and 37.1 (
CH
2Ph); [α]
D −69.4 (
c 1.00, CH
2Cl
2); HRMS (NSI
+): found 312.1596. C
19H
22NO
3 (M + H) requires 312.1600.
3.4.8. (S)-2-(Allyloxy)-N-(1-hydroxy-3-methylbutan-2-yl)benzamide 33
Following the procedure of 3.4.5 using 2-(allyloxy)benzoyl chloride
29 (5.00 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3) and (
S)-2-amino-3-methylbutan-1-ol [
28] (2.61 g, 25.4 mmol) in CH
2Cl
2 (15 cm
3) gave
33 (6.05 g, 90%) as colourless crystals; mp 94−96 °C; ν
max/cm
−1 3343, 2954, 2870, 1612, 1599, 1541, 1233, 1067, 1013, 986, 920, 760, 606 and 598; δ
H (400 MHz) 8.24−8.18 (2H, m, NH and ArH), 7.44 (1H, ddd,
J 8.4, 7.3, 1.9, ArH), 7.10 (1H, ddd,
J 7.9, 1.3, 1.0, ArH), 6.98 (1H, dd,
J 8.4, 1.0, ArH), 6.11 (1H, ddt,
J 17.2, 10.3, 5.9, C
H=CH
2), 5.47 (1H, dq,
J 17.2, 1.4, CH=C
HH), 5.40 (1H, dq,
J 10.3, 1.1, CH=CH
H), 4.68 (2H, dt,
J 5.9, 1.3, OCH
2), 4.01 (1H, tdd,
J 7.2, 6.0, 3.3, C
HCH
2OH), 3.81 (1H, dd,
J 11.1, 3.3, CH
HOH), 3.72 (1H, dd,
J 11.1, 6.8, C
HHOH), 2.00 (1H, m, C
HMe
2), 1.02 (3H, d,
J 6.8, Me) and 0.99 (3H, d,
J 6.8, Me); δ
C (100 MHz) 166.6 (C=O), 156.6 (C-O), 132.9 (
CH=CH
2), 132.4 (CH), 131.9 (CH), 121.5 (CH), 121.4 (C), 120.1 (CH=
CH
2), 112.4 (CH), 70.1 (OCH
2), 65.2 (CH
2OH), 58.1 (CHN), 29.2 (CH), 19.7 (CH
3) and 18.5 (CH
3); [α]
D −24.2 (
c 1.002, CH
2Cl
2); HRMS (NSI
+): found 264.1596. C
15H
22NO
3 (M + H) requires 264.1600.
3.4.9. (S)-2-(2-(Allyloxy)phenyl)-4-phenyl-4,5-dihydrooxazole 34
A solution of (S)-2-(allyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 30 (1.0 g, 3.4 mmol) in CH2Cl2 (20 cm3) was stirred at 0 °C while MsCl (0.31 cm3, 0.46 g, 4.0 mmol) and then Et3N (1.03 cm3, 0.75 g, 7.4 mmol) were added dropwise. The mixture was then heated under reflux for 3 d. It was then cooled and added to water (20 cm3). Separation of the organic layer, extraction of the aqueous layer with CH2Cl2 (2 × 10 cm3), drying and evaporation of the combined organic extracts gave, after purification via flash column chromatography (hexane/EtOAc 7:3) at Rf 0.29, compound 34 (350 mg, 37%) as a pale yellow oil; νmax/cm−1 3381, 3063, 2928, 1728, 1638, 1599, 1449, 1229, 1074, 993, 752, 698 and 525; δH (400 MHz) 7.83 (1H, dd, J 7.7, 1.8, ArH), 7.41 (1H ddd, J 8.4, 7.4, 1.8, ArH), 7.38−7.32 (4H, m, ArH), 7.31−7.26 (1H, m, ArH), 7.04−6.96 (2H, m, ArH), 6.07 (1H, ddt, J 17.3, 10.6, 4.8 CH=CH2), 5.51 (1H, dq, J 17.2, 1.8, CH=CHH), 5.41 (1H, dd, J 10.2, 8.0, CHHOCN), 5.26 (1H, dq, J 10.6, 1.6, CH=CHH), 4.77 (1H, dd, J 10.2, 8.3, CHHOCN), 4.65 (2H, dt, J 4.9, 1.7, ArOCH2) and 4.25 (1H, t, J 8.2, CHPh); δC (100 MHz) 163.8 (C=N), 157.3 (C-O), 142.5 (C), 132.7 (CH=CH2), 132.1 (CH), 131.2 (CH), 128.4 (2CH), 127.3 (CH), 126.6 (2CH), 120.4 (CH), 117.6 (C), 117.1 (=CH2), 113.2 (CH), 74.3 (OCH2), 69.9 (CHN) and 69.3 (OCH2); [α]D −24.5 (c 1.00, CH2Cl2); HRMS (NSI+): found 280.1335. C18H18NO2 (M + H) requires 280.1338.
3.4.10. (R)-2-(2-(Allyloxy)phenyl)-4-phenyl-4,5-dihydrooxazole 35
Following the procedure of 3.4.9 using (R)-2-(allyloxy)-N-(2-hydroxy-1-phenylethyl)benzamide 31 (6.9 g, 22.2 mmol), MsCl (2.06 cm3, 3.05 g, 26.6 mmol) and Et3N (6.80 cm3, 4.93 g, 48.8 mmol) in CH2Cl2 (50 cm3) at 50 °C overnight gave, after purification via flash column chromatography (hexane/EtOAc 7:3) at Rf 0.29, compound 35 (5.67 g, 92%) as a pale yellow oil; νmax/cm−1 3381, 3030, 2932, 2874, 1638, 1599, 1450, 1229, 993, 752, 698 and 525; δH (400 MHz) 7.82 (1H, dd, J 7.7, 1.8, ArH), 7.41 (1H, ddd, J 8.4, 7.4, 1.8, ArH), 7.37–7.33 (4H, m, ArH), 7.30−7.26 (1H, m, ArH), 7.02−6.95 (2H, m, ArH), 6.06 (1H, ddt, J 17.3, 10.6, 4.8, CH=CH2), 5.50 (1H, dq, J 17.3, 1.7, CH=CHH), 5.40 (1H, dd, J 10.2, 7.9, CHHOCN), 5.26 (1H, dq, J 10.6, 1.6, CH=CHH), 4.76 (1H, dd, J 10.2, 8.3, CHHOCN), 4.64 (2H, dt, J 4.8, 1.7, ArOCH2) and 4.24 (1H, t, J 8.1, CHPh); δC (125 MHz) 163.9 (C=N), 157.4 (C-O), 142.6 (C), 132.8 (CH=CH2), 132.2 (CH), 131.3 (CH), 128.5 (2CH), 127.4 (CH), 126.7 (2CH), 120.5 (CH), 117.7 (C), 117.2 (=CH2), 113.3 (CH), 74.5 (OCH2), 70.0 (CHN) and 69.4 (OCH2); [α]D +22.7 (c 0.93, CH2Cl2); HRMS (NSI+): found 280.1334. C18H18NO2 (M + H) requires 280.1338.
