
In Vitro and In Silico Studies of Human Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibition by Stereoisomeric Forms of Lipophilic Nucleosides: The Role of Carbohydrate Stereochemistry in Ligand-Enzyme Interactions

 , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Chemical Synthesis
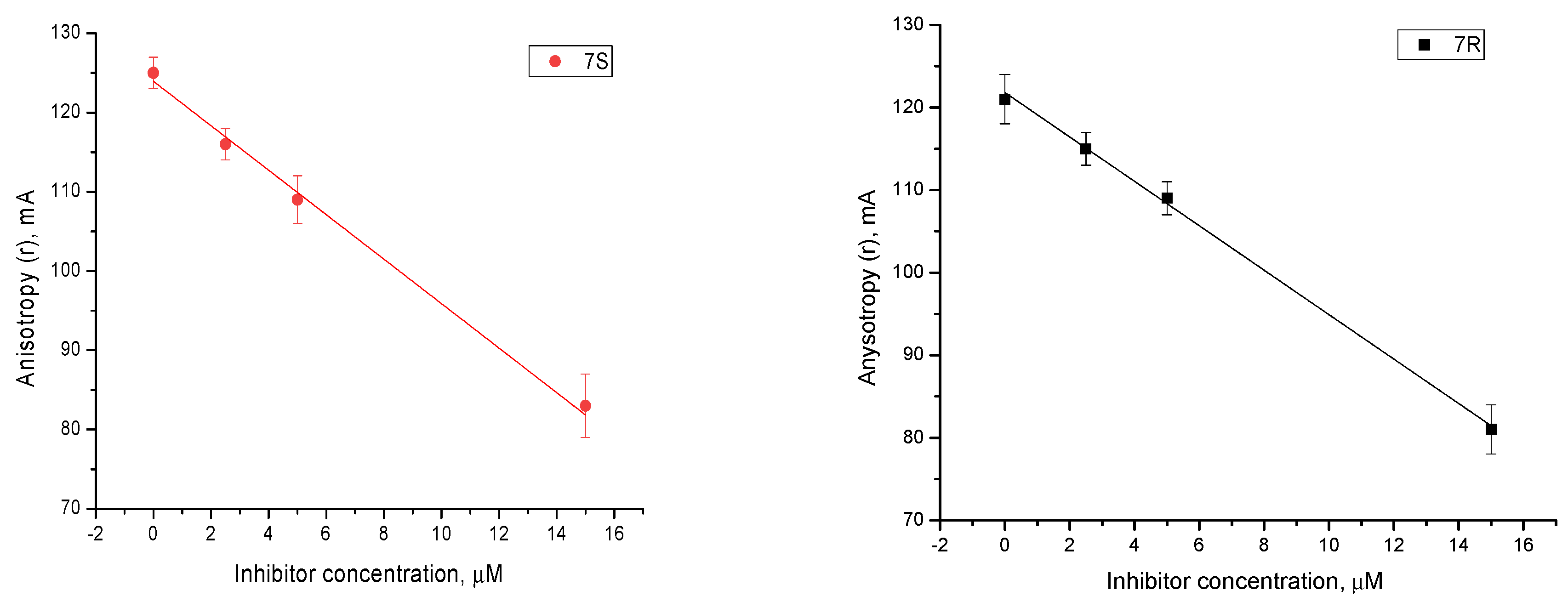
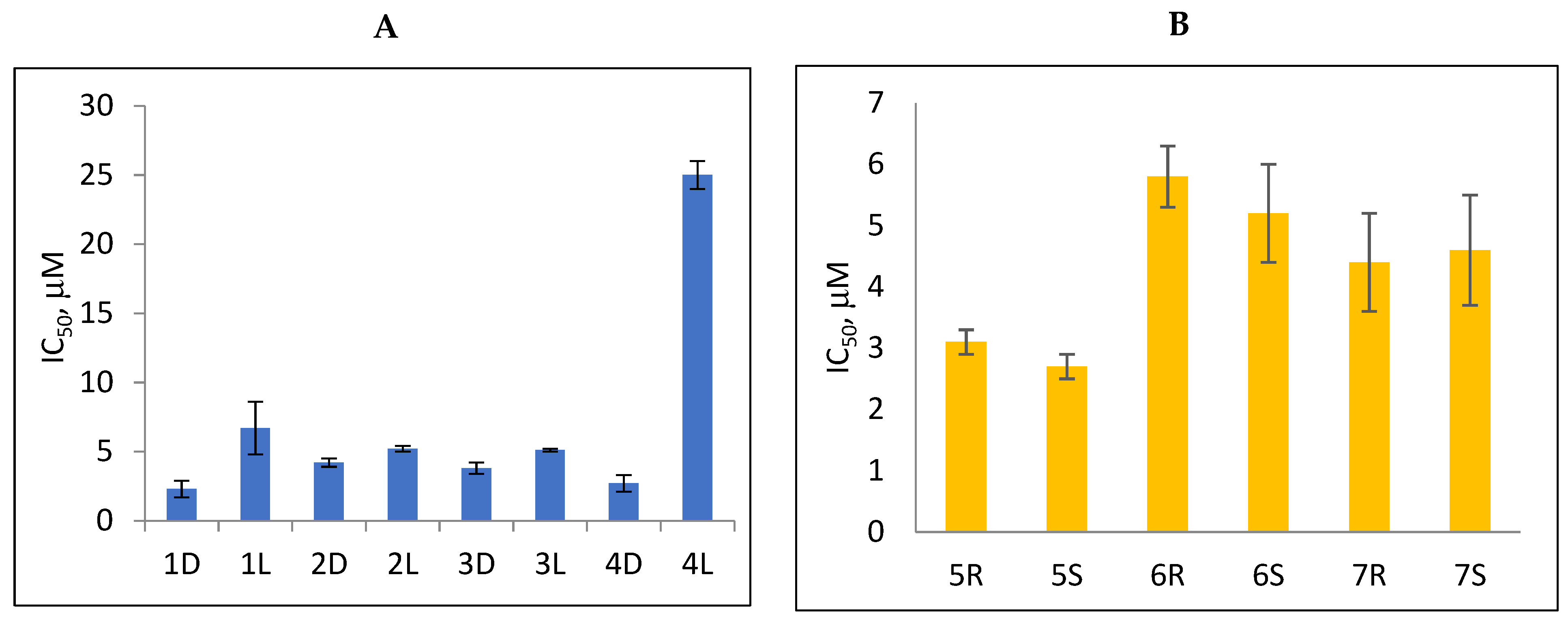
2.2. Tdp1 Inhibition
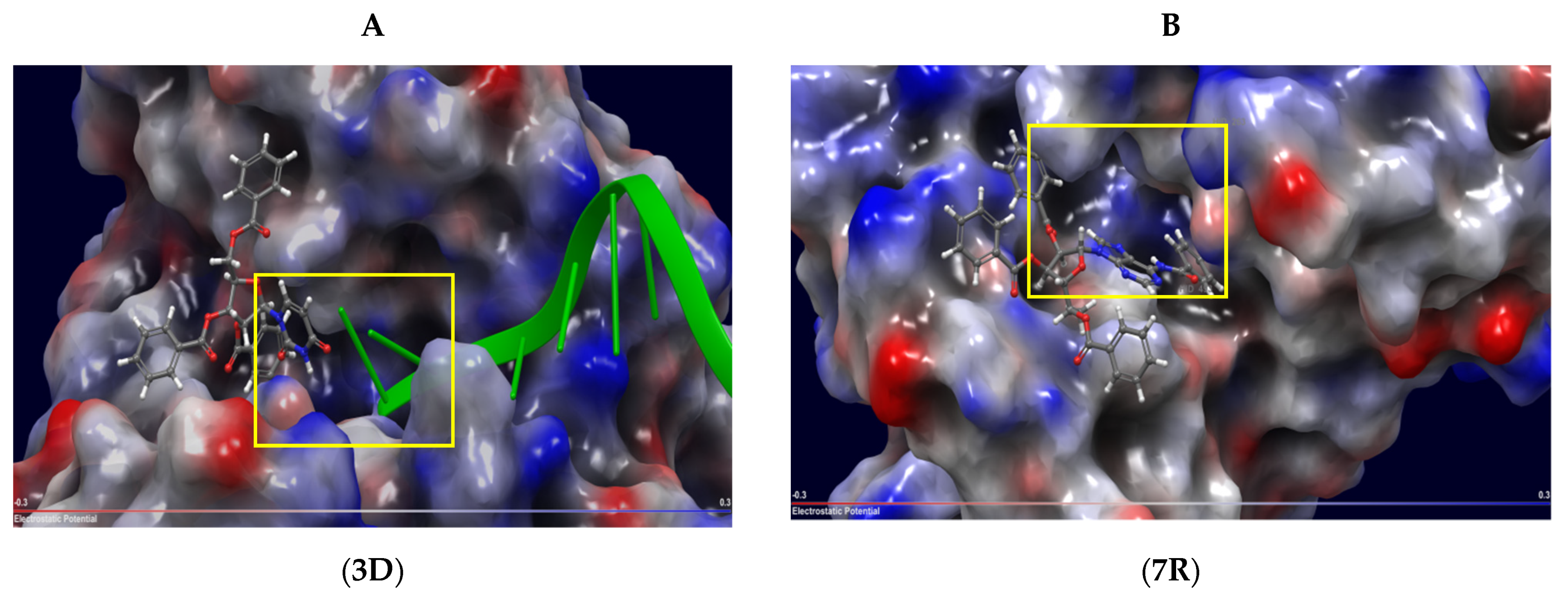
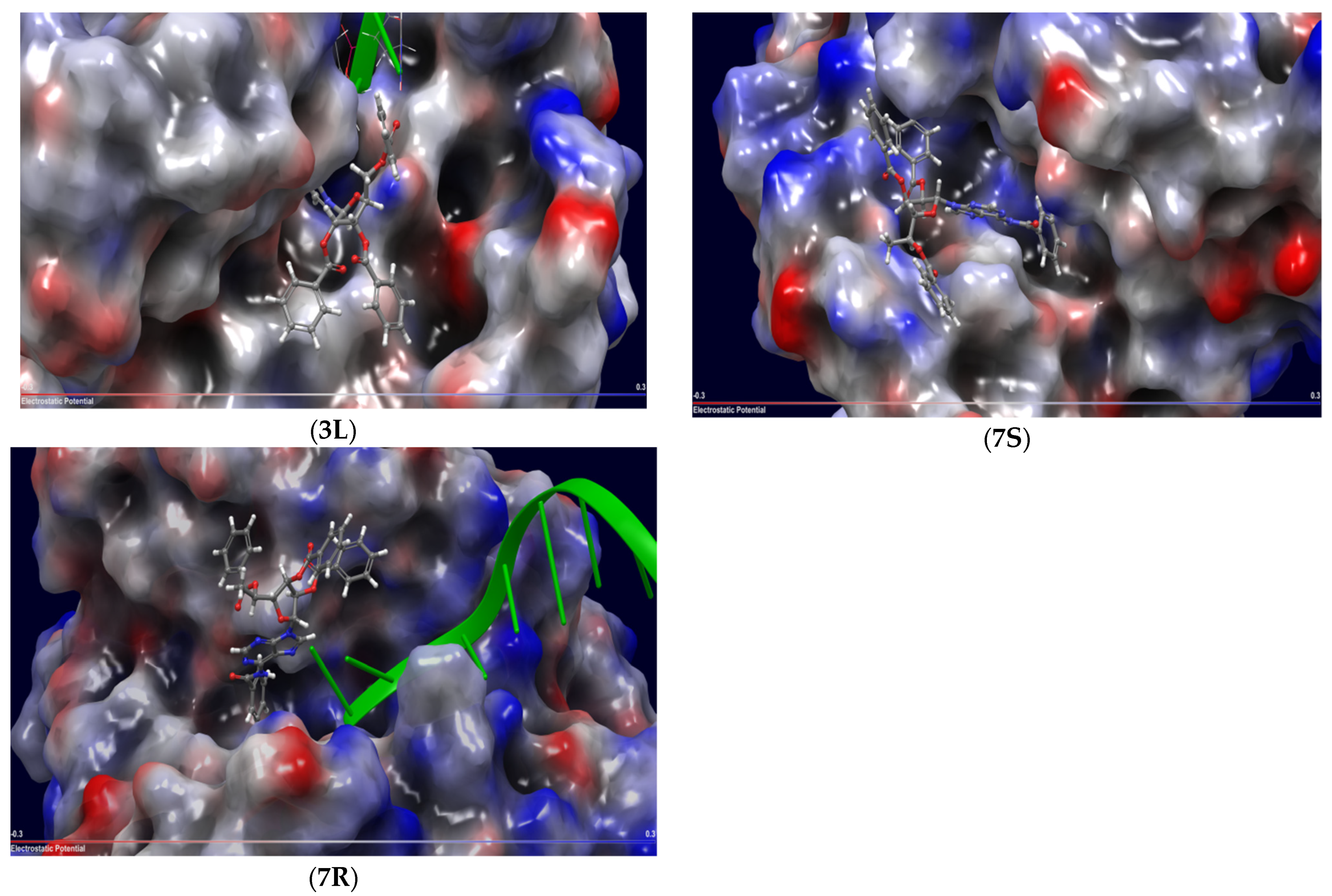
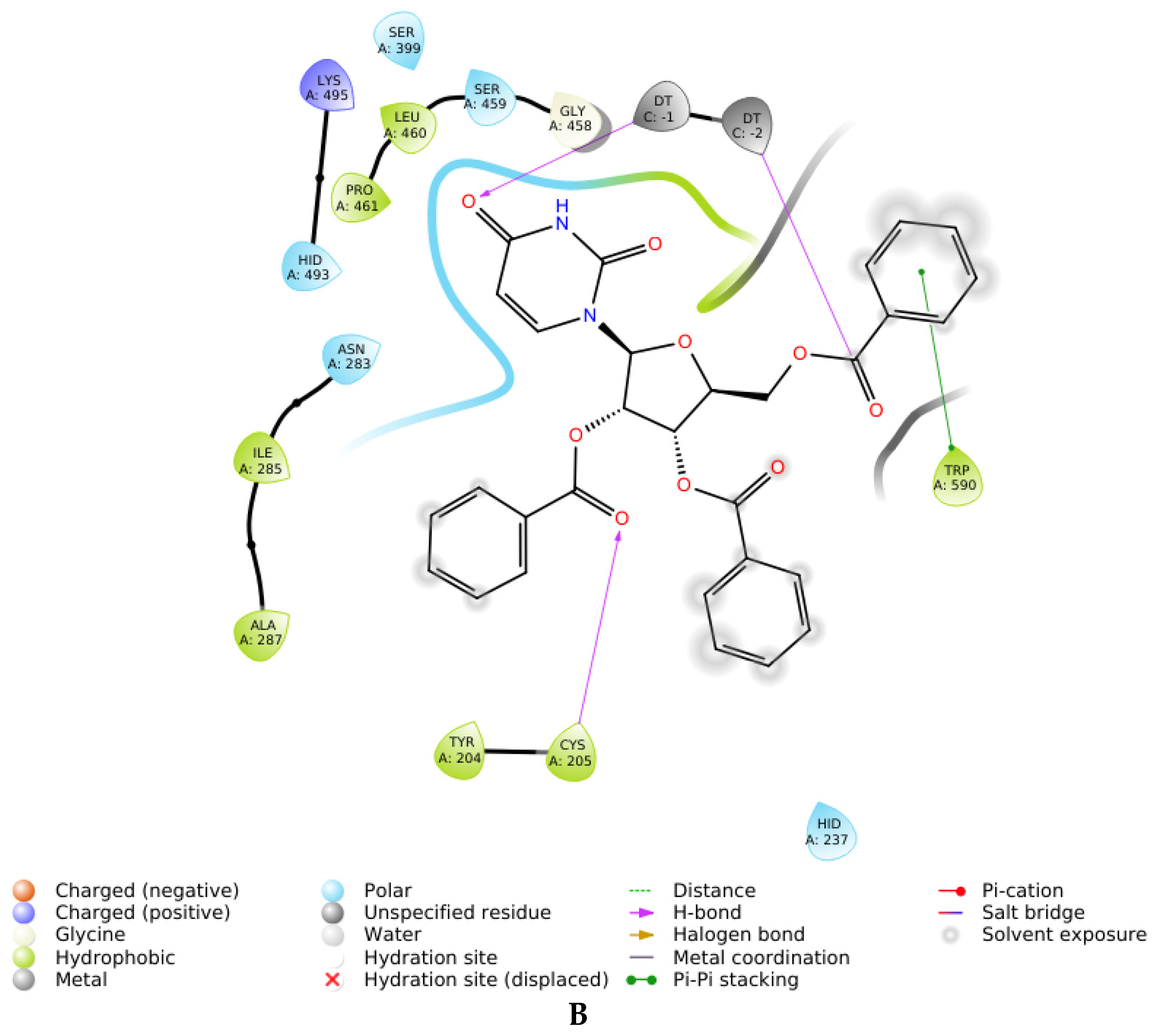
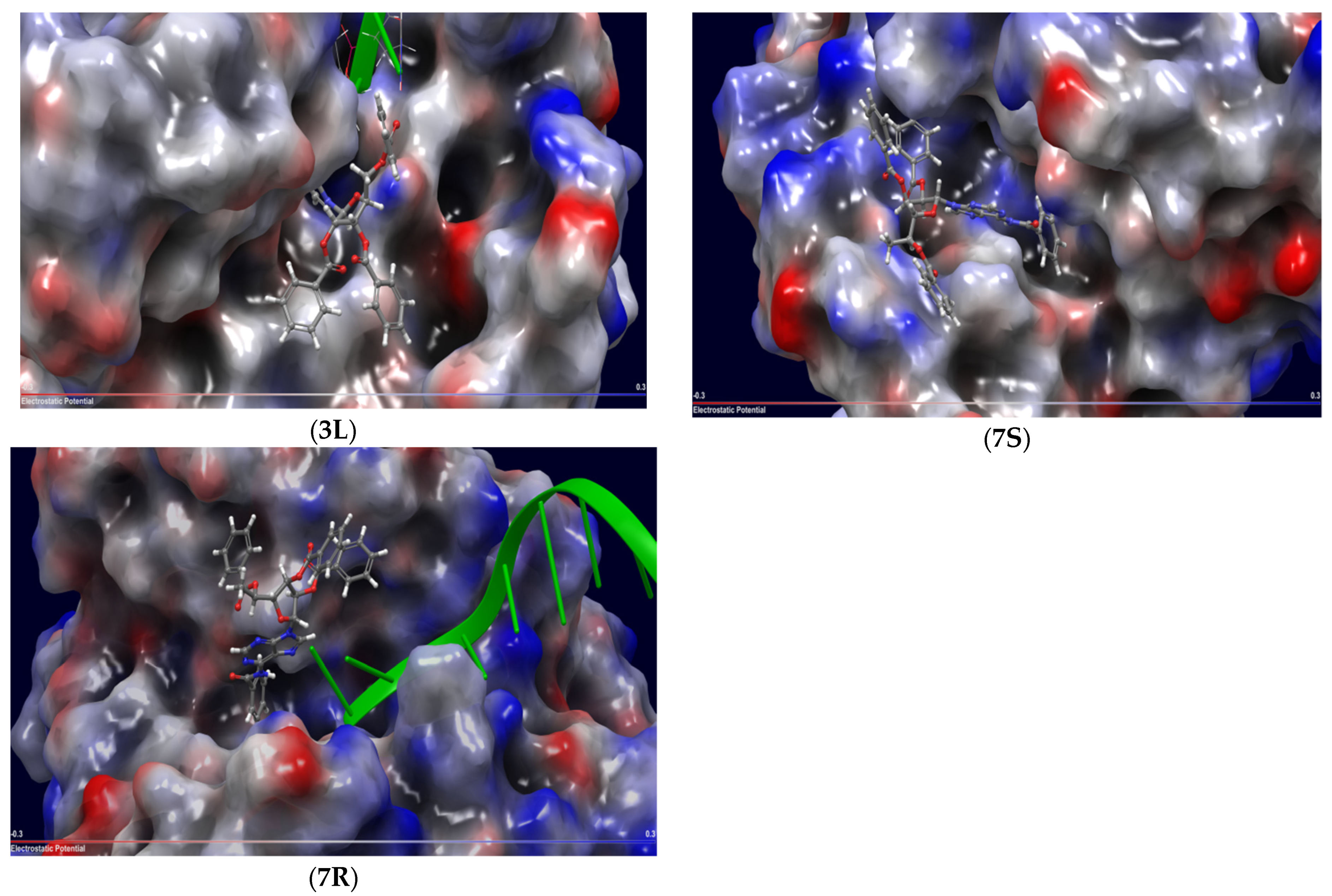
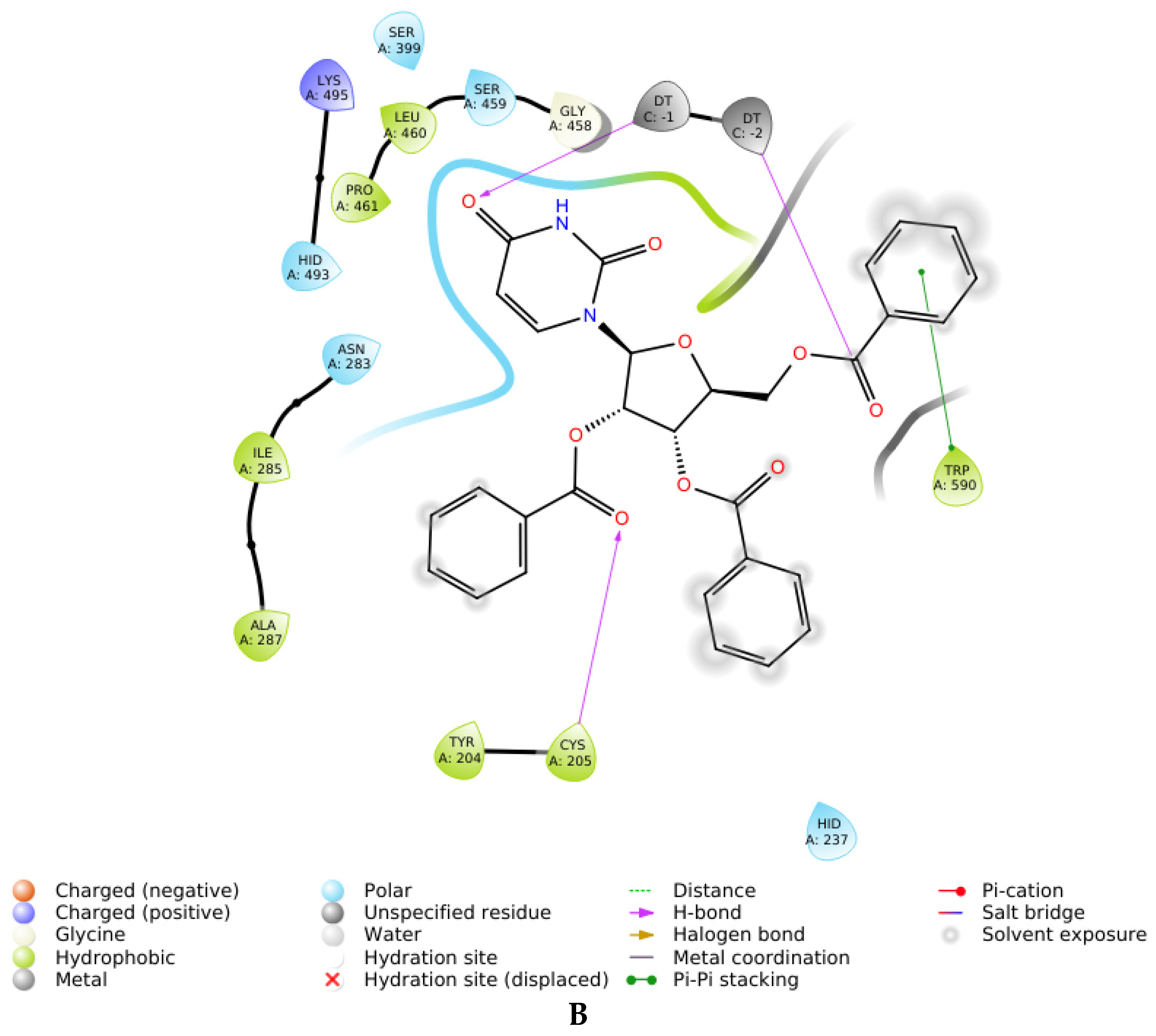
2.3. Molecular Docking
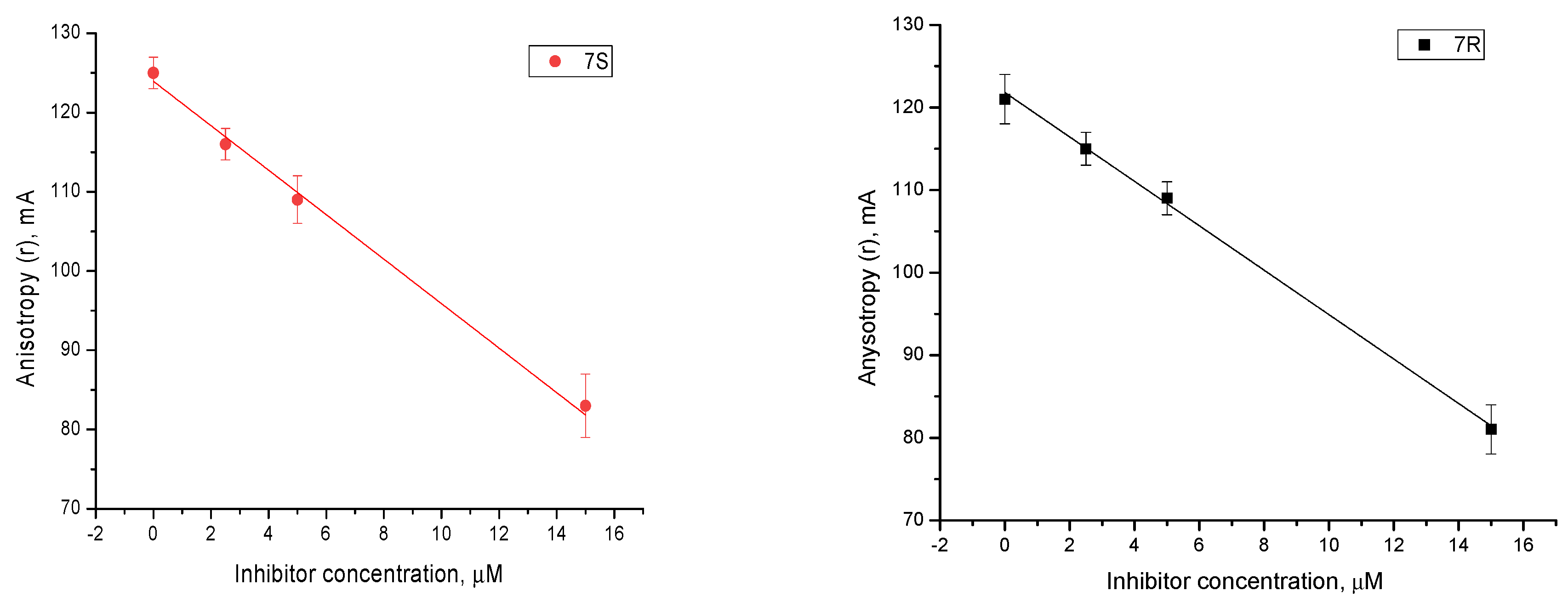
2.4. Mechanism of Action
3. Discussion
4. Materials and Methods
4.1. General Chemistry
4.2. Real-Time Detection of Tdp1 Activity
4.3. Steady-State Kinetic Analysis of Tdp1 Enzymatic Reaction
4.4. Molecular Docking
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hutt, A.J. Drug chirality and its pharmacological consequences. In Introduction to the Principles of Drug Design and Action, 3rd ed.; Smith, H.J., Ed.; Taylor & Francis: Cardiff, UK, 2005; Chapter 4; pp. 98–165. [Google Scholar]
- Munshi, R.; Hansske, F.; Baer, H.P. [125I]N6-(3-Iodo-4-hydroxyphenyl)isopropyladenosine: The use of the diastereomers as ligands for adenosine receptors in rat brain. Eur. J. Pharm. 1985, 111, 107–115. [Google Scholar] [CrossRef]
- Yu, X.; Yao, Z.-P. Chiral recognition and determination of enantiomeric excess by mass spectrometry: A review. Anal. Chim. Acta 2017, 968, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zask, A.; Ellestad, G.A. Recent advances in stereoselective drug targeting. Chirality 2015, 27, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.; Malina, J.; Kasparkova, J.; Brabec, V.; Natile, G. Chiral discrimination in platinum anticancer drugs. Environ. Health Perspect. 