
Inhibition of Aldose Reductase by Ginsenoside Derivatives via a Specific Structure Activity Relationship with Kinetics Mechanism and Molecular Docking Study

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
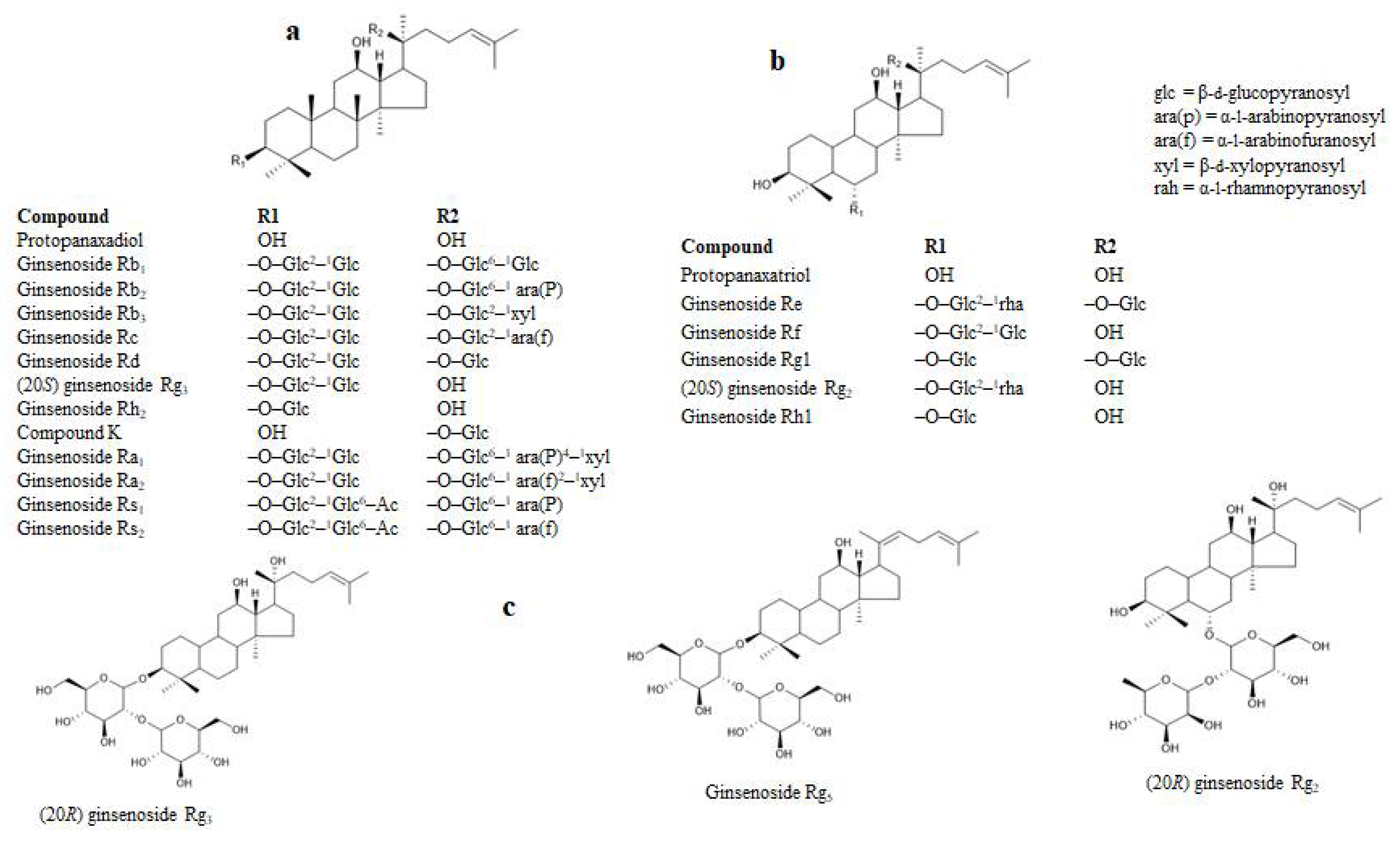
2.1. RLAR Inhibitory Activities of Ginsenoside Derivatives and Analysis of Structure-Activity Relation
2.2. HRAR Inhibitory Activities of Ginsenoside Derivatives and Analysis of Structure-Activity Relation
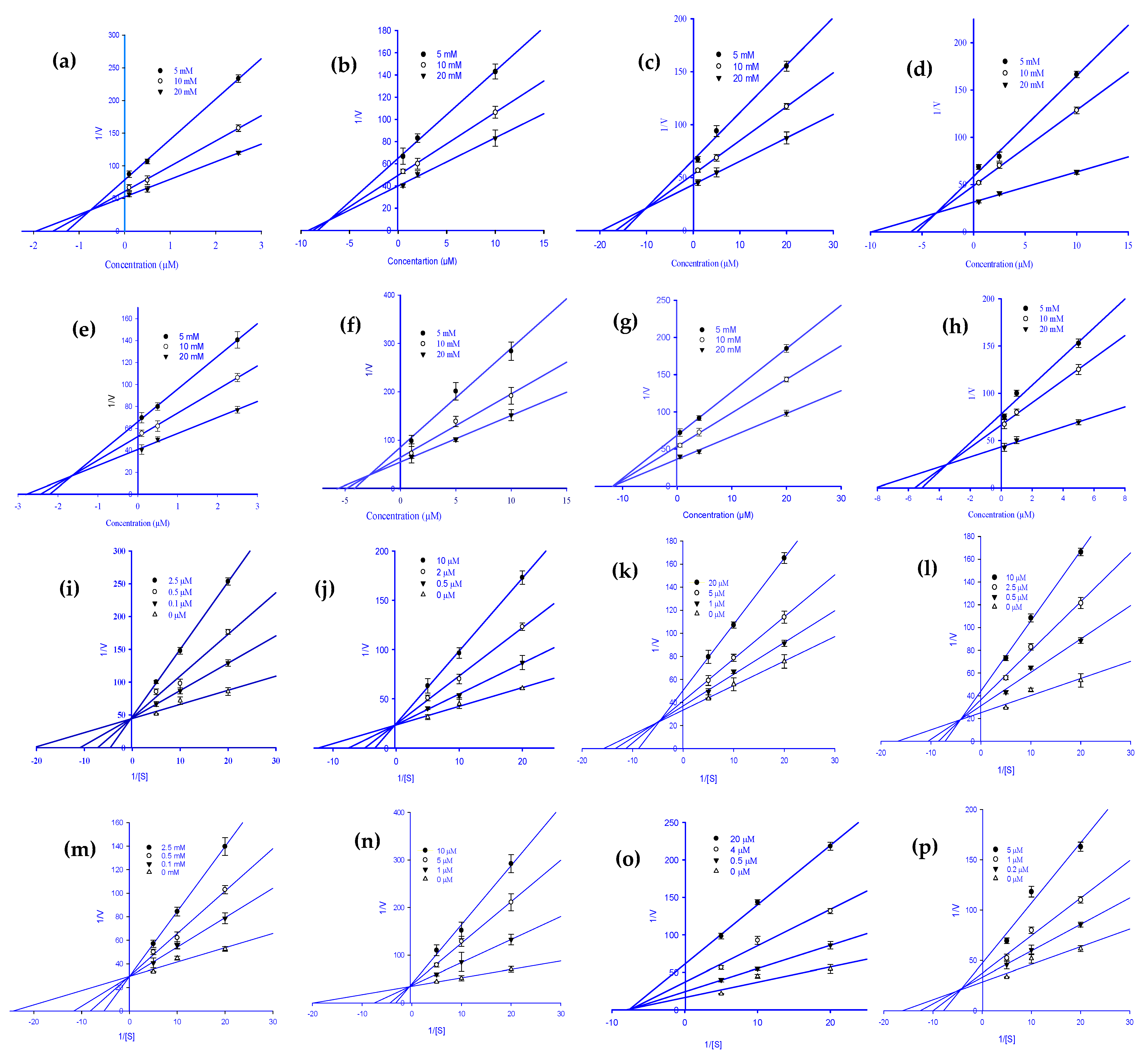
2.3. Enzyme Kinetics Analysis of RLAR and HRAR Inhibition with Ginsenoside Derivatives
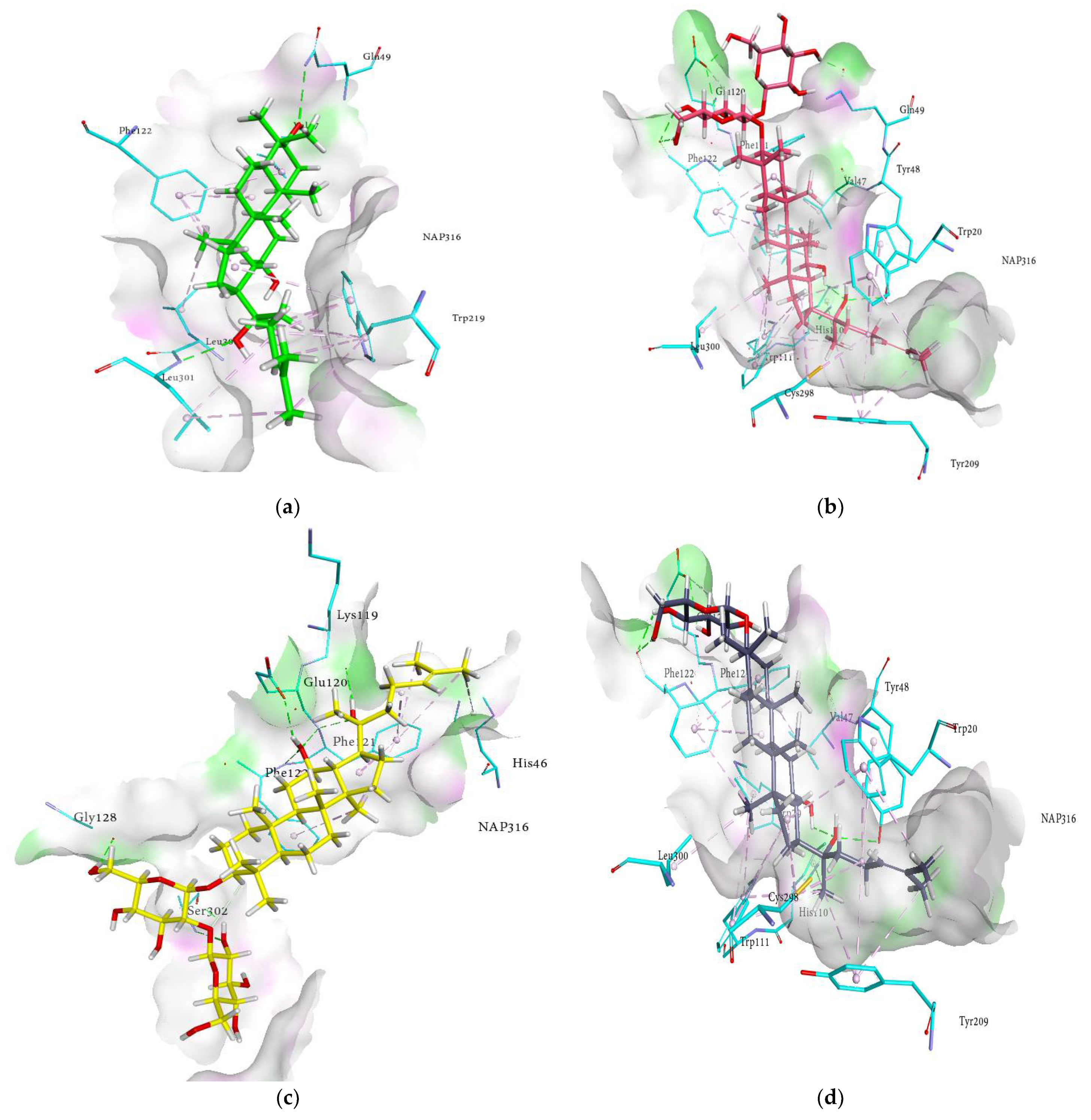
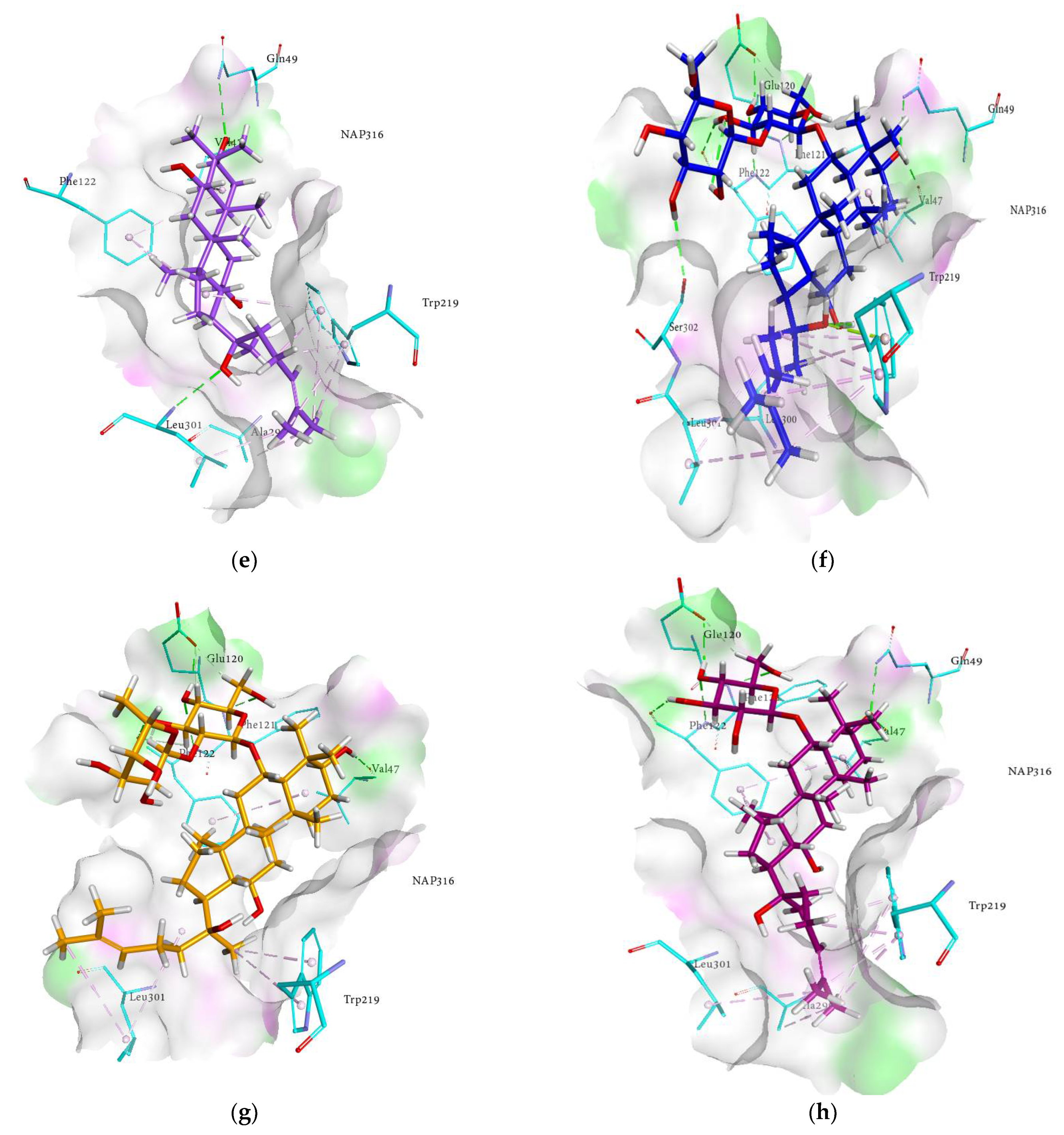
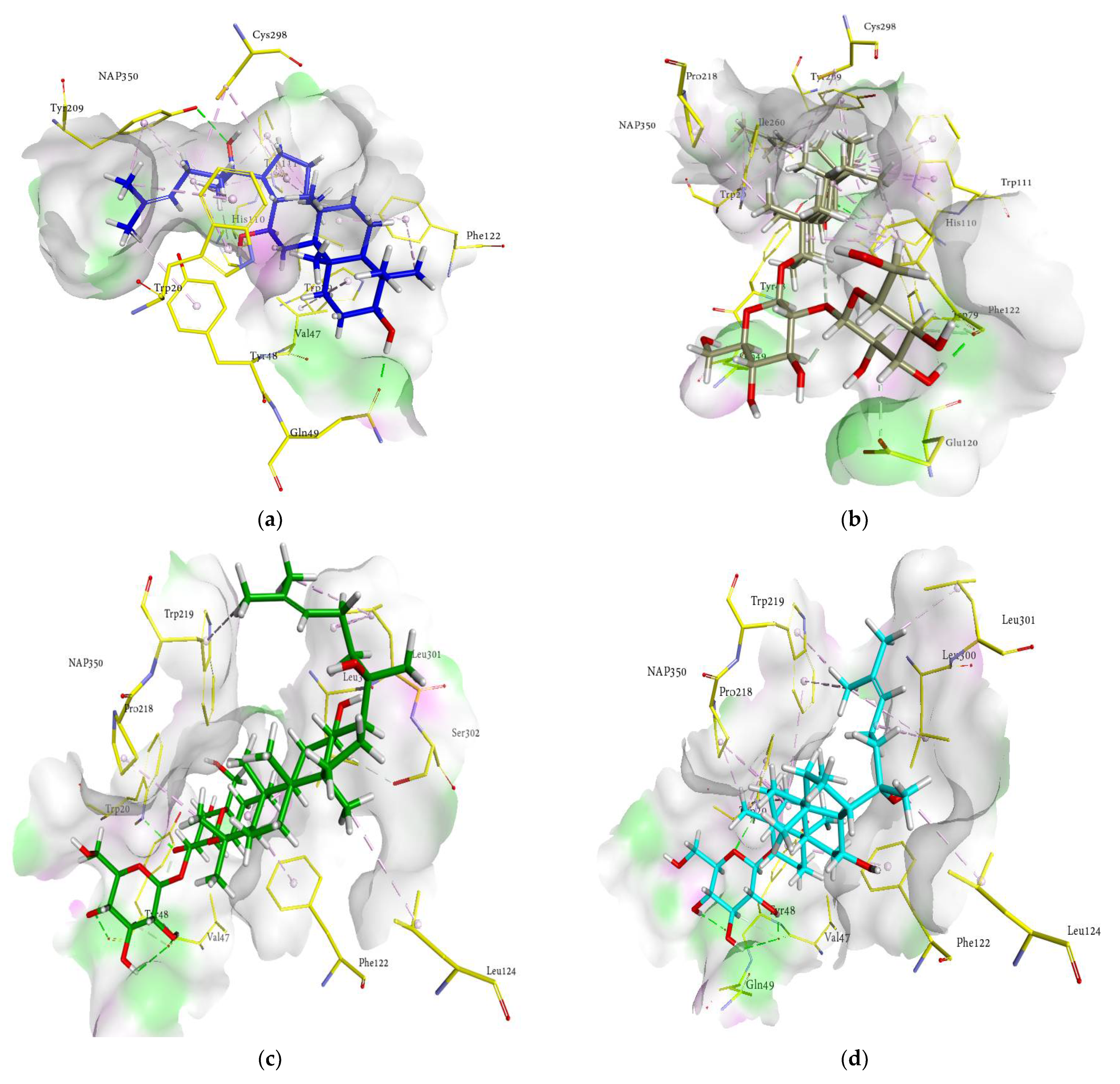
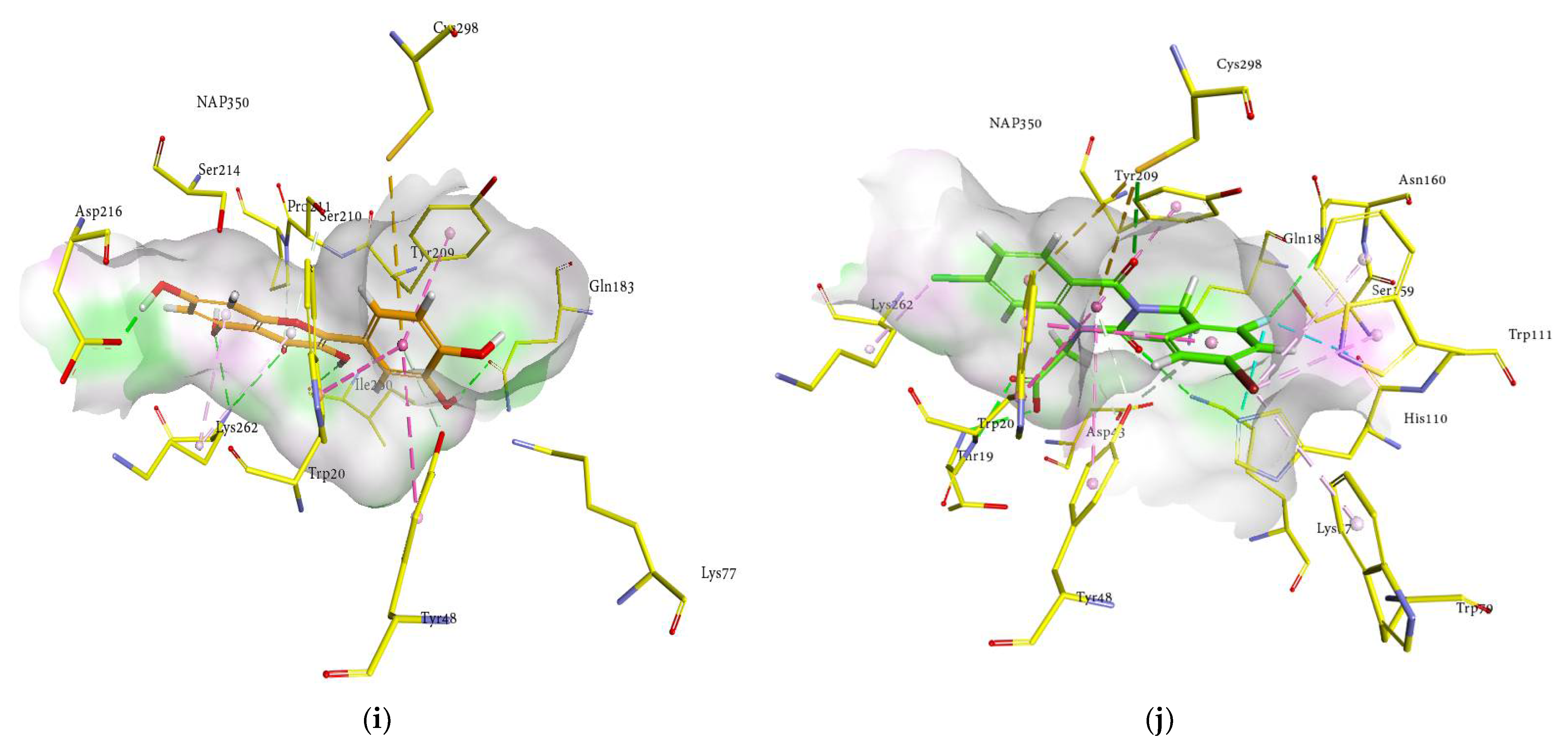
2.4. Molecular Docking Analysis
2.5. Docking Interactions of Ginsenosides and Cognate Ligand with RLAR
2.6. Docking Interactions of Ginsenosides and Cognate Ligand with HRAR
2.7. Inhibitory Activities of Active Ginsenosides on Sorbitol Accumulation
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Assay for RLAR Inhibitory Activity
3.3. HRAR Inhibition Assay
3.4. Determination of Kinetics Parameters of RLAR and HRAR Inhibition via Lineweaver-Burk and Dixon Plots
3.5. Lens Culture and Intracellular Sorbitol Measurement
3.6. Molecular Docking Simulation in RLAR and HRAR Inhibition
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wild, S.H.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care 2004, 27, 2569–2570. [Google Scholar] [CrossRef] [Green Version]
- Sicree, R.; Shaw, J.; Zimmet, P. Diabetes and impaired glucose tolerance. In Diabetes Atlas, 2nd ed.; International Diabetes Federation: Brussels, Belgium, 2006; pp. 15–103. [Google Scholar]