3.4.11. (S)-2-(2-(Allyloxy)phenyl)-4-benzyl-4,5-dihydrooxazole 36
Following the procedure of 3.4.9 using (S)-N-(1-hydroxy-3-phenylpropan-2-yl)-2-propoxybenzamide 32 (1.0 g, 3.2 mmol), MsCl (0.30 cm3, 0.44 g, 3.9 mmol) and Et3N (0.98 cm3, 0.71 g, 7.1 mmol) in CH2Cl2 (20 cm3) at rt overnight gave, after purification via flash column chromatography (hexane/EtOAc 7:3) at Rf 0.34, 36 (220 mg, 24%) as a pale-yellow oil; νmax/cm−1 3393, 3026, 2893, 1643, 1601, 1493, 1450, 1256, 1227, 995, 750 and 698; δH (400 MHz) 7.74 (1H, dd, J 7.7, 1.8, ArH), 7.39 (1H, ddd, J 8.3, 7.4, 1.8, ArH), 7.34−7.27 (4H, m, ArH), 7.25−7.20 (1H, m, ArH), 7.01−6.93 (2H, m, ArH), 6.06 (1H, ddt, J 17.2, 10.6, 4.7, CH=CH2), 5.53 (1H, dq, J 17.2, 1.7, CH=CHH), 5.28 (1H, dq, J 10.6, 1.7, CH=CHH), 4.64 (2H, dt, J 4.7, 1.4, ArOCH2), 4.62−4.56 (1H, m, CHCH2Ph), 4.33 (1H, dd, J 9.4, 8.4, CHHO), 4.11 (1H, dd, J 8.5, 7.4, CHHO), 3.25 (1H, dd, J 13.7, 5.2, CHHPh) and 2.77 (1H, dd, J 13.7, 8.7, CHHPh); δC (100 MHz) 162.7 (C=N), 157.2 (C-O), 137.9 (C), 132.7 (CH), 131.9 (CH), 131.1 (CH), 129.1 (2CH), 128.3 (2CH), 126.2 (CH), 120.3 (CH), 117.6 (C), 116.9 (C=CH2), 113.2 (CH), 71.1 (OCH2), 69.2 (OCH2), 67.8 (CHN) and 41.6 (CH2); [α]D −30.89 (c 1.01, CH2Cl2); HRMS (NSI+): found 294.1491. C19H20NO2 (M + H) requires 294.1494.
3.4.12. (S)-2-(-2(Allyloxy)phenyl)-4-isopropyl-4,5-dihydrooxazole 37
Following the procedure of 3.4.9 using (S)-2-(allyloxy)-N-(1-hydroxy-3-methylbutan-2-yl)benzamide 33 (1.0 g, 3.8 mmol), MsCl (0.38 cm3, 0.56 g, 4.6 mmol) and Et3N (1.17 cm3, 0.85 g, 8.4 mmol) in CH2Cl2 (20 cm3) at rt overnight gave, after purification via flash column chromatography (hexane/EtOAc 7:3) at Rf 0.35, 37 (380 mg, 41%) as a pale-yellow oil; νmax/cm−1 3399, 2961, 2874, 1645, 1599, 1449, 1292, 1227, 1037, 995, 924, 752, 552 and 527; δH (400 MHz) 7.72 (1H, dd, J 7.6, 1.8 ArH), 7.37 (1H, ddd, J 8.3, 7.4, 1.8, ArH), 7.02−6.91 (2H, m, ArH), 6.05 (1H, ddt, J 17.2, 10.6, 4.7, CH=CH2), 5.53 (1H, dq, J 17.2, 1.8, CH=HH), 5.27 (1H, dq, J 10.6, 1.6, CH=CHH), 4.61 (2H, dt, J 4.7, 1.7, ArOCH2), 4.45−4.35 (1H, m, CHCH(CH3)2), 4.14−4.07 (2H, m, CH2OCN), 1.95−1.84 (1H, m, CH(CH3)2), 1.04 (3H, d, J 6.8, CH3) and 0.95 (3H, d, J 6.7, CH3); δC (100 MHz) 162.0 (C=N), 156.9 (C-O), 132.5 (CH=CH2), 131.5 (CH), 130.8 (CH), 120.1 (CH), 117.8 (C), 116.6 (C=CH2), 112.8 (Ar CH), 72.3 (CHN), 69.3 (OCH2), 68.9 (OCH2), 32.4 (CH), 18.4 (CH3) and 17.8 (CH3); [α]D −43.44 (c 1.006, CH2Cl2); HRMS (NSI+): found 246.1486. C15H21NO2 (M + H) requires 246.1494.
3.4.13. (S)-2-(4-Phenyl-4,5-dihydrooxazol-2-yl)phenol 38
A solution of (
S)-2-(2-(allyloxy)phenyl)-4-phenyl-4,5-dihydrooxazole
34 (4.58 g, 16.4 mmol) and KOBu
t (4.05 g, 36.1 mmol) in PhMe (100 cm
3) was stirred at rt under a nitrogen atmosphere while
n-butyllithium (14.4 cm
3, 36.1 mmol) was added. After 2 h, the solution was added to aqueous ammonium chloride solution (50 cm
3) and the mixture was extracted with Et
2O (3 × 50 cm
3). Drying and evaporation of the combined organic extracts gave crude
38 (3.80 g, 97%) as a dark-red oil, which was purified via flash column chromatography (hexane/Et
2O 1:1) to give, at R
f 0.67,
38 (1.65 g, 42%) as a yellow oil; δ
H (400 MHz) 7.72 (1H, dd,
J 7.8, 1.7, ArH), 7.43−7.40 (1H, m, ArH), 7.38−7.36 (2H, m, ArH), 7.34−7.28 (3H, m, ArH), 7.04 (1H, dd,
J 8.4, 1.2, 0.4 ArH), 6.91 (1H, ddd,
J 7.8, 7.3, 1.2, ArH), 5.47 (1H, dd,
J 10.1, 8.3, oxazoline NCH), 4.82 (1H, dd,
J 10.1, 8.3, oxazoline OC
HH) and 4.27 (1H, t,
J 8.3, oxazoline OCH
H); δ
C (100 MHz) 166.4 (C=N), 160.2 (C-O), 141.7 (C), 133.8 (CH), 129.0 (2CH), 128.4 (CH), 128.0 (CH), 126.6 (CH), 118.9 (CH), 117.0 (CH), 110.6 (C), 74.3 (CH
2) and 68.7 (CH); HRMS (NSI
+): found 240.1020. C
15H
14NO
2 (M + H) requires 240.1025. The
1H and
13C spectral data were in accordance with those previously reported [
29].
3.4.14. (R)-2-(4-Phenyl-4,5-dihydrooxazol-2-yl)phenol 39
Following the procedure of 3.4.13 using (
R)-2-(2-(allyloxy)phenyl)-4-phenyl-4,5-dihydrooxazole
35 (3.10 g, 11.1 mmol), KOBu
t (2.74 g, 24.4 mmol) and
n-butyllithium (9.8 cm
3, 24.4 mmol) in PhMe (70 cm
3) at rt for 2 h gave
39 (2.90 g, quant) as a dark-red oil which was used without further purification; δ
H (400 MHz) 12.14 (1H, br s, OH), 7.72 (1H, dd,
J 7.8, 1.7, ArH), 7.46−7.40 (1H, m, ArH), 7.34−7.36 (2H, m, ArH), 7.34−7.28 (3H, m, ArH), 7.06 (1H, ddd,
J 8.4, 1.1, 0.4, ArH), 6.91 (1H, ddd,
J 7.8, 7.3, 1.2, ArH), 5.49 (1H, dd,
J 10.1, 8.3, oxazoline NCH), 4.82 (1H, dd,
J 10.1, 8.3, OC
HH) and 4.27 (1H, t,
J 8.3, OCH
H); δ
C (100 MHz) 166.6 (C=N), 160.2 (C-O), 141.5 (C), 134.0 (CH), 129.0 (2CH), 128.4 (CH), 128.1 (CH), 126.7 (2CH), 118.9 (CH), 117.0 (CH), 110.4 (ArC), 74.3 (CH
2) and 68.7 (CH); HRMS (NSI
+): found 240.1021. C
15H
14NO
2 (M + H) requires 240.1025. The
1H and
13C spectral data were in accordance with those previously reported [
30].
