
Developing CRISPR/Cas9-Mediated Fluorescent Reporter Human Pluripotent Stem-Cell Lines for High-Content Screening

Abstract
1. Introduction
2. Results
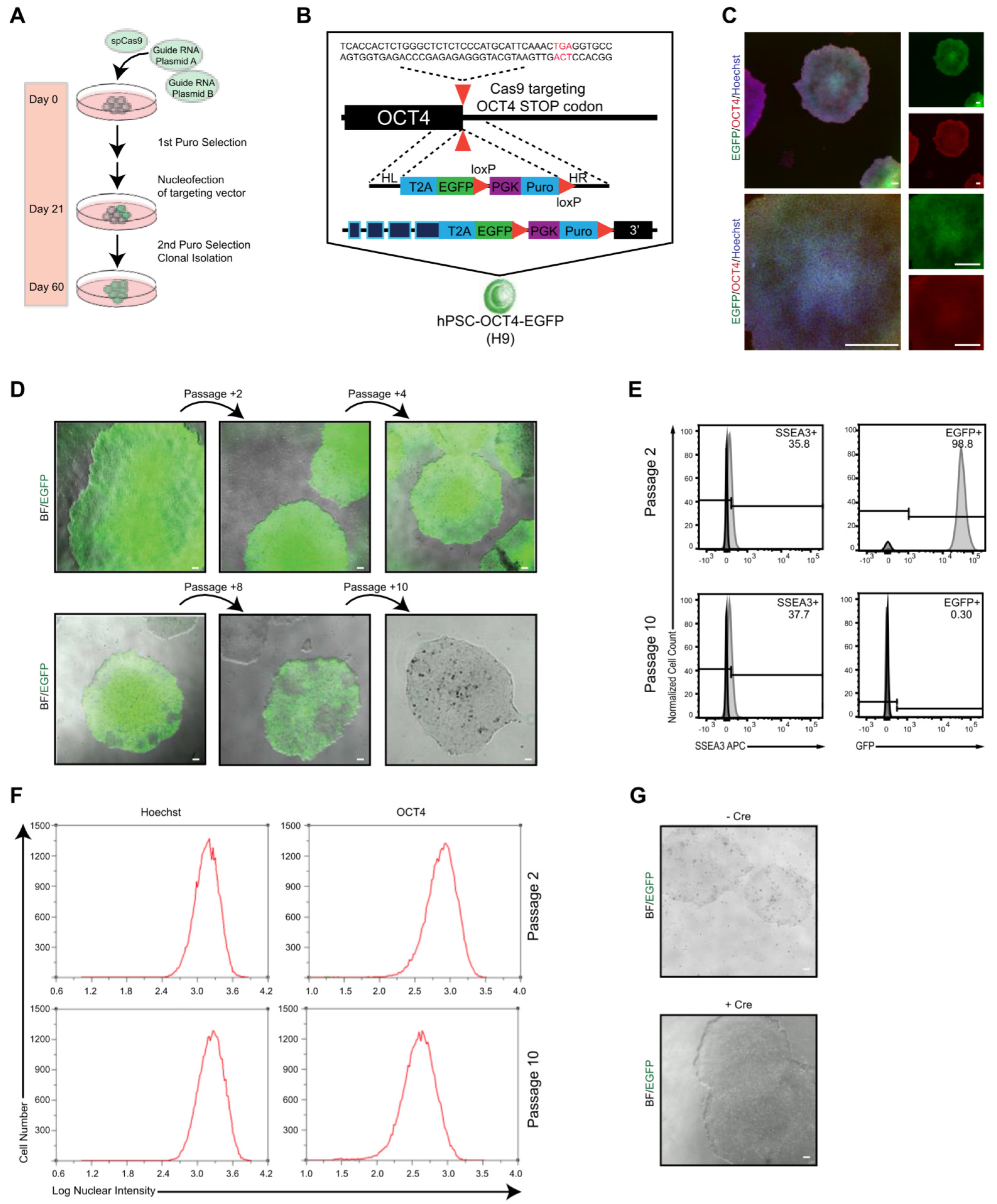
2.1. Generation of OCT4–EGFP Reporter hPSC Line
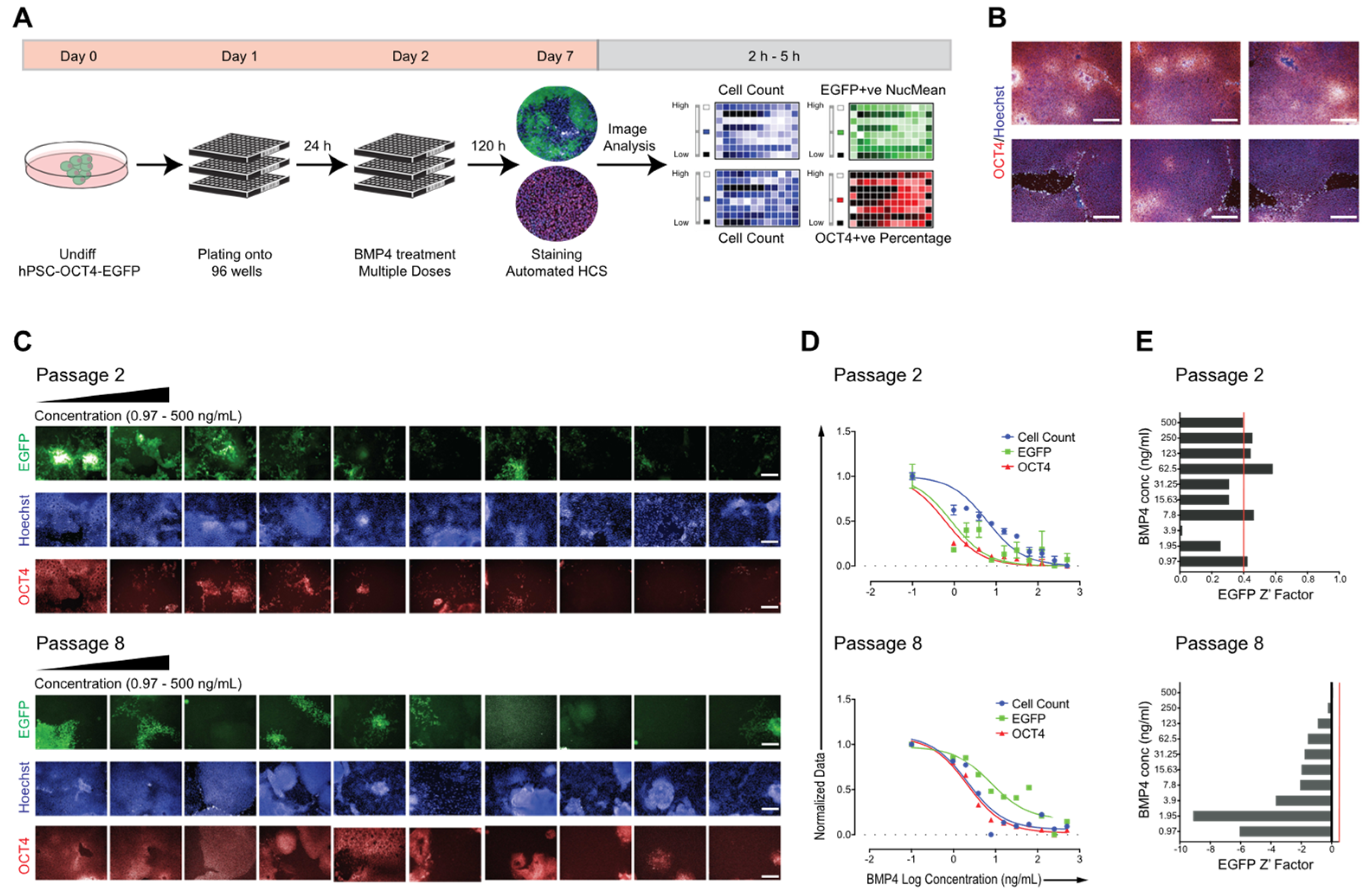
2.2. Adapting hPSC–OCT4–EGFP Culture Conditions for High-Content Screening
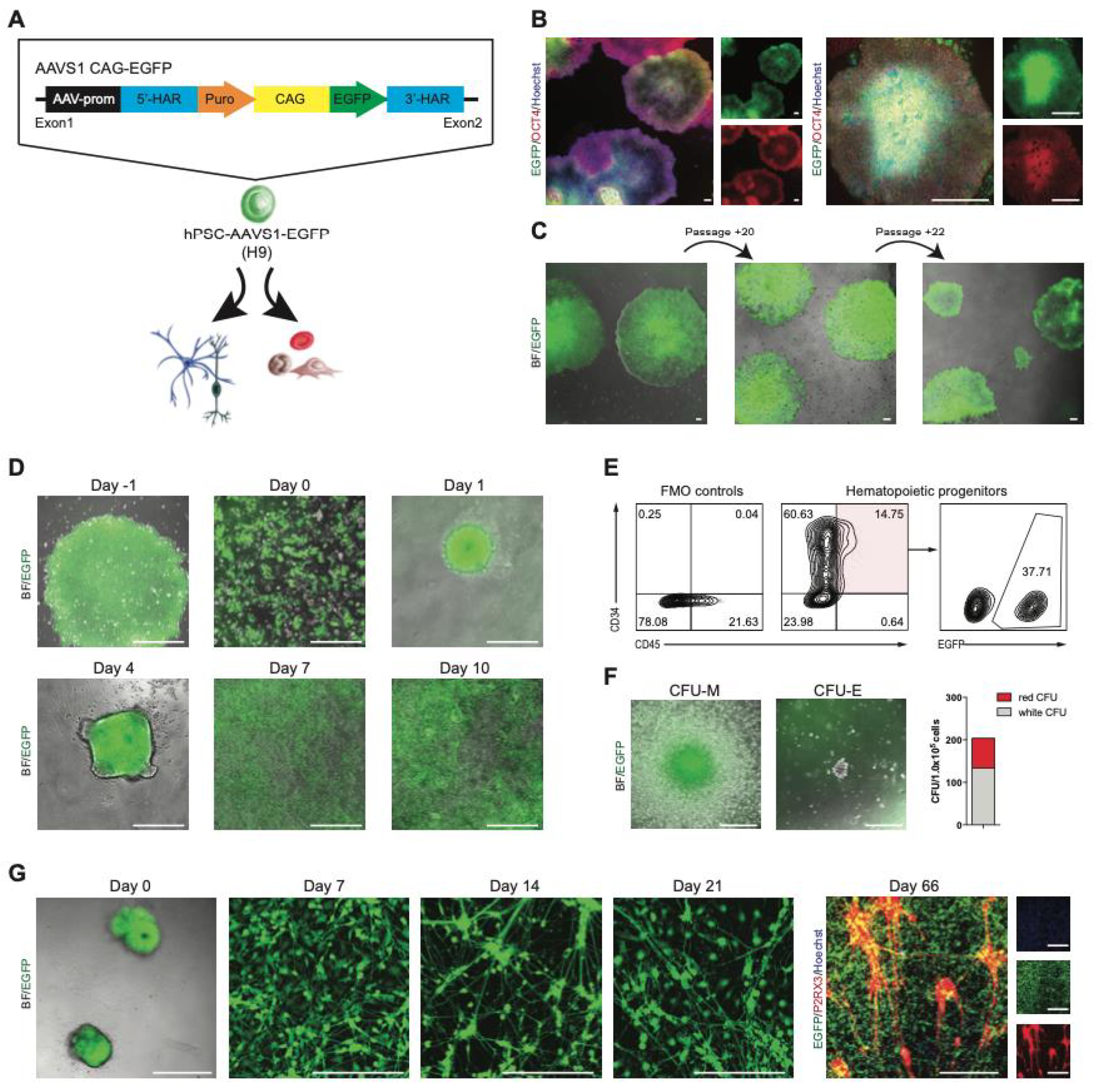
2.3. Development of a Stable EGFP Reporter hPSC Line Targeting the AAVS1 Locus
3. Discussion
4. Materials and Methods
4.1. Pluripotent Stem-Cell Culture and Differentiation
4.2. Genome Editing
4.3. Flow Cytometry
4.4. Immunofluorescence
4.5. BMP4 Differentiation Assay
4.6. Screening with hPSC–AAVS1–EGFP Line
4.7. Image Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Lerou, P.H.; Zhao, R.; Huo, H.; Daley, G.Q. Generation of human-induced pluripotent stem cells. Nat. Protoc. 2008, 3, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Pourquie, O.; Bruneau, B.; Keller, G.; Smith, A. Looking inwards: Opening a window onto human development. Development 2015, 142, 1–2. [Google Scholar] [CrossRef]
- Zhu, Z.; Huangfu, D. Human pluripotent stem cells: An emerging model in developmental biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—Opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Hochedlinger, K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011, 13, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Risueno, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef]
- Odorico, J.S.; Kaufman, D.S.; Thomson, J.A. Multilineage differentiation from human embryonic stem cell lines. Stem. Cells 2001, 19, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bickle, M. The beautiful cell: High-content screening in drug discovery. Anal. Bioanal. Chem. 2010, 398, 219–226. [Google Scholar] [CrossRef]
- Rubin, L.L. Stem cells and drug discovery: The beginning of a new era? Cell 2008, 132, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; Haston, K.M. Stem cell biology and drug discovery. BMC Biol. 2011, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Schultz, P.G. A role for chemistry in stem cell biology. Nat. Biotechnol. 2004, 22, 833–840. [Google Scholar] [CrossRef]
