
Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene


Abstract
:1. Introduction
2. Results and Discussion
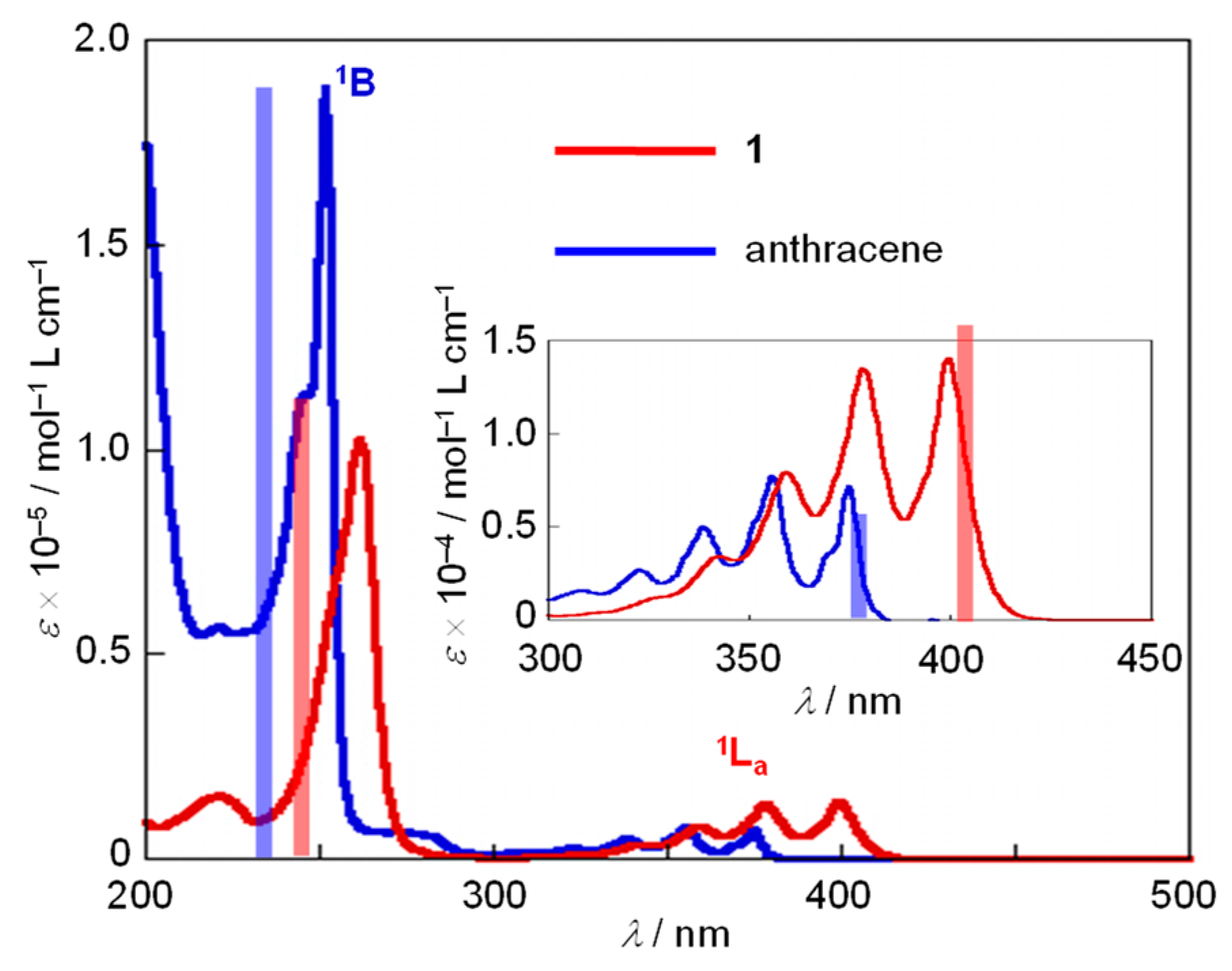
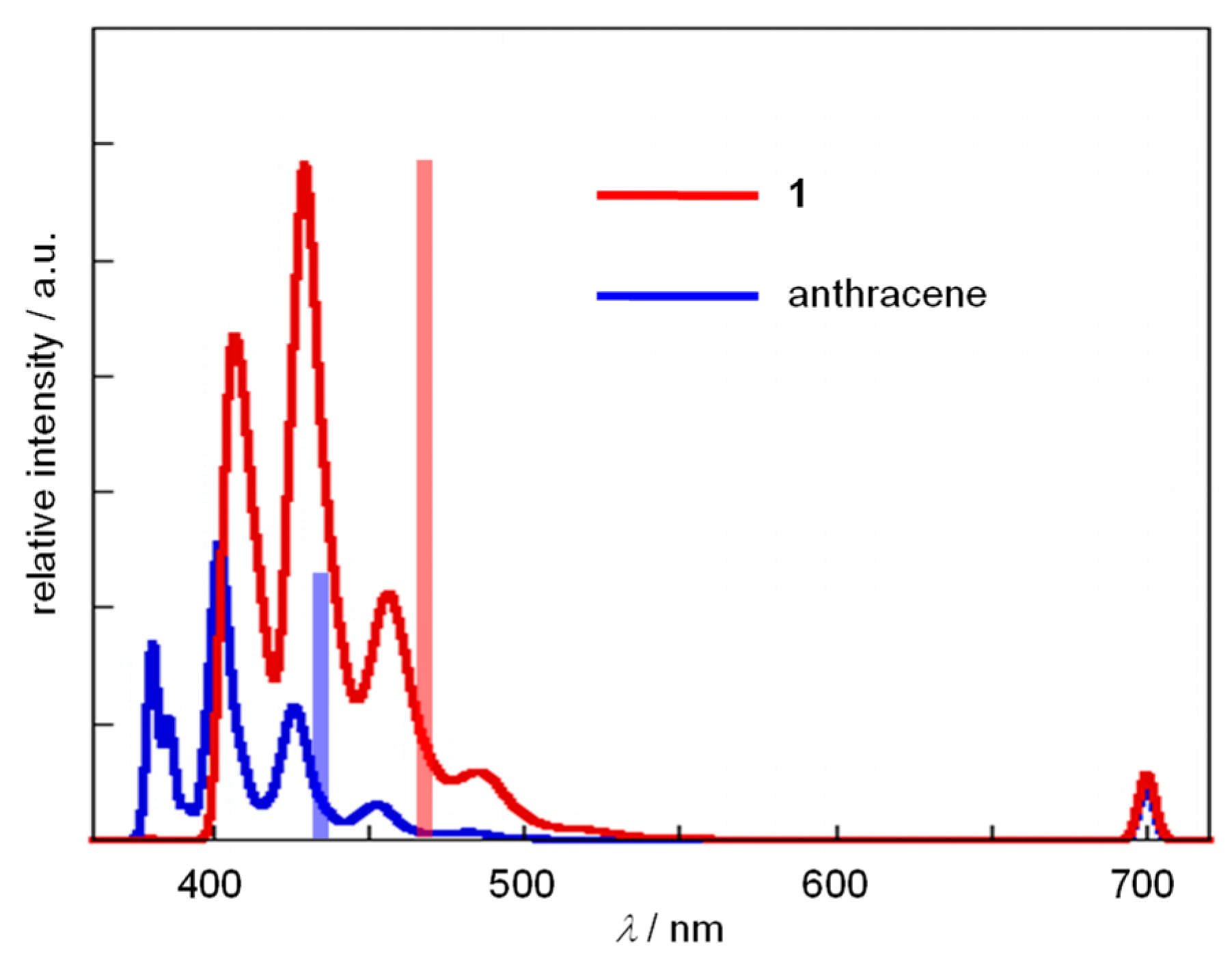
2.1. Measurement of the UV/Vis and Fluorescence Spectra of 1 and Anthracene
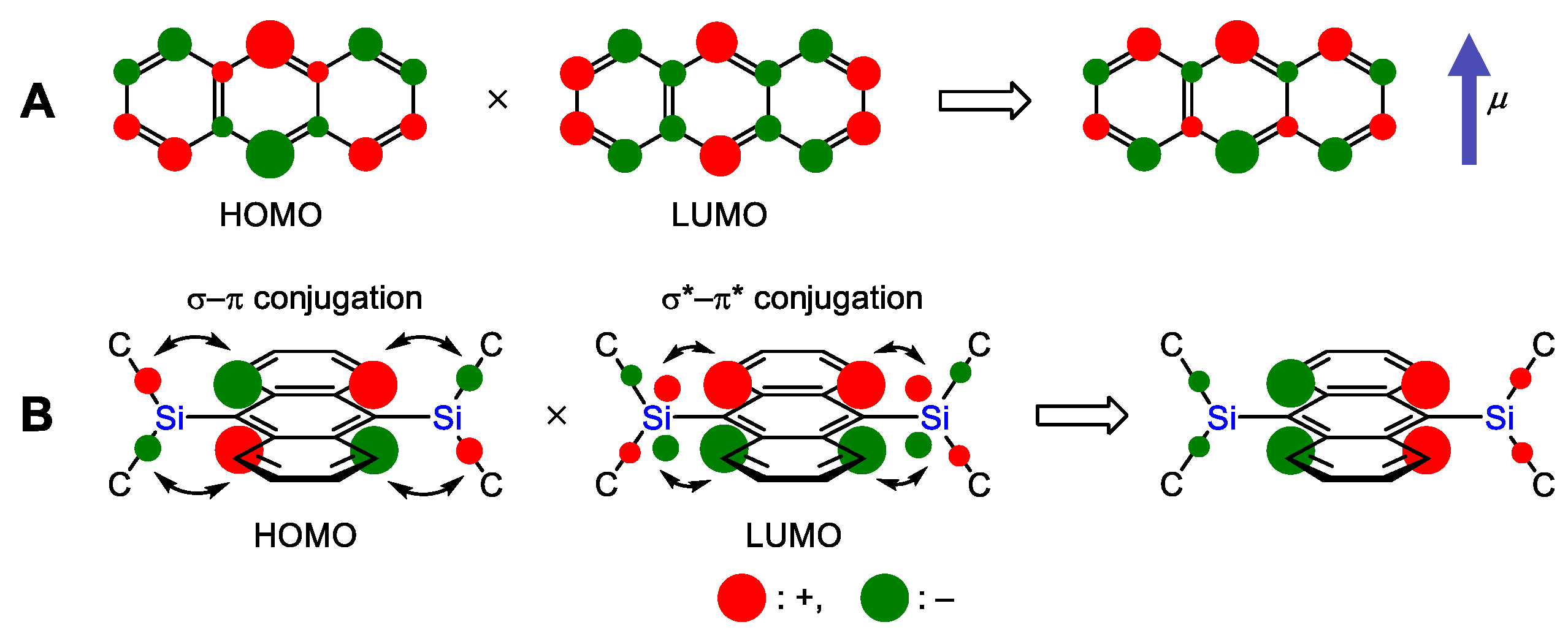
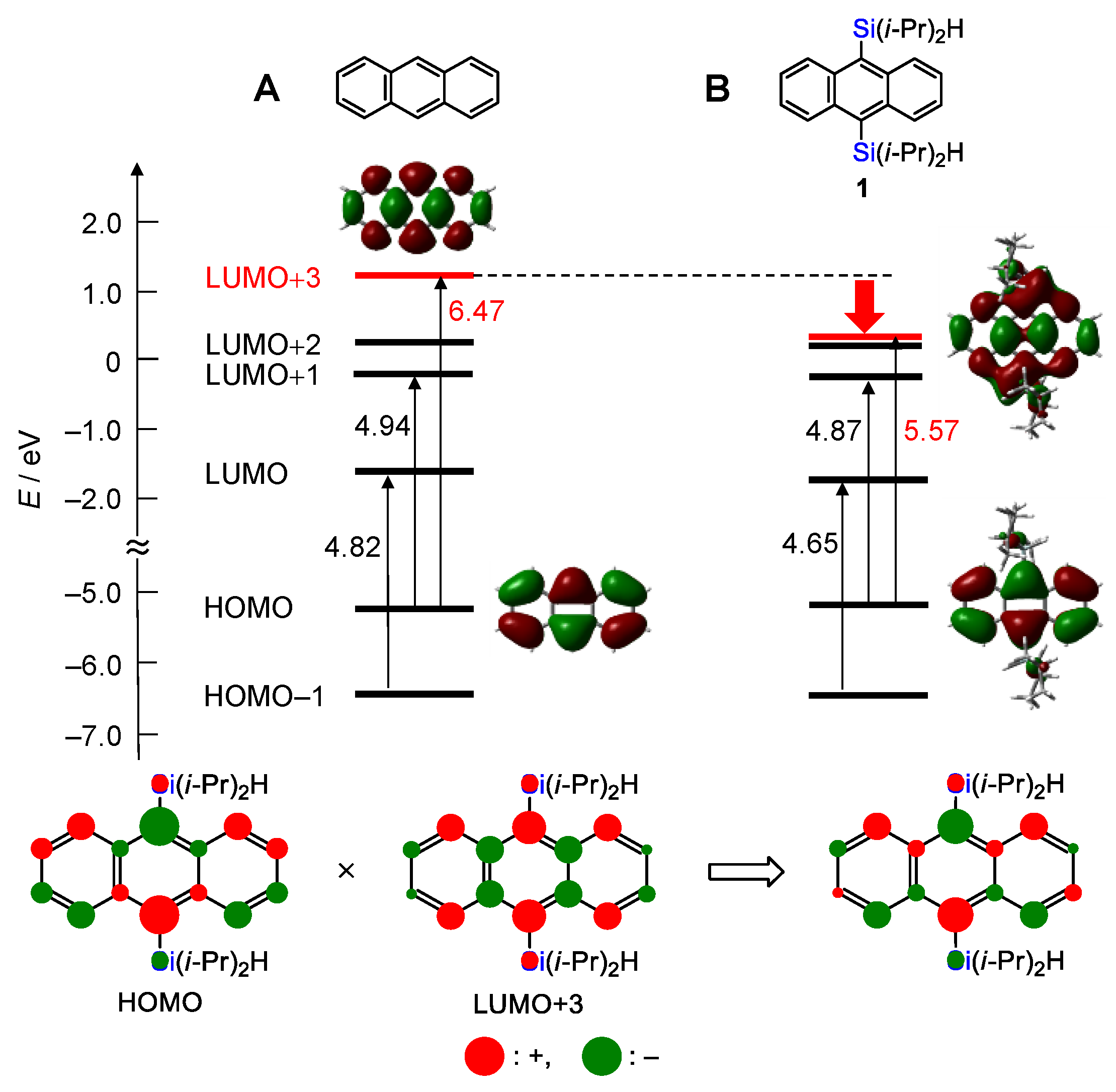
2.2. Theoretical Analysis of the UV/Vis Spectra of 1 and Anthracene
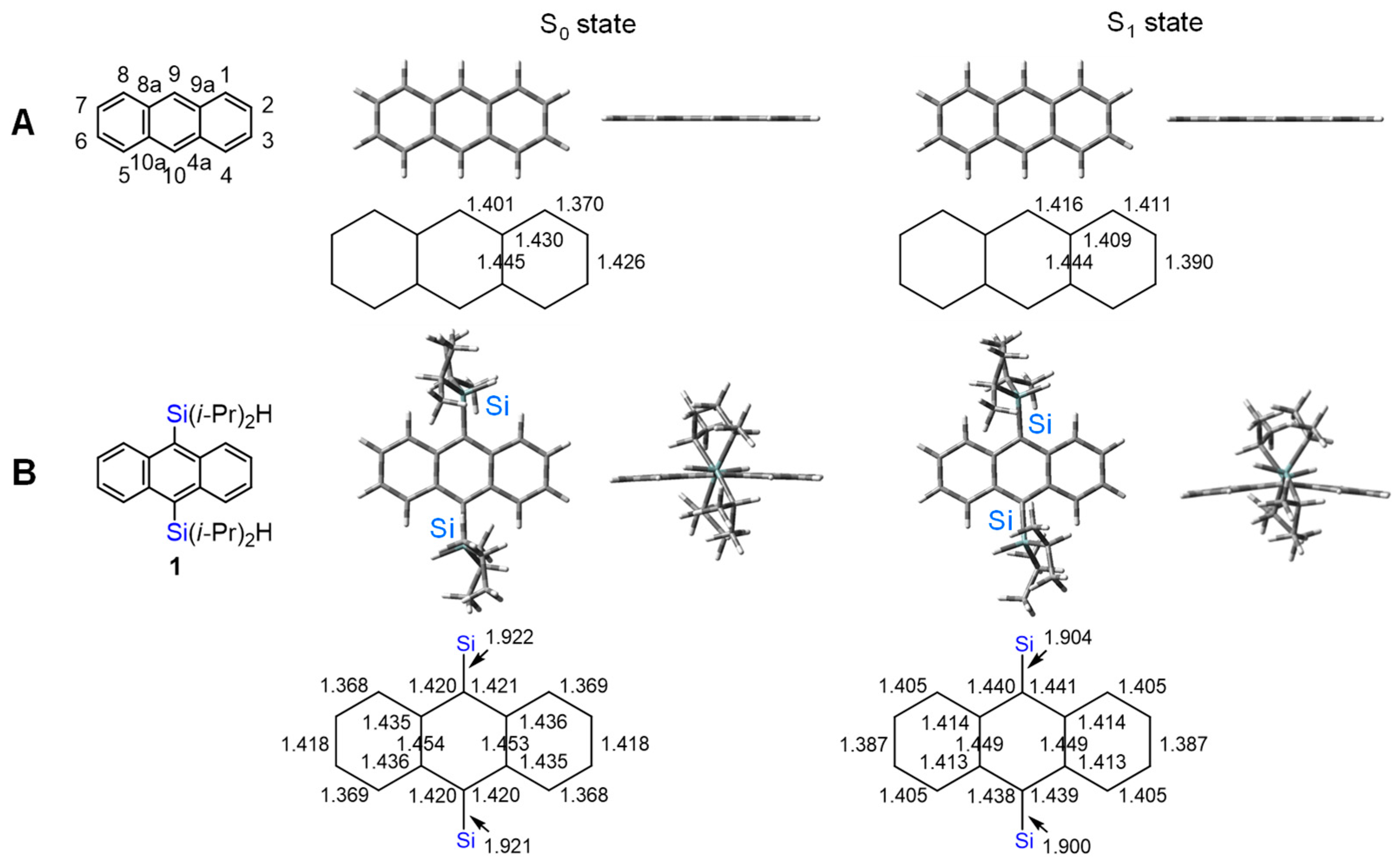
2.3. Optimized Structures of the Excited Singlet States of 1 and Anthracene
2.4. Theoretical Analysis of the Fluorescence Spectra of 1 and Anthracene
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jarvie, A.W.P. Anomalous properties of β-functional organosilicon compounds—β-effect. Organomet. Chem. Rev. A 1970, 6, 153–207. [Google Scholar]
- Sakurai, H.; Kumada, M. The ultraviolet spectra of some polysilanes. Bull. Chem. Soc. Jpn. 1964, 37, 1894–1895. [Google Scholar] [CrossRef]
- Bock, H.; Solouki, B. Photoelectron spectra of silicon compounds. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, UK, 1989; pp. 555–653. [Google Scholar]