3.4.15. (S)-2-(4-Benzyl-4,5-dihydrooxazol-2-yl)phenol 40
Following the procedure of 3.4.13 using (
S)-2-(2-(allyloxy)phenyl)-4-benzyl-4,5-dihydrooxazole
36 (1.59 g, 5.42 mmol), KOBu
t (1.34 g, 11.9 mmol) and
n-butyllithium (4.8 cm
3, 11.9 mmol) in PhMe (30 cm
3) at rt for 2 h gave
40 (1.37 g, quant) as a dark-red oil which was used without further purification; δ
H (400 MHz) 12.18 (1H, br s, OH), 7.62 (1H, dd,
J 7.8, 1.7, ArH), 7.37 (1H, ddd,
J 8.3, 7.3, 1.7, ArH), 7.32–7.27 (2H, m, ArH), 7.27–7.22 (3H, m, ArH), 7.01 (1H, dd,
J 8.4, 1.1, ArH), 6.86 (1H, ddd,
J 8.3, 7.3, 1.1, ArH), 4.65–4.58 (1H, m, OC
HH), 4.38 (1H, dd,
J 9.3, 8.5, OCH
H), 4.13 (1H, dd,
J 8.6, 7.4, NC
HPh), 3.10 (1H, dd,
J 13.7, 6.4, C
HHPh) and 2.81 (1H, dd,
J 13.7, 7.5, CH
HPh); δ
C (100 MHz); 165.4 (C=N), 160.0 (C-O), 137.6 (C), 133.5 (CH), 129.3 (2CH), 128.7 (2CH), 128.1 (CH), 126.8 (CH), 118.7 (CH), 116.8 (CH), 110.7 (C), 71.3 (CH
2), 66.8 (CH) and 42.0 (CH
2); HRMS (NSI
+): found 254.1171. C
16H
16NO
2 (M + H) requires 254.1181. The
1H and
13C spectral data were in accordance with those previously reported [
31].
3.4.16. (S)-2-(4-Isopropyl-4,5-dihydrooxazol-2-yl)phenol 41
Following the procedure of 3.4.13 using (
S)-2-(-2(allyloxy)phenyl)-4-isopropyl-4,5-dihydrooxazole
37 (19.85 g, 80.9 mmol), KOBu
t (19.97 g, 178.0 mmol) and
n-butyllithium (81 cm
3, 178.0 mmol) in PhMe (450 cm
3) at rt for 2 h gave, after purification via flash column chromatography (hexane/Et
2O 9:1) at R
f 0.47,
41 (5.47 g, 33%) as a yellow oil; δ
H (400 MHz)
1H NMR 12.40 (1H, s, OH), 7.67 (1H, dd,
J 7.7, 1.8, ArH), 7.39 (1H, ddd,
J 8.4, 7.3, 1.8, ArH), 7.04 (1H, dd,
J 8.4, 1.0, ArH), 6.89 (1H, ddd,
J 7.7, 7.3, 1.0, ArH), 4.47–4.39 (1H, m OC
HHCHN), 4.17–4.09 (2H, m OCH
HC
HN), 1.86–1.77 (1H, m, C
H(CH
3)
2), 1.02 (3H, d,
J 6.7, CHC
H3CH
3) and 0.95 (3H, d,
J 6.7, CHCH
3C
H3); δ
C (100 MHz) 165.2 (C=N), 160.1 (C-O), 133.3 (CH), 128.1 (CH), 118.6 (CH), 116.8 (CH), 110.8 (C), 71.6 (CH
2), 70.0 (CH), 33.1 (CH), 18.8 (CH
3) and 18.7 (CH
3); [α]
D −30.95, (
c 1.076, CHCl
3); (lit. [
32] −35.4 (
c 1.07, CHCl
3)); HRMS (NSI
+): found 206.1174. C
12H
16NO
2 (M + H) requires 206.1181. The
1H and
13C spectral data were in accordance with those previously reported [
32].
3.4.17. (S)-4-Phenyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 42
To a stirred suspension of NaH (60% dispersion in mineral oil, 0.14 g, pre-washed with hexane, 3.5 mmol) in DMF (15 cm3) at rt was added (S)-2-(4-phenyl-4,5-dihydrooxazol-2-yl)phenol 38 (0.83 g, 3.5 mmol) and the mixture was stirred for 15 min before addition of (1-bromoethyl)benzene (0.48 cm3, 0.65 g, 3.5 mmol). After stirring for 18 h, the mixture was poured into water and extracted with CH2Cl2 followed by Et2O (×3). The combined organic extracts were washed with water (×5), dried and evaporated to give, after purification via column chromatography (hexane/Et2O 1:1) at Rf 0.32, 42 (0.67 g, 56%) as a pale-yellow oil as an inseparable 1:1 mixture of diastereomers; νmax/cm−1 3383, 3028, 2978, 1638, 1599, 1491, 1449, 1067, 1028, 754, 698 and 542; δH (400 MHz) 7.74 (1H, dd, J 7.7, 1.5, ArH), 7.44–7.37 (4H, m, ArH), 7.37–7.32 (1H, m, ArH), 7.32−7.25 (3H, m, ArH), 7.25−7.20 (2H, m, ArH), 6.94−6.88 (1H, m, ArH), 6.83−6.79 (1H, m, ArH), 5.44–5.37 (2H, m, CH(CH3) and OCHHCHN)), 4.79 (1H, app ddd, J 10.2, 8.3, 2.8, OCHHCHN), 4.31−4.25 (1H, m, CHN) and 1.66 (3H, app t, J 6.4, OCHCH3); δC (100 MHz) 164.41 and 164.35 (C=N), 156.67 and 156.64 (C-O), 142.9 and 142.8 (C), 131.8 (CH), 131.1 (CH), 128.6 (2CH), 128.5 (2CH), 127.4 (CH), 126.7 (2CH), 125.6 (2CH), 120.43 and 120.36 (CH), 118.7 and 118.6 (C) 115.1 and 114.9 (CH), 77.1 and 76.9 (CH), 74.62 and 74.56 (CH2), 70.00 and 69.97 (CH) and 24.3 (CH3); HRMS (NSI+): found 344.1649. C23H22NO2 (M + H) requires 344.1651.
3.4.18. (R)-4-Phenyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 43
Following the procedure of 3.4.17 using (R)-2-(4-phenyl-4,5-dihydrooxazol-2-yl)phenol 39 (0.53 g, 2.2 mmol), NaH (89 mg, 2.2 mmol) and (1-bromoethyl)benzene (0.30 cm3, 0.41 g, 2.2 mmol) in DMF (10 cm3) at rt for 18 h gave, after purification via column chromatography (hexane/Et2O 1:1) at Rf 0.32, 43 (0.19 g, 25%) as a pale yellow oil as an inseparable 1:1 mixture of diastereomers; νmax/cm−1 3381, 3030, 1643, 1599, 1491, 1449, 1246, 1067, 1028, 750 and 698; δH (400 MHz); 7.74 (1H, app ddd, J 7.7, 1.9, 0.7, ArH), 7.45–7.20 (11H, m, ArH), 6.90 (1H, app tdd, J 7.5, 2.1, 1.0, ArH), 6.81 (1H, app ddd, J 8.5, 3.5, 1.0, ArH), 5.42−5.33 (2H, m, CH(CH3) and OCHHCHN)), 4.77 (1H, ddd, J 10.2, 8.3, 2.8, OCHHCHN), 4.27 (1H, td, J 8.1, 1.5, CHN) and 1.66 (3H, app t, J 6.4, CH3); δC (100 MHz) 164.34 and 164.29 (C=N), 156.61 and 156.57 (C-O), 142.8 and 142.7 (C), 131.8 (CH), 131.1 (CH), 128.5 (2CH), 128.4 (2CH), 127.3 (CH), 126.7 (2CH), 125.6 (2CH), 120.4 and 120.3 (CH), 118.6 and 118.5 (C), 115.1 and 114.9 (CH), 77.0 and 76.8 (CH), 74.6 and 74.5 (CH2), 69.94 and 69.91 (CH) and 24.3 (CH3); HRMS (NSI+): found 344.1649. C23H22NO2 (M + H) requires 344.1651.