- Dorval, T.; Chanrion, B.; Cattin, M.E.; Stephan, J.P. Filling the drug discovery gap: Is high-content screening the missing link? Curr. Opin. Pharmacol. 2018, 42, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wong, S.T. Concise review: A high-content screening approach to stem cell research and drug discovery. Stem. Cells 2012, 30, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Strovel, J.; Sittampalam, S.; Coussens, N.P.; Hughes, M.; Inglese, J.; Kurtz, A.; Andalibi, A.; Patton, L.; Austin, C.; Baltezor, M.; et al. Early Drug Discovery and Development Guidelines: For Academic Researchers, Collaborators, and Start-up Companies. In Assay Guidance Manual; Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Bejcek, B., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Bethesda: Bethesda, MD, USA, 2004. [Google Scholar]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Majdi, M.; Xia, P.; Wei, K.A.; Talantova, M.; Spiering, S.; Nelson, B.; Mercola, M.; Chen, H.S. Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell-derived cardiomyocytes during differentiation. Stem. Cells Dev. 2010, 19, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Cochran, R.; Cheng, L. Double knockouts in human embryonic stem cells. Cell Res. 2010, 20, 250–252. [Google Scholar] [CrossRef]
- Merkle, F.T.; Eggan, K. Modeling human disease with pluripotent stem cells: From genome association to function. Cell Stem Cell 2013, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Sandoe, J.; Eggan, K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013, 16, 780–789. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism. 2017, 8, 11. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Beumer, K.; Trautman, J.K.; Carroll, D. Enhancing gene targeting with designed zinc finger nucleases. Science 2003, 300, 764. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.A.; Li, T.; Yang, B.; Spalding, M.H. TALEN-mediated genome editing: Prospects and perspectives. Biochem. J. 2014, 462, 15–24. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Jaenisch, R. Gene targeting in human pluripotent cells. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 201–209. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Maeder, M.L.; Mali, P.; Pruett-Miller, S.M.; Thibodeau-Beganny, S.; Chou, B.K.; Chen, G.; Ye, Z.; Park, I.H.; Daley, G.Q.; et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 2009, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, 763. [Google Scholar] [CrossRef]
- Wood, A.J.; Lo, T.W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333, 307. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Zhu, Z.; Shi, Z.D.; Lelli, K.; Verma, N.; Li, Q.V.; Huangfu, D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 2014, 15, 215–226. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2, e00471. [Google Scholar] [CrossRef]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F. CRISPR/Cas9 genome editing in human pluripotent stem cells: Harnessing human genetics in a dish. Dev. Dyn. 2016, 245, 788–806. [Google Scholar] [CrossRef] [PubMed]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Nunez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Wang, Q.; Zou, Y.; Nowotschin, S.; Kim, S.Y.; Li, Q.V.; Soh, C.L.; Su, J.; Zhang, C.; Shu, W.; Xi, Q.; et al. The p53 Family Coordinates Wnt and Nodal Inputs in Mesendodermal Differentiation of Embryonic Stem Cells. Cell Stem Cell 2017, 20, 70–86. [Google Scholar] [CrossRef]
- Hunt, C.P.J.; Pouton, C.W.; Haynes, J.M. Characterising the developmental profile of human embryonic stem cell-derived medium spiny neuron progenitors and assessing mature neuron function using a CRISPR-generated human DARPP-32(WT/eGFP-AMP) reporter line. Neurochem. Int. 2017, 106, 3–13. [Google Scholar] [CrossRef]
- Merkle, F.T.; Neuhausser, W.M.; Santos, D.; Valen, E.; Gagnon, J.A.; Maas, K.; Sandoe, J.; Schier, A.F.; Eggan, K. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015, 11, 875–883. [Google Scholar] [CrossRef]