2002, 110, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Zenchenko, A.A.; Drenichev, M.S.; Il’icheva, I.A.; Mikhailov, S.N. Antiviral and Antimicrobial Nucleoside Derivatives: Structural Features and Mechanisms of Action. Rus. J. Mol. Biol. 2021, 55, 786–812. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef]
- Drenichev, M.S.; Oslovsky, V.E.; Mikhailov, S.N. Cytokinin Nucleosides—Natural compounds with a unique spectrum of biological activities. Curr. Top. Med. Chem. 2016, 16, 2562–2576. [Google Scholar] [CrossRef]
- Enkvist, E.; Raidaru, G.; Vaasa, A.; Pehk, T.; Lavogina, D.; Uri, A. Carbocyclic 3-deoxyadenosine-based highly potent bisubstrate-analog inhibitor of basophilic protein kinases. Bioorg. Med. Chem. Lett. 2007, 17, 5336–5339. [Google Scholar] [CrossRef]
- Chu, C.K.; Ahn, S.K.; Kim, H.O.; Beach, W.J.; Alves, A.J.; Jeong, L.S.; Islam, Q.; Van Roey, P.; Schinazi, R.F. Asymmetric synthesis of enantiomerically pure (−)-(1′R, 4′R)-dioxolane-thymine and its anti-HIV activity. Tetrahedron Lett. 1991, 32, 3791–3794. [Google Scholar] [CrossRef]
- Eckstein, F. Nucleoside phosphorothioates. Ann. Rev. Biochem. 1985, 54, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Gumina, G.; Song, G.-Y.; Chu, C.K. L-Nucleosides as chemotherapeutic agents. FEMS Microbiol. Lett. 2001, 202, 9–15. [Google Scholar] [CrossRef]
- Comeaux, E.Q.; Waardenburg, R.C. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef]