3.4.19. (S)-4-Benzyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 44
Following the procedure of 3.4.17 using (R)-2-(4-phenyl-4,5-dihydrooxazol-2-yl)phenol 40 (0.28 g, 1.1 mmol), NaH (44 mg, 1.1 mmol) and (1-bromoethyl)benzene (0.15 cm3, 0.20 g, 1.1 mmol) in DMF (10 cm3) at rt for 18 h gave, after purification via column chromatography (hexane/Et2O 1:1) at Rf 0.27, 44 (0.12 g, 31%) as a pale yellow oil as an inseparable 1:1 mixture of diastereomers; νmax/cm−1 3028, 2928, 1643, 1599, 1491, 1450, 1240, 1066, 1028, 750, 698 and 519; δH (400 MHz) 7.68 (1H, dd, J 7.8, 1.8, ArH), 7.40 (2H, dt, J 8.0, 1.7, ArH), 7.33–7.27 (6H, m, ArH), 7.25–7.18 (3H, m, ArH), 6.89 (1H, tdd, J 7.5, 1.9, 1.0, ArH), 6.78 (1H, dd, J 8.4, 3.6, 1.0 ArH), 5.33 (1H, app quintet, J 6.2, OCHCH3), 4.60 (1H, ddddd, J 9.3, 8.4, 7.3, 5.4, 0.9, CHN), 4.35 (1H, dd, J 9.4, 8.4, OCHHCN), 4.13 (1H, ddd, J 8.5, 7.3, 1.4, OCHHCN), 3.25 (1H, dd, J 13.7, 5.3, CH2Ph), 2.80 (1H, ddd, J 13.8, 10.0, 8.4, CH2Ph) and 1.63 and 1.62 (3H, 2xd, J 6.4, OCHCH3); δC (100 MHz) 163.57 and 163.50 (C=N), 156.6 (C-O), 142.9 (C), 138.09 and 138.06 (C), 131.7 (CH), 131.0 (CH), 129.28 and 129.25 (2CH), 128.4 (4CH), 127.3 (CH), 126.3 (CH), 125.6 (2CH), 120.47 and 120.42 (CH), 118.9 and 118.8 (C), 115.4 and 115.3 (CH), 77.2 and 77.1 (CHN), 71.54 and 71.46 (CH2O), 67.7 (CH), 41.7 (CH2Ph) and 24.2 (CH3); HRMS (NSI+): found 358.1803. C24H24NO2 (M + H) requires 358.1807.
3.4.20. (S)-4-Isopropyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 45
Following the procedure of 3.4.17 using (S)-2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenol 41 (1.0 g, 4.87 mmol), NaH (0.20 g, 4.87 mmol) and (1-bromoethyl)benzene (0.66 cm3, 0.89 g, 4.87 mmol) in DMF (20 cm3) at rt for 18 h gave, after purification via column chromatography (hexane/Et2O 1:1) at Rf 0.28, 45 (0.53 g, 35%) as a colourless oil and an inseparable 1:1 mixture of diastereomers; νmax/cm−1 2959, 2928, 2872, 1643, 1599, 1490, 1479, 1449, 1236, 1067, 752 and 698; δH (400 MHz) 7.64 (1H, dt, J 7.6, 1.9, ArH), 7.43–7.36 (2H, m, ArH), 7.32–7.25 (2H, m, ArH), 7.24–7.14 (1H, m, ArH), 7.13–7.08 (1H, m, ArH), 6.86 (1H, tdd, J 7.6, 2.9, 1.0, ArH), 6.75 (1H, dd, J 8.4, 2.9, 1.0, ArH), 5.34 (1H, dq, J 9.3, 6.3,OCH), 4.43−4.34 (1H, m, OCHHCHN), 4.17–4.07 (2H, m, OCHHCHN), 1.93−1.83 (1H, m, CH(CH3)2), 1.62 (3H, app t, J 6.3, OCHCH3), 1.061 and 1.055 (3H, 2 × d, J 6.8, CHCH3CH3), and 0.989 and 0.983 (3H, 2 × d, J 6.8, CHCH3CH3); δC (100 MHz) 162.8 and 162.7 (C=N), 156.33 and 156.30 (C-O), 142.8 (C), 131.3 (CH), 130.80 and 130.78 (CH), 128.3 (2CH), 127.2 (CH), 125.48 and 125.45 (2CH), 120.24 and 120.16 (CH), 119.0 and 118.9 (C), 115.0 and 114.8 (CH), 76.8 and 76.6 (CHO), 72.41 and 72.36 (CH), 69.7 (CH2), 32.62 and 32.57 (CH), 24.16 and 24.13 (CH3), 18.55 and 18.48 (CH3) and 18.11 and 18.06 (CH3); HRMS (NSI+): found 310.1805. C20H24NO2 (M + H) requires 310.1807.
3.4.21. Methyl 2-(1-phenylethoxy)benzoate 46
Following the procedure of 3.4.17 using methyl 2-hydroxybenzoate (17.04 cm
3, 131.4 mmol), sodium hydride (5.27 g, 131.4 mmol) and (1-bromoethyl)benzene (17.93 cm
3, 24.31 g, 131.4 mmol) in DMF (400 cm
3) gave
46 (29.98 g, 89%) as a yellow oil; δ
H (300 MHz) 7.75 (1H, dd,
J 7.8, 1.8, ArH), 7.46–7.19 (6H, m, ArH), 6.88 (1H, td,
J 7.5, 1.0, ArH), 6.83–6.77 (1H, m, ArH), 5.37 (1H, q,
J 6.4, OCH), 3.91 (3H, s, OCH
3) and 1.66 (3H, d,
J 6.4 CH
CH
3); δ
C (100 MHz) 166.2 (C=O), 156.7 (C-O), 142.3 (C), 132.4 (CH), 130.9 (CH), 128.1 (2CH), 127.0 (CH), 125.1 (2CH), 120.9 (C), 119.7 (CH), 114.8 (CH), 76.3 (OCHPh), 51.2 (O
CH
3) and 23.7 (CH
3); The
1H spectral data were in accordance with those previously reported [
33]. The
13C spectral data are reported for the first time.
3.4.22. 2-(1-Phenylethoxy)benzoic Acid 47
Following a literature procedure [
34], NaOH (13.58 g, 339.45 mmol) was added to a stirred solution of methyl 2-(2-phenylethoxy)benzoate
46 (29.00 g, 113.15 mmol) in an EtOH/water mixture (9:1, 140 cm
3) and the reaction mixture was heated at reflux for 3 h. After cooling to rt, the mixture was acidified to pH 1 by addition of 2 M HCl and extracted with PhMe (3 × 100 cm
3). The combined organic layers were dried over MgSO
4 and concentrated to give
47 (21.13 g, 77%) as a pale-yellow oil which slowly crystallised to give colourless crystals; mp 57−59 °C; ν
max/cm
−1 3238, 2978, 1730, 1692, 1601, 1454, 1375, 1240, 1217, 1065, 750, 698 and 527; δ
H (400 MHz) 8.12 (1H, dd,
J 7.7, 1.9, ArH), 7.36–7.31 (5H, m, ArH), 7.29−7.25 (1H, m, ArH), 6.98 (1H, td,
J 7.7, 1.0, ArH), 6.90 (1H, dd,
J 8.5, 1.0, ArH), 5.56 (1H, q,
J 6.4, OCH) and 1.74 (3H, d,
J 6.4, CH
3); δ
C (100 MHz) 165.7 (C=O), 156.5 (C-O), 140.4 (C), 134.7 (CH), 133.5 (CH), 129.0 (2CH), 128.4 (CH), 125.3 (2CH), 122.0 (CH), 118.2 (C), 114.4 (CH), 79.1 (OC
HPh) and 23.9 (CH
3); HRMS (NSI
−): found 241.0871, C
15H
13O
3 (M–H) requires 241.0865.