- Traylor, T.G.; Hanstein, W.; Berwin, H.J.; Clinton, N.A.; Brown, R.S. Vertical stabilization of cations by neighboring σ bonds. General considerations. J. Am. Chem. Soc. 1971, 93, 5715–5725. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tamao, K. Theoretical study of the electronic structure of 2,2′-bisilole in comparison with 1,1′-bi-1,3-cyclopentadiene: σ*–π* conjugation and a low-lying LUMO as the origin of the unusual optical properties of 3,3′,4,4′-tetraphenyl-2,2′-bisilole. Bull. Chem. Soc. Jpn. 1996, 69, 2327–2334. [Google Scholar] [CrossRef]
- Kyushin, S.; Ikarugi, M.; Goto, M.; Hiratsuka, H.; Matsumoto, H. Synthesis and electronic properties of 9,10-disilylanthracenes. Organometallics 1996, 15, 1067–1070. [Google Scholar] [CrossRef]
- Fang, M.-C.; Watanabe, A.; Matsuda, M. Synthesis of σ–π conjugated organosilicon copolymers and their optical properties. Jpn. J. Appl. Phys. 1995, 34 (Suppl. 1), 98–100. [Google Scholar] [CrossRef]
- Suzuki, H.; Satoh, S.; Kimata, Y.; Kuriyama, A. Synthesis and properties of poly(methylphenylsilane) containing anthracene units. Chem. Lett. 1995, 24, 451–452. [Google Scholar] [CrossRef]
- Fang, M.-C.; Watanabe, A.; Matsuda, M. Emission spectra of σ–π-conjugated organosilicon copolymers consisting of alternating dimethylsilylene and aromatic units. Macromolecules 1996, 29, 6807–6813. [Google Scholar] [CrossRef]
- Satoh, S.; Suzuki, H.; Kimata, Y.; Kuriyama, A. Optical and electroluminescence properties of poly(methylphenylsilane) containing an anthracene unit. Synth. Met. 1996, 79, 97–102. [Google Scholar] [CrossRef]