- Kearns, N.A.; Genga, R.M.; Enuameh, M.S.; Garber, M.; Wolfe, S.A.; Maehr, R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 2014, 141, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ma, D.; Huang, R.; Ming, J.; Ye, M.; Kee, K.; Xie, Z.; Na, J. An inducible CRISPR-ON system for controllable gene activation in human pluripotent stem cells. Protein Cell 2017, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutskov, K.; Jouhilahti, E.M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human pluripotent reprogramming with CRISPR activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Gao, F.; Han, S.; Cheah, K.S.; Tse, H.F.; Lian, Q. CRISPR/Cas9 Genome-Editing System in Human Stem Cells: Current Status and Future Prospects. Mol. Ther. Nucleic Acids 2017, 9, 230–241. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Avior, Y.; Lezmi, E.; Yanuka, D.; Benvenisty, N. Modeling Developmental and Tumorigenic Aspects of Trilateral Retinoblastoma via Human Embryonic Stem Cells. Stem Cell Rep. 2017, 8, 1354–1365. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, D.; Liu, H.; Gao, G.; Liu, Y.; Zhuang, Y.; Yang, F.; Wang, K.; Zhou, T.; Qin, D.; Hong, L.; et al. Creating a patient carried Men1 gene point mutation on wild type iPSCs locus mediated by CRISPR/Cas9 and ssODN. Stem Cell Res. 2017, 18, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Gerwen, B.V.; van der Ven, M.; Wetering, M.V.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife 2016, 5, e18489. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [PubMed]
- De Ravin, S.S.; Li, L.; Wu, X.; Choi, U.; Allen, C.; Koontz, S.; Lee, J.; Theobald-Whiting, N.; Chu, J.; Garofalo, M.; et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017, 9, eaah3480. [Google Scholar] [CrossRef] [PubMed]
- Chia, N.Y.; Chan, Y.S.; Feng, B.; Lu, X.; Orlov, Y.L.; Moreau, D.; Kumar, P.; Yang, L.; Jiang, J.; Lau, M.S.; et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 2010, 468, 316–320. [Google Scholar] [CrossRef]