3.4.23. N-((S)-1-Hydroxy-3-methylbutan-2-yl)-2-(1-phenylethoxy)benzamide 48
To a stirred solution of 2-(1-phenylethoxy)benzoic acid 47 (21.00 g, 86.7 mmol) in PhMe (190 cm3) was added SOCl2 (12.57 cm3, 20.61 g, 173.4 mmol) and the mixture heated to reflux for 2 h then cooled to rt and concentrated.
To a stirred solution of the acid chloride prepared as above (12.65 g, 48.5 mmol) in CH2Cl2 at 0 °C was added Et3N (6.76 cm3, 4.91 g, 48.5 mmol) and (S)-2-amino-3-methylbutan-1-ol (5.00 g, 48.5 mmol) and the mixture stirred at rt for 18 h. The reaction mixture was poured into water (100 cm3) and extracted with CH2Cl2 (2 × 100 cm3), the combined organic layers were dried over MgSO4 and concentrated to give, after purification via flash column chromatography (Et2O/hexane 4:1) at Rf 0.18, 48 (9.13 g, 58%) as a colourless oil as an inseparable 1:1 mixture of diastereomers; νmax/cm−1 3375, 2961, 2874, 1632, 1599, 1531, 1477, 1225, 1065, 932, 752, 700 and 583; δH (400 MHz) 8.40 (1H, d, J 8.3, NH), 8.18 (1H, dd, J 7.8, 1.9, ArH), 7.34 (4H, d, J 4.4, ArH), 7.32−7.26 (1H, m, ArH), 7.22 (1H, ddd, J 8.3, 7.3, 1.9, ArH), 6.96 (1H, dd, J 8.1, 7.3, 1.0, ArH), 6.78 (1H, dd, J 8.6, 1.0, ArH), 5.48 (1H, q, J 6.4, OCHCH3), 4.07 (1H, dtd, J 8.3, 5.9, 3.8, CHN), 3.84–3.73 (2H, m, CH2OH), 3.62 (1H, s, OH), 2.00 (1H, septet, J 6.8, (CH(CH3)2), 1.74 (3H, d, J 6.4, CH(CH3)Ph), 1.02 (3H, d, J 6.8, CH(CH3)(CH3)) and 0.99 (3H, d, J 6.8, CH(CH3)(CH3)); δC (125 MHz) 166.3 (C=O), 155.8 (C-O), 141.4 (C), 132.5 (CH), 132.2 (CH), 128.9 (2CH), 128.0 (CH), 125.2 (2CH), 121.5 (C), 121.0 (CH), 113.8 (CH), 77.3 (OCHPh), 64.2 (CH2OH), 57.5 (CHN), 29.2 (CH3), 24.2 (CH), 19.5 (CH3) and 18.6 (CH3); HRMS (NSI+): found 328.1911. C20H27NO3 (M + H) requires 328.1913
3.4.24. (S)-4-Isopropyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 45 from 48
Following the procedure of 3.4.9 using N-((S)-1-Hydroxy-3-methylbutan-2-yl)-2-(1-phenylethoxy)benzamide 48 (9.13 g, 28.4 mmol), MsCl (2.61 cm3, 3.86 g, 33.7 mmol) and Et3N (8.62 cm3, 6.26 g, 62.0 mmol) in CH2Cl2 (80 cm3) gave 45 (7.37 g, 84%) as a colourless oil as an inseparable 1:1 mixture of diastereomers, spectroscopic data as in 3.4.20.
3.4.25. 1-Phenylethyl 2-hydroxybenzoate 49
To a stirred solution of 2-(1-phenylethoxy)benzoic acid
47 (1.00 g, 4.1 mmol) in CH
2Cl
2 (10 cm
3) at 0 °C was added (COCl)
2 (0.43 cm
3, 0.63 g, 5.0 mmol) and the solution stirred at rt for 2 h. The reaction was concentrated and the crude residue, purified via flash column chromatography (hexane/EtOAc 4:1) to give, at R
f 0.83,
49 (0.51g, 51%) as colourless crystals; mp 57−60 °C; δ
H (400 MHz) 10.78 (1H, s, OH), 7.94 (1H, ddd,
J 8.0, 1.8. 0.5, ArH), 7.47−7.41 (3H, m, ArH), 7.40−7.35 (2H, m, ArH), 7.34−7.29 (1H, m, ArH), 6.96 (1H, ddd,
J 8.4, 1.2, 0.5, ArH), 6.89 (1H, ddd,
J 8.2, 7.2, 1.2, ArH), 6.14 (1H, q,
J 6.6, OC
HCH
3) and 1.69 (3H, d,
J 6.6, OCHC
H3); δ
C (100 MHz) 169.4 (C=O), 161.7 (C-O), 141.1 (C), 135.7 (CH), 129.9 (CH), 128.6 (2CH), 128.1 (CH), 126.0 (2CH), 119.1 (CH), 117.6 (CH), 112.7 (C), 73.6 (O
CH) and 22.3 (CH
3). The
1H spectral data were in accordance with those previously reported [
16], and the
13C data are reported for the first time.