- Kyushin, S.; Kitahara, T.; Matsumoto, H. Benzo[1,2:4,5]bis(1,1,2,2-tetraisopropyldisilacyclobutene). Chem. Lett. 1998, 27, 471–472. [Google Scholar] [CrossRef]
- Maeda, H.; Inoue, Y.; Ishida, H.; Mizuno, K. UV absorption and fluorescence properties of pyrene derivatives having trimethylsilyl, trimethylgermyl, and trimethylstannyl groups. Chem. Lett. 2001, 30, 1224–1225. [Google Scholar] [CrossRef]
- Kyushin, S.; Takemasa, N.; Matsumoto, H.; Horiuchi, H.; Hiratsuka, H. 2,3,6,7,10,11-Hexakis(dimethylsilyl)triphenylene. Chem. Lett. 2003, 32, 1048–1049. [Google Scholar] [CrossRef]
- Kyushin, S.; Ishikita, Y.; Matsumoto, H.; Horiuchi, H.; Hiratsuka, H. Yellow-green fluorescence of 5,11- and 5,12-bis(diisopropylsilyl)naphthacenes. Chem. Lett. 2006, 35, 64–65. [Google Scholar] [CrossRef]
- Kyushin, S.; Matsuura, T.; Matsumoto, H. 2,3,4,5-Tetrakis(dimethylsilyl)thiophene: The first 2,3,4,5-tetrasilylthiophene. Organometallics 2006, 25, 2761–2765. [Google Scholar] [CrossRef]
- Shimizu, M.; Oda, K.; Bando, T.; Hiyama, T. Preparation, structure, and properties of tris(trimethylsilyl)silyl-substituted anthracenes: Realization of ideal conformation for σ–π conjugation involving eclipse of Si–Si σ-bond with p-orbital of aromatic ring. Chem. Lett. 2006, 35, 1022–1023. [Google Scholar] [CrossRef]