- Ali, Y.; Jung, H.A.; Jannat, S.; Choi, J.S. Dihydroxanthyletin-type coumarins from Angelica decursiva that inhibits the formation of advanced glycation end products and human recombinant aldose reductase. Arch. Pharmacal Res. 2017, 41, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Petropoulos, I.N.; Alam, U.; Malik, R.A. Treatment of painful diabetic neuropathy. Ther. Adv. Chronic Dis. 2015, 6, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Kotwani, A. Implication of oxidative stress in progression of diabetic retinopathy. Surv. Ophthalmol. 2016, 61, 187–196. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.; Zaib, S.; Rahman, M.M.; Jannat, S.; Iqbal, J.; Park, S.K.; Chang, M.S. Poncirin, an orally active flavonoid exerts antidiabetic complications and improves glucose uptake activating PI3K/Akt signaling pathway in insulin resistant C2C12 cells with anti-glycation capacities. Bioorganic Chem. 2020, 102, 104061. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2014, 58, 2047–2067. [Google Scholar] [CrossRef]
- Veeresham, C.; Rao, A.R.; Asres, K. Aldose Reductase Inhibitors of Plant Origin. Phytother. Res. 2014, 28, 317–333. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Ali, Y.; Jung, H.A.; Choi, J.S. Anti-diabetic and anti-Alzheimer’s disease activities of Angelica decursiva. Arch. Pharmacal Res. 2015, 38, 2216–2227. [Google Scholar] [CrossRef]