3.6. Synthesis of Further More Highly Substituted Oxazolines
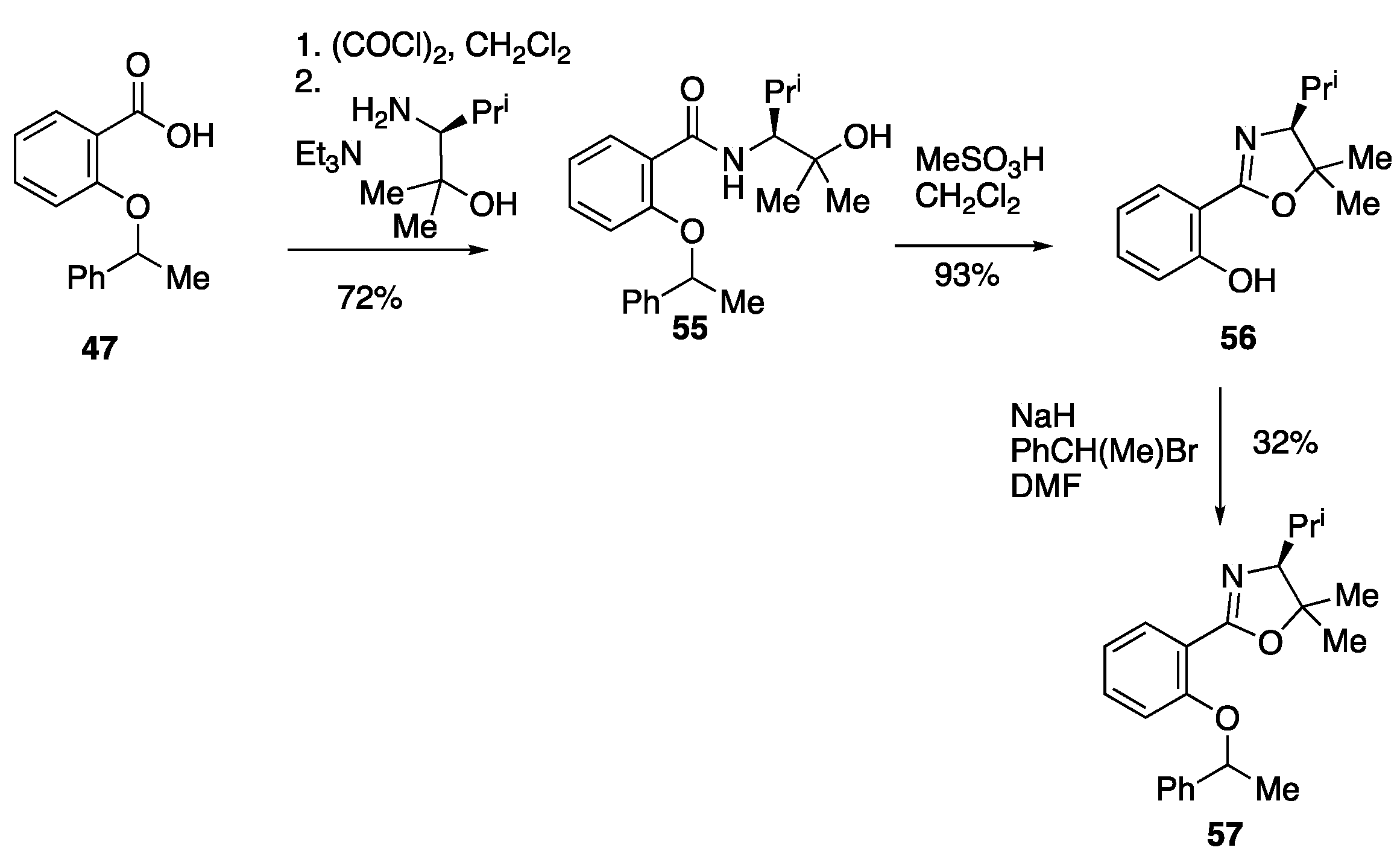
3.6.1. N-((S)-2-Hydroxy-2,4-dimethylpentan-3-yl)-2-(1-phenylethoxy)benzamide 55
To a stirred solution of 2-(1-phenylethoxy)benzoic acid 47 (4.0 g, 16.5 mmol) in CH2Cl2 (165 cm3) was added (COCl)2 (2.97 cm3, 4.40 g, 34.65 mmol) and two drops of DMF, and the mixture stirred at rt for 2 h, then cooled to rt and concentrated.
To a stirred solution of the resulting acid chloride (1.99 g, 7.62 mmol) in CH
2Cl
2 (20 cm
3) at 0 °C was added Et
3N (1.06 cm
3, 0.77 g, 7.62 mmol) and (
S)-3-amino-2,4-dimethylpentan-2-ol [
36] (1.0 g, 7.62 mmol) and the mixture stirred at rt for 18 h. The reaction mixture was poured into H
2O (50 cm
3) and extracted with CH
2Cl
2 (2 × 50 cm
3), the combined organic layers were dried over MgSO
4 and concentrated. The crude residue was purified via flash column chromatography (Et
2O/hexane 4:1) to give, at R
f 0.42,
55 (1.95 g, 72%) as a colourless oil as an inseparable mixture 2:1 of diastereomers whose spectra were sufficiently different to be separately identified; ν
max/cm
−1 3385, 3356, 2972, 2957, 1690, 1624, 1533, 1479, 1219, 1177, 1161, 752, 702, 675 and 529; δ
H (400 MHz, major diastereomer) 8.52 (1H, d,
J 9.8, NH), 8.24 (1H, ddd,
J 7.8, 1.9, 1.0, ArH), 7.42−7.37 (3H, m, ArH), 7.32−7.26 (3H, m, ArH), 7.05−6.98 (1H, m, ArH), 6.83 (1H, d,
J 8.2, ArH), 5.57 (1H, q,
J 6.4, CH
3C
HOPh), 4.17 (1H, m, CHN), 2.25 (1H, octet,
J 6.8, C
H(CH
3)
2), 1.77 (3H, d,
J 6.5, C
H3CH(OAr)Ph), 1.36 (3H, s,
gem dimethyl), 1.32 (3H, s,
gem dimethyl) and 1.04−1.01 (6H, m, CH(C
H3)
2); δ
H (400 MHz, minor diastereomer) 8.44 (1H, d,
J 10.0, NH), 8.24 (1H, ddd,
J 7.8, 1.9, 1.0, ArH), 7.42−7.37 (3H, m, ArH), 7.32−7.26 (3H, m, ArH), 7.05−6.98 (1H, m, ArH), 6.80 (1H, d,
J 8.3, ArH), 5.53 (1H, q,
J 6.4, CH
3C
HOPh), 4.18 (1H, m, CHN), 2.27 (1H, octet,
J 6.8, C
H(CH
3)
2), 1.76 (3H, d,
J 6.5, C
H3CH(OAr)Ph), 1.35 (3H, s,
gem dimethyl), 1.31 (3H, s,
gem dimethyl) and 1.04−1.01 (6H, m, CH(C
H3)
2); δ
C (125 MHz, major diastereomer) 166.1 (C=O), 155.8 (C-O), 141.5 (C), 132.4 (CH), 132.3 (CH), 128.9 (2CH), 128.0 (CH), 125.5 (2CH), 121.73 (C), 120.93 (CH), 113.55 (CH), 76.6 (OCH), 73.95 (COH), 60.8 (NHCH), 29.32 (CH
3), 28.42 (CH), 27.3 (CH
3), 24.5 (CH
3), 22.5 (CH
3) and 17.5 (CH
3); δ
C (125 MHz, minor diastereomer) 166.2 (C=O), 155.9 (C-O), 141.7 (C), 132.6 (CH), 132.4 (CH), 128.95 (2CH), 128.0 (CH), 125.2 (2CH), 121.67 (C), 120.98 (CH), 113.65 (CH), 77.0 (OCH), 73.92 (COH), 60.9 (NHCH), 29.26 (CH
3), 28.38 (CH), 27.1 (CH
3), 24.8 (CH
3), 22.6 (CH
3) and 17.6 (CH
3); HRMS (NSI
+): found 356.2223. C
22H
31NO
3 (M + H) requires 356.2226.
3.6.2. (S)-2-(4-Isopropyl-5,5-dimethyl-4,5-dihydrooxazol-2-yl)phenol 56
A stirred solution of N-((S)-2-hydroxy-2,4-dimethylpentan-3-yl)-2-(1-phenylethoxy)benzamide 55 (0.80 g, 2.25 mmol) and MeSO3H (0.89 cm3, 1.31 g, 13.7 mmol) in CH2Cl2 (30 cm3) was heated to reflux using a Soxhlet extractor with CaH2 in the thimble for 12 h. The reaction mixture was cooled to rt, poured into H2O (10 cm3), extracted with CH2Cl2 (3 × 20 cm3), dried over MgSO4 and concentrated. The crude residue was purified via flash column chromatography (9:1 Hexane: EtOAc) to give, at Rf 0.45, 56 (480 mg, 93%) as a colourless oil; νmax/cm−1 2972, 2872, 1640, 1491, 1350, 1261, 1236, 1069, 1040, 820, 752, 696, 681 and 540; δH (400 MHz) 12.63 (1H, br, OH), 7.60 (1H, dd, J 7.8, 1.8, ArH), 7.34 (1H, ddd, J 8.3, 7.3, 1.8, ArH), 6.98 (1H, dd, J 8.3, 1.1, ArH), 6.84 (1H, ddd, J 7.8, 7.3, 1.1, ArH), 3.50 (1H, d, J 8.6, CH-N), 1.87 (1H, d of septets, J 8.6, 6.6, CH(CH3)2), 1.54 (3H, s, CH3), 1.38 (3H, s, CH3), 1.11 (3H, d, J 6.6, CH3), 1.01 (3H, d, J 6.6, CH3); δC (100 MHz) 163.7 (C=N), 160.1 (C-O), 133.0 (CH), 127.9 (CH), 118.3 (CH), 116.6 (CH), 111.1 (C), 86.5 (C), 79.2 (CH-N), 28.93 (CH3), 28.88 (CH3), 21.2 (CH), 20.8 (CH3), 20.7 (CH3); HRMS (NSI+): found 234.1910. C14H21NO2 (M + H) requires M, 234.1914.