- Karatsu, T. Photochemistry and photophysics of organomonosilane and oligosilanes: Updating their studies on conformation and intramolecular interactions. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 111–137. [Google Scholar] [CrossRef]
- Kyushin, S.; Yoshimura, K.; Sato, K.; Matsumoto, H. Hyperchromic effect of silyl groups on the UV–visible spectrum of 5,10,15,20-tetraphenylporphyrin. Chem. Lett. 2009, 38, 324–325. [Google Scholar] [CrossRef]
- Otsuka, K.; Ishida, S.; Kyushin, S. A light-emitting liquid crystal containing p-terphenyl and an alkylsilyl group. Chem. Lett. 2012, 41, 307–309. [Google Scholar] [CrossRef]
- Maeda, H.; Maeda, T.; Mizuno, K. Absorption and fluorescence spectroscopic properties of 1- and 1,4-silyl-substituted naphthalene derivatives. Molecules 2012, 17, 5108–5125. [Google Scholar] [CrossRef] [Green Version]
- Toma, Y.; Kuribara, T.; Iizuka, T.; Nagashima, H.; Kyushin, S. Synthesis, structure, and electronic properties of benzohexasilabicyclo[2.2.2]octene. Chem. Lett. 2013, 42, 250–252. [Google Scholar] [CrossRef]