- Brettrager, E.J.; van Waardenburg, R.C.A.M. Targeting tyrosyl-DNA phosphodiesterase I to enhance toxicity of phosphodiester linked DNA adducts. Cancer Drug Resist. 2019, 2, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Zakharenko, A.; Dyrkheeva, N.; Lavrik, O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef]
- Komarova, A.O.; Drenichev, M.S.; Dyrkheeva, N.S.; Kulikova, I.V.; Oslovsky, V.E.; Zakharova, O.D.; Zakharenko, A.L.; Mikhailov, S.N.; Lavrik, O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enzym. Inhib. Med. Chem. 2018, 33, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Zakharenko, A.L.; Drenichev, M.S.; Dyrkheeva, N.S.; Ivanov, G.A.; Oslovsky, V.E.; Ilina, E.S.; Chernyshova, I.A.; Lavrik, O.I.; Mikhailov, S.N. Inhibition of tyrosyl-DNA phosphodiesterase 1 by lipophilic pyrimidine nucleosides. Molecules 2020, 25, 3694. [Google Scholar] [CrossRef]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.X.; Morrell, A.; Conda-Sheridan, M.; Marchand, C.; Agama, K.; Bermingam, A.; Stephen, A.G.; Chergui, A.; Naumova, A.; Fisher, R.; et al. Synthesis and biological evaluation of the first dual tyrosyl-DNA phosphodiesterase I (Tdp1)–topoisomerase I (Top1) inhibitors. J. Med. Chem. 2012, 55, 4457–4478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexheimer, T.S.; Gediya, L.K.; Stephen, A.G.; Weidlich, I.; Antony, S.; Marchand, C.; Interthal, H.; Nicklaus, M.; Fisher, R.J.; Njar, V.C.; et al. 4-Pregnen-21-ol-3, 20-dione-21-(4-bromobenzenesufonate)(NSC 88915) and related novel steroid derivatives as tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors. J. Med. Chem. 2009, 52, 7122–7131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, S.; Marchand, C.; Stephen, A.G.; Thibaut, L.; Agama, K.K.; Fisher, R.J.