- Jung, H.A.; Yoon, N.Y.; Kang, S.S.; Kim, Y.S.; Choi, J.S. Inhibitory activities of prenylated flavonoids from Sophora fla-vescens against aldose reductase and generation of advanced glycation endproducts. J. Pharm. Pharmacol. 2008, 60, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, K.; Ueda, H.; Moriyasu, M. Aldose reductase inhibitors from the nature. Curr. Med. Chem. 2003, 10, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Manzanaro, S.; Salvá, J.; de la Fuente, J. Phenolic Marine Natural Products as Aldose Reductase Inhibitors. J. Nat. Prod. 2006, 69, 1485–1487. [Google Scholar] [CrossRef]
- Shin, B.-K.; Kwon, S.W.; Park, J.H. Chemical diversity of ginseng saponins from Panax ginseng. J. Ginseng Res. 2015, 39, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H. Chemical Diversity of Panax ginseng, Panax quinquifolium, and Panax notoginseng. J. Ginseng Res. 2012, 36, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Park, J.D.; Rhee, D.K.; Lee, Y.H. Biological activities and chemistry of saponins from Panax ginseng CA Meyer. Phytochem. Rev. 2015, 4, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H. Cardiovascular Diseases and Panax ginseng: A Review on Molecular Mechanisms and Medical Applications. J. Ginseng Res. 2012, 36, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Lee, D.; Lee, H.L.; Kim, C.-E.; Jung, K.; Kang, K.S. Beneficial effects of Panax ginseng for the treatment and prevention of neurodegenerative diseases: Past findings and future directions. J. Ginseng Res. 2018, 42, 239–247. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Liu, J.-G.; Li, H.; Yang, H.-M. Pharmacological Effects of Active Components of Chinese Herbal Medicine in the Treatment of Alzheimer’s Disease: A Review. Am. J. Chin. Med. 2016, 44, 1525–1541. [Google Scholar] [CrossRef]