- Horiuchi, H.; Hosaka, M.; Mashio, H.; Terata, M.; Ishida, S.; Kyushin, S.; Okutsu, T.; Takeuchi, T.; Hiratsuka, H. Silylation improves the photodynamic activity of tetraphenylporphyrin derivatives in vitro and in vivo. Chem. Eur. J. 2014, 20, 6054–6060. [Google Scholar] [CrossRef] [PubMed]
- Kyushin, S.; Saito, Y.; Yoshimura, K.; Horiuchi, H.; Hiratsuka, H. Synthesis and properties of 5,10,15,20-tetrakis(4’-trimethylsilylphenyl)chlorin. Heteroatom Chem. 2014, 25, 514–517. [Google Scholar] [CrossRef]
- Murai, M.; Okada, R.; Nishiyama, A.; Takai, K. Synthesis and properties of sila[n]helicenes via dehydrogenative silylation of C–H bonds under rhodium catalysis. Org. Lett. 2016, 18, 4380–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurusaki, A.; Kobayashi, A.; Kyushin, S. Synthesis, structures, and electronic properties of dithienosiloles bearing bulky aryl groups: Conjugation between a π-electron system and “perpendicular” aryl groups. Asian J. Org. Chem. 2017, 6, 737–745. [Google Scholar] [CrossRef]
- Maeda, H.; Suzuki, T.; Segi, M. Effects of substituents in silyl groups on the absorption, fluorescence and structural properties of 1,3,6,8-tetrasilylpyrenes. Photochem. Photobiol. Sci. 2018, 17, 781–792. [Google Scholar] [CrossRef]
- Maeda, H.; Horikoshi, R.; Yamaji, M.; Furuyama, T.; Segi, M. Photophysical properties of silyl-substituted stilbene derivatives. Eur. J. Org. Chem. 2020, 2020, 3410–3422. [Google Scholar] [CrossRef]