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Aid. Res. 2007, 35, 4474–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, C.; Lea, W.A.; Jadhav, A.; Dexheimer, T.S.; Austin, C.P.; Inglese, J.; Pommier, Y.; Simeonov, A. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol. Cancer Ther. 2009, 8, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conda-Sheridan, M.; Reddy, P.N.; Morrell, A.; Cobb, B.T.; Marchand, C.; Agama, K.; Chergui, A.; Renaud, A.; Stephen, A.G.; Bindu, L.; et al. Synthesis and biological evaluation of indenoisoquinolines that inhibit both tyrosyl-DNA phosphodiesterase I (Tdp1) and topoisomerase I (Top1). J. Med. Chem. 2013, 56, 182–200. [Google Scholar] [CrossRef] [Green Version]
- Sirivolu, V.R.; Vernekar, S.K.V.; Marchand, C.; Naumova, A.; Chergui, A.; Renaud, A.; Stephen, A.G.; Chen, F.; Sham, Y.Y.; Pommier, Y.; et al. 5-Arylidenethioxothiazolidinones as inhibitors of tyrosyl–DNA phosphodiesterase I. J. Med. Chem. 2012, 55, 8671–8684. [Google Scholar] [CrossRef]
- Takagi, M.; Ueda, J.Y.; Hwang, J.H.; Hashimoto, J.; Izumikawa, M.; Murakami, H.; Sekido, Y.; Shin-ya, K. Tyrosyl-DNA phosphodiesterase 1 inhibitor from an anamorphic fungus. J. Nat. Prod. 2012, 75, 764–767. [Google Scholar] [CrossRef]
- Tian, L.W.; Feng, Y.; Tran, T.D.; Shimizu, Y.; Pfeifer, T.; Forster, P.I.; Quinn, R.J. Tyrosyl-DNA phosphodiesterase I inhibitors from the Australian plant Macropteranthes leichhardtii. J. Nat. Prod. 2015, 78, 1756–1760. [Google Scholar] [CrossRef]