- Yun, T.K.; Choi, S.Y.; Yun, H.Y. Epidemiological Study on Cancer Prevention by Ginseng: Are All Kinds of Cancers Preventable by Ginseng? J. Korean Med. Sci. 2001, 16, S19–S27. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.-H.; Seog, H.-M.; Choi, I.-W.; Choi, H.-D.; Cho, H.-Y. Effects of wild ginseng (Panax ginseng C.A. Meyer) leaves on lipid peroxidation levels and antioxidant enzyme activities in streptozotocin diabetic rats. J. Ethnopharmacol. 2005, 98, 245–250. [Google Scholar] [CrossRef]
- Babiker, L.B.; Gadkariem, E.A.; Alashban, R.M.; Aljohar, H.I. Investigation Of Stability Of Korean Ginseng In Herbal Drug Product. Am. J. Appl. Sci. 2014, 11, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Kang, K.S.; Yamabe, N.; Nagai, R.; Yokozawa, T. Protective Effect of Heat-Processed American Ginseng against Diabetic Renal Damage in Rats. J. Agric. Food Chem. 2007, 55, 8491–8497. [Google Scholar] [CrossRef]
- Kang, K.S.; Kim, H.Y.; Yamabe, N.; Nagai, R.; Yokozawa, T. Protective Effect of Sun Ginseng against Diabetic Renal Damage. Biol. Pharm. Bull. 2006, 29, 1678–1684. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.A.; Qin, J.; Wang, W.; Wang, M.-H.; Wang, H.; Zhang, R. Ginsenosides as anticancer agents: In vitro and in vivo activities, structure–activity relationships, and molecular mechanisms of action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Park, S.K.; Hyun, S.H.; In, G.; Park, C.-K.; Kwak, Y.-S.; Jang, Y.-J.; Kim, B.; Kim, J.-H.; Han, C.-K. The antioxidant activities of Korean Red Ginseng (Panax ginseng) and ginsenosides: A systemic review through in vivo and clinical trials. J. Ginseng Res. 2021, 45, 41–47. [Google Scholar] [CrossRef]