- Haruki, R.; Sasaki, Y.; Masutani, K.; Yanai, N.; Kimizuka, N. Leaping across the visible range: Near-infrared-to-violet photon upconversion employing silyl-substituted anthracene. Chem. Commun. 2020, 56, 7017–7020. [Google Scholar] [CrossRef]
- Terada, N.; Uematsu, K.; Higuchi, R.; Tokimaru, Y.; Sato, Y.; Nakano, K.; Nozaki, K. Synthesis and properties of spiro-double sila[7]helicene: The LUMO spiro-conjugation. Chem. Eur. J. 2021, 27, 9342–9349. [Google Scholar] [CrossRef]
- Benkyi, I.; Tapavicza, E.; Fliegl, H.; Sundholm, D. Calculation of vibrationally resolved absorption spectra of acenes and pyrene. Phys. Chem. Chem. Phys. 2019, 21, 21094–21103. [Google Scholar] [CrossRef] [Green Version]
- Sambursky, S.; Wolfsohn, G. On the fluorescence and absorption spectra of anthracene and phenanthrene in solutions. Trans. Faraday Soc. 1940, 35, 427–432. [Google Scholar] [CrossRef]
- Jones, N. The ultraviolet absorption spectra of anthracene derivatives. Chem. Rev. 1947, 41, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Schoof, S.; Güsten, H.; von Sonntag, C. Fluoreszenz und Fluoreszenzlöschung von meso-substituierten Anthracenderivaten in Lösung. Ber. Bunsenges. Phys. Chem. 1978, 82, 1068–1073. [Google Scholar] [CrossRef]