3.6.3. (S)-4-Isopropyl-5,5-dimethyl-2-(2-(1-phenylethoxy)phenyl)-4,5-dihydrooxazole 57
Following the procedure of 3.4.17 using (S)-2-(4-isopropyl-5,5-dimethyl-4,5-dihydrooxazol-2-yl)phenol 56 (390 mg, 1.67 mmol), NaH (67 mg, 1.67 mmol) and (1-bromoethyl)benzene (0.23 cm3, 0.31 g, 1.67 mmol) in DMF (10 cm3) at rt for 18 h gave, after purification via flash column chromatography (hexane/Et2O 3:1) at Rf 0.25, 57 (180 mg, 32%) as a slightly yellow oil as an inseparable 3:2 mixture of diastereomers; νmax/cm−1 2972, 1643, 1601, 1489, 1450, 1242, 1042, 1069, 750 and 700; δH (400 MHz) 7.59 (1H, app dt, J 7.6, 2.0, ArH), 7.43−7.38 (2H, m, ArH), 7.34−7.28 (2H, m, ArH), 7.25−7.19 (1H, m, ArH), 7.18−7.14 (2H, m, ArH), 6.85 (1H, app tdd, J 7.5, 2.9, 1.0, ArH), 6.72 (1H, app dd, J 8.5, 1.0, ArH), 5.35 (1H, quintet, J 6.4, OCHCH3), 3.50 (1H, d, J 7.6, oxazoline CH), 1.93 (1H, app d of septets, J 7.5, 6.5, CH(CH3)2), 1.626 and 1.620 (3H, 2xd, J 6.4, OCHCH3), 1.54 (3H, s, gem dimethyl), 1.44 (3H, s, gem dimethyl), 1.182 and 1.180 (3H, 2xd, J 6.5, CH(CH3)CH3) and 1.06 (3H, d, J 6.5, CH(CH3)CH)3); δC (100 MHz) 161.7 (C=N), 156.41 and 156.38 (C-O), 143.0 (C), 131.2 (CH), 130.8 (CH), 128.42 (CH), 128.41 (CH), 128.3 (CH), 127.28 (CH), 127.27 (CH), 125.60 and 125.56 (CH), 125.3 (CH), 120.2 and 120.1 (CH), 119.7 and 119.6 (C) 114.6 and 114.4 (CH), 86.2 (C), 80.19 and 80.15 (CH), 76.6 and 76.4 (CH), 29.16 and 29.14 and 29.11 and 29.09 (CH and CH3), 24.37 and 24.34 (CH3), 21.2 (2CH3) and 20.3 (CH3); HRMS (NSI+): found 338.2120. C22H29NO2 (M + H) requires 338.2120.
3.6.4. (S)-4-Isopropyl-2-(2-(1-phenylpropoxy)phenyl)-4,5-dihydrooxazole 58
Following the procedure of 3.4.17 using (
S)-2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenol
41 (1.0 g, 4.87 mmol), (1-bromopropyl)benzene [
37] (970 mg, 4.87 mmol) and NaH (195 mg, 4.87 mmol) in DMF (20 cm
3) gave, after purification via flash column chromatography (hexane/Et
2O 4:1) at R
f 0.21,
58 (670 mg, 43%) as a pale-yellow oil as an inseparable 1:1 mixture of diastereomers; ν
max/cm
−1 3061, 3030, 2961, 2932, 2876, 1645, 1601, 1450, 1491, 1250, 1238, 1040, 976, 748, 733 and 700; δ
H (400 MHz) 7.62 (1H, app ddd,
J 7.6, 1.9, 1.0, ArH), 7.39−7.27 (4H, m, ArH), 7.27−7.20 (1H, m, ArH), 7.16 (1H, app ddd,
J 8.4, 7.4, 1.8, ArH), 6.85 (1H, app tdd,
J 7.5, 2.1, 1.0, ArH), 6.70 (1H, app ddd,
J 8.4, 3.1, 1.0, ArH), 5.12 and 5.11 (1H, 2x t,
J 7.2, PhC
H(O)CH
2CH
3), 4.46−4.38 (1 H, m, OC
HHCHN), 4.20−4.10 (2 H, m, OCH
HCHN), 2.07−1.79 (3H, m, C
H(CH
3)
2 and CH(OAr)C
H2CH
3), 1.07 (3H, d,
J 6.8, CH(C
H3)CH
3), 1.01 (3H, d,
J 6.4, CH(CH
3)C
H3) and 0.98 (3H, t,
J 7.2, CH
2C
H3); δ
C (100 MHz) 163.2 and 163.1 (C=N), 156.73 and 156.65 (C-O), 141.5 and 141.4 (C), 131.4 (CH), 130.97 and 130.92 (CH), 128.35 and 128.34 (2CH), 127.3 (CH), 126.15 and 126.13 (2CH), 120.06 and 120.04 (CH), 118.87 and 118.84 (C), 114.48 and 114.43 (CH), 81.7 and 81.6 (OCH), 72.5 (CH), 70.0 and 69.9 (CH
2), 32.78 and 32.76 (CH), 31.37 and 31.33 (CH
2), 18.75 and 18.73 (CH
3), 18.22 and 18.17 (CH
3) and 9.65 and 9.63 (CH
3); HRMS (ESI
+): found 324.1950. C
21H
26NO
2 (M + H) requires 324.1950.
3.6.5. 2-(1-Bromo-2-methylpropyl)thiophene
A solution of 2-methyl-1-(thiophen-2-yl)-propan-1-ol [
38] (1.00 g, 6.4 mmol) and pyridine (cat.) in CH
2Cl
2 (30 cm
3) was stirred at 0 °C while PBr
3 (0.30 cm
3, 0.87 g, 3.2 mmol) was added dropwise. After 1 h, aqueous Na
2CO
3 was added dropwise and the mixture was warmed to RT before being separated, with the aqueous layer being further extracted with CH
2Cl
2, and the combined organic extracts were dried and evaporated to give the title product (1.30 g, 93%) as a dark brown oil, which was used without further purification; δ
H (400 MHz) 7.27 (1H, ddd,
J 5.1, 1.3, 0.5, ArH), 7.03 (1H, ddd,
J 3.5, 1.3, 0.5, ArH), 6.91 (1H, dd,
J 5.1, 3.5, ArH), 5.10 (1H, d,
J 7.5, ArC
H(Br)
iPr), 2.29 (1H, d of septets,
J 7.6, 6.6, C
H(CH
3)
2), 1.18 (3 H, d,
J 6.6, CH
3) and 0.99 (3 H, d,
J 6.6, CH
3).