- Dean, R.A.; Fam, H.K.; An, J.; Choi, K.; Shimizu, Y.; Jones, S.J.; Boerkoel, C.F.; Interthal, H.; Pfeifer, T.A. Identification of a putative Tdp1 inhibitor (CD00509) by in vitro and cell-based assays. J. Biomol. Screen. 2014, 19, 1372–1382. [Google Scholar] [CrossRef] [Green Version]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Aivazashvili, V.A.; Mikhailov, S.N.; Padyukova, N.S.H.; Bibilashvili, R.S.H.; Karpeiskii, M.Y.A. 5′-C-methylnucleoside triphosphates in the reaction of RNA synthesis catalyzed by Escherichia coli RNA polymerase. Rus. J. Mol. Biol. 1987, 21, 1080–1091. [Google Scholar]
- Ganeshpurkar, A.; Gutti, G.; Singh, S.K. RNA-Dependent RNA Polymerases and Their Emerging Roles in Antiviral Therapy. In Viral Polymerases Structures, Functions and Roles as Antiviral Drug Targets; Gupta, S.P., Ed.; Academic Press: Cambridge, MA, USA, 2018; Chapter 1; pp. 1–42. [Google Scholar]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA phosphodiesterase 1 inhibitors: Usnic acid enamines enhance the cytotoxic effect of camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef] [PubMed]
- Korbukh, I.A.; Abramova, L.N.; Preobrazhenskaya, M.N. Anomers of 1-O-acetyl-2,3,5-tri-O-benzoyl-d-arabinose. In Nucleic Acid chemistry. Improved and New Synthetic Procedures, Methods and Techniques (Part I); Townsend, L.B., Stuart Tipson, R., Eds.; John Wiley and Sons: New York, NY, USA, 1978; pp. 151–152. [Google Scholar]
- Niedballa, U.; Vorbruggen, H. A general synthesis of N-glycosides. IV. Synthesis of nucleosides of hydroxyl and mercapto N-heterocycles. J. Org. Chem. 1977, 39, 3668–3671. [Google Scholar] [CrossRef] [PubMed]
- Vorbrüggen, H.; Bennua, B. A new simplified nucleoside synthesis. Chem. Ber. 1981, 114, 1279–1286. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley & Sons: New York, NY, USA, 2001; Volume 60, pp. 10–29. [Google Scholar]