- Hirayama, S.; Lampert, R.A.; Phillips, D. Photophysics of meso-substituted anthracenes. Part 1.—Solutions and isolated vapours. J. Chem. Soc. Faraday Trans. 2 1985, 81, 371–382. [Google Scholar] [CrossRef]
- Klevens, H.B.; Platt, J.R. Spectral resemblances of cata-condensed hydrocarbons. J. Chem. Phys. 1949, 17, 470–481. [Google Scholar] [CrossRef]
- Platt, J.R. Classification of spectra of cata-condensed hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Pariser, R. Theory of the electronic spectra and structure of the polyacenes and of alternant hydrocarbons. J. Chem. Phys. 1956, 24, 250–268. [Google Scholar] [CrossRef]
- Heinrich, G.; Güsten, H. Deuterium-Isotopieeffekt auf die strahlende und strahlungslose Desaktivierung von Triplettzuständen polycyclischer aromatischer Kohlenwasserstoffe. Z. Phys. Chem. (Wiesb.) 1979, 118, 31–41. [Google Scholar] [CrossRef]
- Brittain, E.F.H.; George, W.O.; Wells, C.H.J. Introduction of Molecular Spectroscopy: Theory and Experiment; Academic Press: London, UK; New York, NY, USA, 1970. [Google Scholar]
- Orchin, M.; Jaffé, H.H. Symmetry, Orbitals, and Spectra (S.O.S.); Wiley-Interscience: New York, NY, USA, 1971. [Google Scholar]