- Ahn, S.; Siddiqi, M.H.; Noh, H.-Y.; Kim, Y.-J.; Kim, Y.-J.; Jin, C.-G.; Yang, D.-C. Anti-inflammatory activity of ginsenosides in LPS-stimulated RAW 264.7 cells. Sci. Bull. 2015, 60, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Xin, Y.; Li, Y.; Xu, F.; Xi, X.; Guo, H.; Cui, X.; Cao, H.; Zhang, X.; Han, C. Ginsenosides: A potential neuro-protective agent. BioMed. Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Razgonova, M.P.; Veselov, V.V.; Zakharenko, A.M.; Golokhvast, K.S.; Nosyrev, A.E.; Cravotto, G.; Tsatsakis, A.; Spandidos, D.A. Panax ginseng components and the pathogenesis of Alzheimer’s disease. Mol. Med. Rep. 2019, 19, 2975–2998. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Cao, J.; Bi, X.; Xia, X.; Li, W.; Zhao, Y. New dammarane-type triterpenoids from the leaves of Panax notoginseng and their protein tyrosine phosphatase 1B inhibitory activity. J. Ginseng Res. 2014, 38, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.J.; Kim, G.W.; Chung, S.H. AMP-activated protein kinase: An emerging target for ginseng. J. Ginseng Res. 2014, 38, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.Y.; Jannat, S.; Rahman, M.M. Ginsenoside derivatives inhibit advanced glycation end-product formation and glu-cose-fructose mediated protein glycation in vitro via a specific structure-activity relationship. Bioorg. Chem. 2021, 111, 104844. [Google Scholar]
- Ali, Y.; Zaib, S.; Jannat, S.; Khan, I. Inhibition of Angiotensin-I Converting Enzyme by Ginsenosides: Structure–Activity Relationships and Inhibitory Mechanism. J. Agric. Food Chem. 2021, 69, 6073–6086. [Google Scholar] [CrossRef]