- Vorbrüggen, H. Adventures in silicon-organic chemistry. Acc. Chem. Res. 1995, 28, 509–520. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Efimtseva, E.V.; Rodionov, A.A.; Bobkov, G.V.; Kulikova, I.V.; Herdewijn, P. Synthesis of 2′-O-β-d-ribofuranosylnucleosides. Curr. Prot. Nucl. Acid Chem. 2006, 27, 1.14.1–1.14.18. [Google Scholar] [CrossRef]
- Reist, E.J.; Goodman, L.; Baker, B.R. Potential anticancer agents. VIII. Synthesis of nucleosides derived from L-talofuranose. J. Am. Chem. Soc. 1958, 80, 5775–5779. [Google Scholar] [CrossRef]
- Prasad, A.K.; Kumar, V.; Malhotra, S.; Ravikumar, V.T.; Sanghvi, Y.S.; Parmar, V.S. ‘Green’ methodology for efficient and selective benzoylation of nucleosides using benzoyl cyanide in an ionic liquid. Bioorg. Med. Chem. 2005, 13, 4467–4472. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | clogP 1 | IC50 μM | Eut/Dis | ER 2 | EI 3 |
|---|---|---|---|---|---|---|
| 1D | 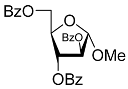 | 5.83 | 2.3 ± 0.6 | Eut | 0.343 | 0.465 |
| 1L | 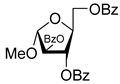 | 5.83 | 6.7 ± 1.9 | Dis | ||
| 2D | 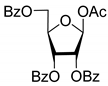 | 5.62 | 4.2 ± 0.3 | Eut | 0.808 | 0.09 |
| 2L | 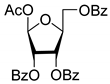 | 5.62 | 5.2 ± 0.2 | Dis | ||
| 3D | 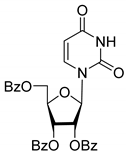 | 5.07 | 3.8 ± 0.4 | Eut | 0.745 | 0.128 |
| 3L | 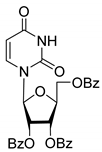 | 5.07 | 5.1 ± 0.1 | Dis | ||
| 4D | 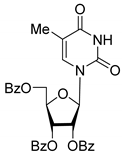 | 5.47 | 2.7 ± 0.6 | Eut | 0.108 | 0.967 |
| 4L | 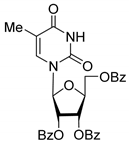 | 5.47 | 25 ± 1 | Dis | ||
| 4′D | 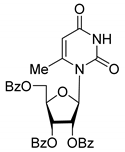 | 5.27 | 3.4 ± 0.2 | - | - | - |