- Klessinger, M.; Michl, J. Excited States and Photochemistry of Organic Molecules; VCH: New York, NY, USA, 1995. [Google Scholar]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Franck, J.; Dymond, E.G. Elementary processes of photochemical reactions. Trans. Faraday Soc. 1926, 21, 536–542. [Google Scholar] [CrossRef]
- Condon, E. A theory of intensity distribution in band systems. Phys. Rev. 1926, 28, 1182–1201. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λa/nm 1 (ε/mol–1 L cm–1) | λf/nm 2 |
|---|---|---|
| 1 | 221 (15,400) 262 (102,000) 343 (3400) 359 (7900) 378 (13,500) 400 (14,000) | 406 429 456 485 |
| anthracene | 252 (190,000) 323 (2700) 339 (5000) 356 (7700) 375 (7200) | 380 402 426 453 482 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyushin, S.; Suzuki, Y. Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene. Molecules 2022, 27, 2241. https://doi.org/10.3390/molecules27072241
Kyushin S, Suzuki Y. Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene. Molecules. 2022; 27(7):2241. https://doi.org/10.3390/molecules27072241
Chicago/Turabian StyleKyushin, Soichiro, and Yuya Suzuki. 2022. "Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene" Molecules 27, no. 7: 2241. https://doi.org/10.3390/molecules27072241
APA StyleKyushin, S., & Suzuki, Y. (2022). Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene. Molecules, 27(7), 2241. https://doi.org/10.3390/molecules27072241