- Theunissen, T.W.; Powell, B.E.; Wang, H.; Mitalipova, M.; Faddah, D.A.; Reddy, J.; Fan, Z.P.; Maetzel, D.; Ganz, K.; Shi, L.; et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 2014, 15, 471–487. [Google Scholar] [CrossRef]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef]
- Bukhari, H.; Muller, T. Endogenous Fluorescence Tagging by CRISPR. Trends Cell Biol. 2019, 29, 912–928. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed]
- Sluch, V.M.; Chamling, X.; Wenger, C.; Duan, Y.; Rice, D.S.; Zack, D.J. Highly efficient scarless knock-in of reporter genes into human and mouse pluripotent stem cells via transient antibiotic selection. PLoS ONE 2018, 13, e0201683. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Igai, K.; Hagihara, Y.; Hashimoto, R.; Hanawa, M.; Sakuma, T.; Tachibana, M.; Sakurai, F.; Yamamoto, T.; Mizuguchi, H. Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res. 2017, 45, 5198–5207. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Um, E.; Cho, S.R.; Jung, C.; Kim, H.; Kim, J.S. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods 2011, 8, 941–943. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Cho, S.W.; Kim, S.; Song, M.; Gopalappa, R.; Kim, J.S.; Kim, H. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat. Commun. 2014, 5, 3378. [Google Scholar] [CrossRef]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef]
- Koehler, K.R.; Tropel, P.; Theile, J.W.; Kondo, T.; Cummins, T.R.; Viville, S.; Hashino, E. Extended passaging increases the efficiency of neural differentiation from induced pluripotent stem cells. BMC Neurosci. 2011, 12, 82. [Google Scholar] [CrossRef]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Vojnits, K.; Mahammad, S.; Collins, T.J.; Bhatia, M. Chemotherapy-Induced Neuropathy and Drug Discovery Platform Using Human Sensory Neurons Converted Directly from Adult Peripheral Blood. Stem Cells Transl. Med. 2019, 8, 1180–1191. [Google Scholar] [CrossRef]