| 5R |  | 5.49 | 3.1 ± 0.2 | Dis | 0.871 | 0.06 |
| 5S | 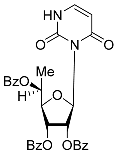 | 5.49 | 2.7 ± 0.2 | Eut | ||
| 6R | 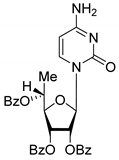 | 5.10 | 5.8 ± 0.5 | Dis | 0.896 | 0.048 |
| 6S | 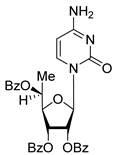 | 5.10 | 5.2 ± 0.8 | Eut | ||
| 7R | 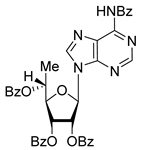 | 7.73 | 4.4 ± 0.8 | Eut | 0.956 | 0.0193 |
| 7S | 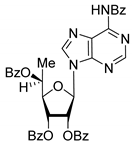 | 7.73 | 4.6 ± 0.9 | Dis | ||
| 8D | 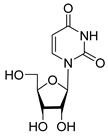 | −2.42 | >50 | - | - | - |
| 8L |  | −2.42 | >50 | - |
| Compound | Inhibition Type | Docking_score 1 | Interactions with Amino Acids in Enzyme |
|---|---|---|---|
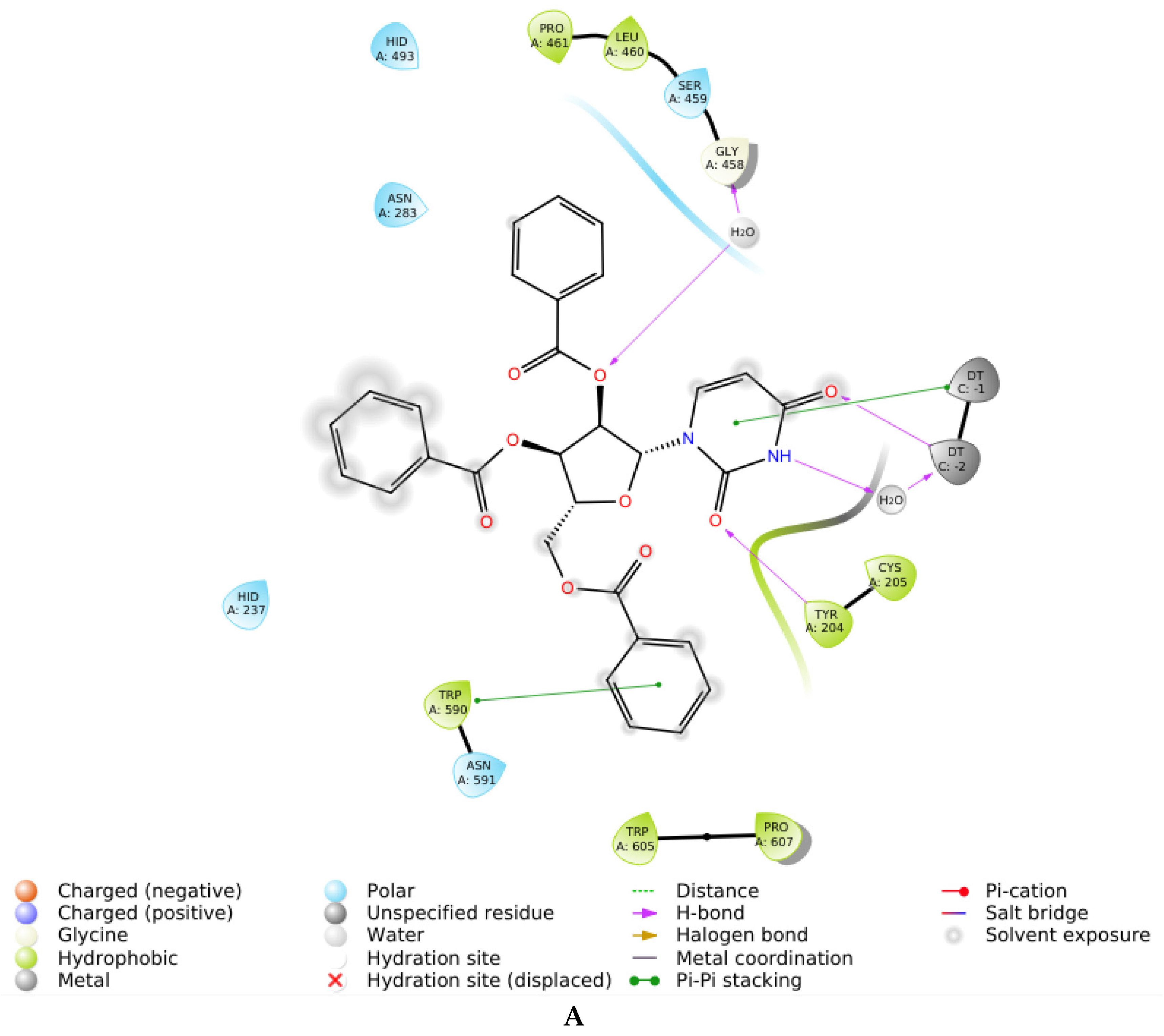
| 3D | Uncompetitive (Tdp1-DNA) | −5.216 | Tyr204-Cys205 (mediated by H2O), Gly458 (mediated by H2O), Trp590 |
| 3L | Uncompetitive (Tdp1-DNA) | −5.228 | Tyr204, Cys-205, Trp590 |
| 4D | Uncompetitive (Tdp1-DNA) | −4.670 | Gly458 (mediated by H2O), Trp590, H2O (near Asp230) |
| 4L | Uncompetitive (Tdp1-DNA) | −5.362 | Tyr204, Cys205, Asn283, H2O (near Gly-458) |
| 7S | Competitive (Tdp1) | −2.506 | Tyr204, Thr466, Trp590 |
| 7R | Competitive (Tdp1) | −1.299 | Tyr204, Ser462, Ser462 (mediated by H2O), Trp590 |
| 7R | Uncompetitive (Tdp1-DNA) | −6.490 | Trp590, Gly458 (mediated by H20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyrkheeva, N.S.; Chernyshova, I.A.; Ivanov, G.A.; Porozov, Y.B.; Zenchenko, A.A.; Oslovsky, V.E.; Zakharenko, A.L.; Nasyrova, D.I.; Likhatskaya, G.N.; Mikhailov, S.N.; et al. In Vitro and In Silico Studies of Human Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibition by Stereoisomeric Forms of Lipophilic Nucleosides: The Role of Carbohydrate Stereochemistry in Ligand-Enzyme Interactions. Molecules 2022, 27, 2433. https://doi.org/10.3390/molecules27082433