3.6.6. (S)-4-Isopropyl-2-(2-(2-methyl-1-(thiophen-2-yl)propoxy)phenyl)-4,5-dihydrooxazole 59
Following the procedure of 3.4.17 using (S)-2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenol 41 (1.0 g, 4.87 mmol), 2-(1-bromo-2-methylpropyl)thiophene (1.07 g, 4.87 mmol) and NaH (195 mg, 4.87 mmol) in DMF (20 cm3) gave, after purification via flash column chromatography (hexane/Et2O 4:1) at Rf 0.13, 59 (70 mg, 4%) as a pale-yellow oil as an inseparable 1:1 mixture of diastereoisomers; νmax/cm−1 2959, 2930, 2872, 1645, 1601, 1491, 1352, 1250, 1233, 1040, 1001, 961, 750 and 696; δH (400 MHz) 7.63 (1H, app dd, J 7.6, 1.8, ArH), 7.24−7.18 (2H, m, ArH), 6.98−6.94 (1H, m, ArH), 6.94−6.85 (2H, m, ArH), 6.85−6.81 (1H, m, ArH), 5.21 and 5.20 (1H, 2x d, J 5.4, ArCHO(Ar), 4.46−4.37 (1H, m, OCHHCHN), 4.18−4.08 (2H, m, OCHHCHN), 2.26−2.17 (1H, m, CH(CH3)2), 1.95−1.85 (1H, m, oxazoline CH(CH3)2), 1.10−1.05 (6H, m, CH(CH3)2) and 1.01−0.96 (6H, m, oxazoline CH(CH3)2); δC (100 MHz) 163.33 and 163.25 (C=N), 156.7 and 156.6 (4ry, ArC-O), 143.2 and 143.1 (C), 131.5 (CH), 131.1 and 131.0 (CH), 126.3 (CH), 125.45 and 125.41 (CH), 124.6 (CH), 120.4 (CH), 119.0 (C), 114.2 (CH), 81.8 and 81.7 (CH(OAr), 72.5 (oxazoline CH), 70.1 and 70.0 (CH2), 35.6 and 35.5 (CH), 32.8 (CH), 18.86 and 18.85 (CH3), 18.4 (CH3), 18.22 and 18.17 (CH3) and 18.07 and 18.02 (CH3); HRMS (ESI+): found 366.1489. C20H2NaNO25S (M + Na) requires 366.1504.
3.6.7. (S)-4-Isopropyl-2-(2-((1-phenyl-3-(trimethylsilyl)prop-2-yn-1-yl)oxy)phenyl)-4,5-dihydrooxazole 60
Following the procedure of 3.4.17 using (
S)-2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenol
41 (1.0 g, 4.87 mmol), (3-bromo-3-phenylprop-1-yn-1-yl)trimethylsilane [
39] (1.30 g, 4.87 mmol) and NaH (195 mg, 4.87 mmol) in DMF (20 cm
3) gave, after purification via flash column chromatography (hexane/Et
2O 4:1) at R
f 0.26,
60 (210 mg, 11%) as a pale-yellow oil as an inseparable 1:1 mixture of diastereomers; ν
max/cm
−1 2959, 2899, 1645, 1601, 1493, 1450, 1354, 1248, 1036, 908, 841, 754, 731 and 696; δ
H (400 MHz) 7.87 (1H, ddd,
J 7.7, 2.8, 1.8, ArH), 7.84−7.79 (2H, m, ArH), 7.52−7.44 (4H, m, ArH), 7.42−7.37 (1H, m, ArH), 7.16 (1H, tt,
J 7.5, 1.2, ArH), 6.03 and 6.01 (1H, s, ArC
HO(Ar)), 4.49−4.41 (1H, m, OC
HHCHN), 4.24−4.12 (2H, m, OCH
HCHN), 1.99−1.89 (1H, m, C
H(CH
3)
2, 1.11 (3H, d,
J 6.7, CH(C
H3)CH
3), 1.03 and 1.02 (3H, 2 × d,
J 6.7, CH(CH
3)C
H3) and 0.27 (9H, s, SiMe
3); δ
C (100 MHz) 162.4 and 162.1 (C=N), 156.1 and 156.0 (C), 137.7 and 137.6 (C), 131.4 (CH), 130.9 (CH), 128.40 (CH), 128.2 (2CH), 127.2 (2CH), 121.8 (CH), 119.7 and 119.6 (C), 117.6 and 117.5 (CH), 102.39 and 102.35 (≡
C-CHOAr), 94.02 and 93.98 (≡C-SiMe
3), 72.7 and 72.5 (CH), 72.1 and 72.0 (CH), 69.8 and 69.7 (CH
2), 32.68 and 32.61 (CH), 18.8 (CH
3), 18.2 and 18.1 (CH
3) and –0.4 (SiMe
3); HRMS (ESI
+): found 414.1850. C
24H
29NaNO
2Si (M + Na) requires 414.1865.
3.6.8. (S)-4-Isopropyl-2-(2-((1-phenylbut-3-en-1-yl)oxy)phenyl)-4,5-dihydrooxazole 61
Following the procedure of 3.4.17 using (
S)-2-(4-isopropyl-4,5-dihydrooxazol-2-yl)phenol
41 (1.0 g, 4.87 mmol), (1-bromobut-3-en-1-yl)benzene [
40] (1.03 g, 4.87 mmol) and NaH (195 mg, 4.87 mmol) in DMF (20 cm
3) gave, after purification via flash column chromatography (hexane/Et
2O 4:1) at R
f 0.19,
61 (50 mg, 3%) as a pale-yellow oil as an inseparable 1:1 mixture of diastereomers; ν
max/cm
−1 2956, 1643, 1601, 1582, 1450, 1491, 1385, 1250, 1040, 750 and 700; δ
H (400 MHz) 7.63 (1H, app dt,
J 7.6, 1.5, ArH), 7.40−7.34 (2H, m, ArH), 7.34−7.28 (2H, m, ArH), 7.24–7.20 (1H, m, ArH), 7.19−7.14 (1H, m, ArH), 6.86 (1H, app tdd,
J 7.5, 18.8, 1.0, ArH), 6.69 (1H, app ddd,
J 8.4, 3.9, 1.0, ArH), 5.90 (1H, ddt,
J 17.2, 10.3, 7.0, C
H=CH
2), 5.20 (1H, q,
J 6.2, OC
HPh), 5.08−5.01 (2H, m, CH=C
H2), 4.45−4.38 (1H, m, OC
HHCHN), 4.19−4.10 (2H, m, OCH
HC
HN), 2.84−2.72 (1H, m, C
HHCH=CH
2), 2.67−2.57 (1H, m, CH
HCH=CH
2), 1.96−1.86 (1H, m, C
H(CH
3)
2), 1.07 (3H, d,
J 6.8, CH(C
H3)CH
3) and 1.00 (3H, d,
J 6.8, CH(CH
3)C
H3)); δ
C (100 MHz) 163.2 and 163.1 (C=N), 156.5 and 156.4 (ArC-O), 140.94 and 140.88 (ArC), 134.1 and 134.0 (CH=), 131.45 and 131.43 (CH), 131.05 and 130.96 (CH), 128.41 and 128.39 (2CH), 127.5 (CH), 126.20 and 126.17 (2CH), 120.3 (CH), 118.96 and 118.90 (C), 117.48 and 117.42 (=CH
2), 114.64 and 114.61 (CH), 80.4 and 80.3 (CH), 72.5 (CH), 70.0 and 69.9 (CH
2), 42.81 and 42.78 (CH
2), 32.78 and 32.74 (CH), 18.8 (CH
3) and 18.24 and 18.18 (CH
3); HRMS (ESI
+): found 358.1768. C
22H
25NaNO
2 (M + Na) requires 358.1783.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