- Lee, J.; Dykstra, B.; Spencer, J.A.; Kenney, L.L.; Greiner, D.L.; Shultz, L.D.; Brehm, M.A.; Lin, C.P.; Sackstein, R.; Rossi, D.J. mRNA-mediated glycoengineering ameliorates deficient homing of human stem cell-derived hematopoietic progenitors. J. Clin. Investig. 2017, 127, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Kearns, N.A.; Maehr, R. Transcriptional Regulation with CRISPR/Cas9 Effectors in Mammalian Cells. Methods Mol. Biol. 2016, 1358, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Teshigawara, R.; Hirano, K.; Nagata, S.; Ainscough, J.; Tada, T. OCT4 activity during conversion of human intermediately reprogrammed stem cells to iPSCs through mesenchymal-epithelial transition. Development 2016, 143, 15–23. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Passage 2 EC50 (ng/mL) | Passage 8 EC50 (ng/mL) | |
|---|---|---|
| Cell count | 5.6 | 3.75 |
| EGFP | 1.23 | 7.8 |
| OCT4 | 0.78 | 3.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojnits, K.; Nakanishi, M.; Porras, D.; Kim, Y.; Feng, Z.; Golubeva, D.; Bhatia, M. Developing CRISPR/Cas9-Mediated Fluorescent Reporter Human Pluripotent Stem-Cell Lines for High-Content Screening. Molecules 2022, 27, 2434. https://doi.org/10.3390/molecules27082434
Vojnits K, Nakanishi M, Porras D, Kim Y, Feng Z, Golubeva D, Bhatia M. Developing CRISPR/Cas9-Mediated Fluorescent Reporter Human Pluripotent Stem-Cell Lines for High-Content Screening. Molecules. 2022; 27(8):2434. https://doi.org/10.3390/molecules27082434
Chicago/Turabian StyleVojnits, Kinga, Mio Nakanishi, Deanna Porras, Yeonjoon Kim, Zhuohang Feng, Diana Golubeva, and Mick Bhatia. 2022. "Developing CRISPR/Cas9-Mediated Fluorescent Reporter Human Pluripotent Stem-Cell Lines for High-Content Screening" Molecules 27, no. 8: 2434. https://doi.org/10.3390/molecules27082434
APA StyleVojnits, K., Nakanishi, M., Porras, D., Kim, Y., Feng, Z., Golubeva, D., & Bhatia, M. (2022). Developing CRISPR/Cas9-Mediated Fluorescent Reporter Human Pluripotent Stem-Cell Lines for High-Content Screening. Molecules, 27(8), 2434. https://doi.org/10.3390/molecules27082434