- Fatmawati, S.; Ersam, T.; Yu, H.; Zhang, C.; Jin, F.; Shimizu, K. 20(S)-Ginsenoside Rh2 as aldose reductase inhibitor from Panax ginseng. Bioorganic Med. Chem. Lett. 2014, 24, 4407–4409. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, C.; Tian, Q.; Zhang, Y.; Zhang, G.; Guan, Y.; Tong, S.; Yan, J. Screening and characterization of aldose reductase inhibitors from Traditional Chinese medicine based on ultrafiltration-liquid chromatography mass spectrometry and in silico molecular docking. J. Ethnopharmacol. 2021, 264, 113282. [Google Scholar] [CrossRef]
- Kador, P.F.; Kinoshita, J.H.; Tung, W.H.; Chylack, L.T. Differences in the susceptibility of various aldose reductases to inhibition. II. Investig. Ophthalmol. Vis. Sci. 1980, 19, 980–982. [Google Scholar]
- Nishimura, C.; Yamaoka, T.; Mizutani, M.; Yamashita, K.; Akera, T.; Tanimoto, T. Purification and characterization of the recombinant human aldose reductase expressed in baculovirus system. Biochim. Et Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1991, 1078, 171–178. [Google Scholar] [CrossRef]
- Kinoshita, T.; Miyake, H.; Fujii, T.; Takakura, S.; Goto, T. The structure of human recombinant aldose reductase complexed with the potent inhibitor zenarestat. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 622–626. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, L.; Zhai, J.; Chen, Y.; Luo, H.; Hu, X. The molecular basis for inhibition of sulindac and its metabolites towards human aldose reductase. FEBS Lett. 2012, 586, 55–59. [Google Scholar] [CrossRef] [Green Version]
- LeadIT Version 2.3.2; BioSolveIT GmbH: Sankt Augustin, Germany, 2017; Available online: www.biosolveit.de/LeadIT (accessed on 23 July 2021).