Dyrkheeva NS, Chernyshova IA, Ivanov GA, Porozov YB, Zenchenko AA, Oslovsky VE, Zakharenko AL, Nasyrova DI, Likhatskaya GN, Mikhailov SN, et al. In Vitro and In Silico Studies of Human Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibition by Stereoisomeric Forms of Lipophilic Nucleosides: The Role of Carbohydrate Stereochemistry in Ligand-Enzyme Interactions. Molecules. 2022; 27(8):2433. https://doi.org/10.3390/molecules27082433
Chicago/Turabian StyleDyrkheeva, Nadezhda S., Irina A. Chernyshova, Georgy A. Ivanov, Yuri B. Porozov, Anastasia A. Zenchenko, Vladimir E. Oslovsky, Alexandra L. Zakharenko, Darina I. Nasyrova, Galina N. Likhatskaya, Sergey N. Mikhailov, and et al. 2022. "In Vitro and In Silico Studies of Human Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibition by Stereoisomeric Forms of Lipophilic Nucleosides: The Role of Carbohydrate Stereochemistry in Ligand-Enzyme Interactions" Molecules 27, no. 8: 2433. https://doi.org/10.3390/molecules27082433
APA StyleDyrkheeva, N. S., Chernyshova, I. A., Ivanov, G. A., Porozov, Y. B., Zenchenko, A. A., Oslovsky, V. E., Zakharenko, A. L., Nasyrova, D. I., Likhatskaya, G. N., Mikhailov, S. N., Lavrik, O. I., & Drenichev, M. S. (2022). In Vitro and In Silico Studies of Human Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibition by Stereoisomeric Forms of Lipophilic Nucleosides: The Role of Carbohydrate Stereochemistry in Ligand-Enzyme Interactions. Molecules, 27(8), 2433. https://doi.org/10.3390/molecules27082433