- Hwang, S.H.; Wang, Z.; Quispe, Y.N.G.; Lim, S.S.; Yu, J.M. Evaluation of aldose reductase, protein glycation, and anti-oxidant inhibitory activities of bioactive flavonoids in Matricaria recutita L. and their structure-activity relationship. J. Diabetes Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, S.H.; Jung, S.H.; Kim, J.K.; Pan, C.-H.; Lim, S.S. Aldose Reductase Inhibitory Compounds from Glycyrrhiza uralensis. Biol. Pharm. Bull. 2010, 33, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Shehzad, M.T.; Hameed, A.; Al-Rashida, M.; Imran, A.; Uroos, M.; Asari, A.; Mohamad, H.; Islam, M.; Iftikhar, S.; Shafiq, Z.; et al. Exploring antidiabetic potential of adamantyl-thiosemicarbazones via aldose reductase (ALR2) inhibition. Bioorganic Chem. 2019, 92, 103244. [Google Scholar] [CrossRef]
- Hayman, S.; Kinoshita, J.H. Isolation and Properties of Lens Aldose Reductase. J. Biol. Chem. 1965, 240, 877–882. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The Determination of Enzyme Dissociation Constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Dixon, M. The Determination of Enzyme Inhibitor Constants; Springer: New York, NY, USA, 1953; Volume 55. [Google Scholar]
- Kwang-Hyok, S.; Ui-Nam, P.; Sarkar, C.; Bhadra, R. A sensitive assay of red blood cell sorbitol level by high performance liquid chromatography: Potential for diagnostic evaluation of diabetes. Clin. Chim. Acta 2005, 354, 41–47. [Google Scholar] [CrossRef]
- Chemical Computing Group’s Molecular Operating Environment (MOE) MOE 2019. 0201. Available online: http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm (accessed on 23 July 2021).
- Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry, Chemical Computing Group. 2007. Available online: http://www.chemcomp.com/journal/proton.htm (accessed on 23 July 2021).
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: Methods behind the HYDE scoring function. J. Comput. Mol. Des. 2012, 27, 15–29. [Google Scholar] [CrossRef]
- BIOVIA Discovery Studio Client v19.1.0.18287. Accelrys Discovery Studio; Accelrys Software Inc.: San Diego, CA, USA, 2019. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | RLAR | HRAR | ||||
|---|---|---|---|---|---|---|
| IC50 (µM) a | Ki (µM) b | Mode of Inhibition c | IC50 (µM) a | Ki (µM) b | Mode of Inhibition c | |
| Protopanaxadiol | 21.38 ± 2.45 | 18.77 (Kic), 37.39 (Kiu) | Mixed type | 0.36 ± 0.1 | 0.72 | Competitive type |
| Ginsenoside Rb1 | 103.11 ± 7.45 | 93.32 ± 5.76 | ||||
| Ginsenoside Rb2 | 121.12 ± 3.43 | >100 | ||||
| Ginsenoside Rb3 | 117.43 ± 6.89 | 78.99 ± 4.55 | ||||
| Ginsenoside Rc | >200 | 56.56 ± 2.19 | ||||
| Ginsenoside Rd | 89.58 ± 2.99 | 37.45 ± 1.33 | ||||
| (20S) ginsenoside Rg3 | 1.25 ± 0.28 | 1.97 | Competitive type | 9.92 ± 0.56 | 8.06 | Competitive type |
| (20R) ginsenoside Rg3 | 4.28 ± 0.31 | 3.69 | Competitive type | 8.67 ± 0.87 | 11.08 (Kic), 17.99 (Kiu) | Mixed type |
| Ginsenoside Rg5 | 54.47 ± 1.22 | 38.56 ± 2.91 | ||||
| Ginsenoside Rh2 | 0.67 ± 0.01 | 0.43 | Competitive type | 7.44 ± 0.55 | 3.63 (Kic), 9.22 (Kiu) | Mixed type |
| Compound K | 41.48 ± 3.99 | 2.23 ± 0.54 | 1.59 | Competitive type | ||
| Ginsenoside Ra1 | >200 | 75.55 ± 4.33 | ||||
| Ginsenoside Ra2 | 178.39 ± 7.39 | 82.43 ± 0.44 | ||||
| Ginsenoside Rs1 | 142.77 ± 3.77 | >100 | ||||
| Ginsenoside Rs2 | 149.34 ± 4.22 | >100 | ||||
| Protopanaxatriol | 27.88 ± 1.19 | 28.88 | Non-competitive type | 1.43 ± 0.14 | 2.61 | Competitive type |
| Ginsenoside Re | 81.27 ± 2.18 | 43.45 ± 3.11 | ||||
| Ginsenoside Rf | 11.29 ± 1.49 | 11.17(Kic), 18.60 (Kiu) | Mixed type | 19.45 ± 1.55 | ||
| Ginsenoside Rg1 | 49.48 ± 1.88 | 27.56 ± 2.12 | ||||
| (20S) ginsenoside Rg2 | 14.38 ± 0.99 | 10.77 (Kic), 17.04 (Kiu) | Mixed type | 15.67 ± 1.05 | ||
| (20R) ginsenoside Rg2 | 29.38 ± 2.33 | 13.66 ± 0.99 | 11.67 | Non-competitive | ||
| Ginsenoside Rh1 | 7.28 ± 0.27 | 5.39 | Competitive type | 4.66 ± 0.34 | 3.40 (Kic), 8.31 (Kiu) | Mixed type |
| Quercetin d | 4.88 ± 0.71 | - | - | 3.11 ± 0.22 | - | |
| Zenaresta e | - | - | - | 0.69 ± 0.11 | - | |
| Compounds | Docked Energy (kcal/mol) | Hydrogen Bond Interactions (No. of H-Bond) | Hydrophobic Interactions |
|---|---|---|---|
| (20R) ginsenoside Rg3 | −6.10 | Lys119 (2.15 Å), Glu120 (2.29 Å), Phe121 (2.44 and 2.45 Å), Phe122 (2.66 Å), Gly128 (2.08 Å), Ser302 (3.14 and 1.91 Å) | His46 (π-alkyl 5.05 Å), Phe121 (π-alkyl 5.21, 4.38, 5.26 and 5.34 Å), Phe122 (π-alkyl 5.19 Å) |
| (20S) ginsenoside Rg2 | −10.31 | Val47 (2.09 Å), Glu120 (1.94 Å), Phe121 (2.84 Å), Phe122 (3.04 and 2.11 Å) | Phe122 (π-alkyl 4.84, 4.47, 3.10 and 5.20 Å), Leu301 (Alkyl 5.14 Å) |
| (20S) ginsenoside Rg3 | −14.56 | Tyr48 (2.88 Å), Gln49 (1.65 Å), Glu120 (2.55 and 1.54 Å), Phe122 (2.15 and 2.09 Å) | Trp20 (π-alkyl 4.73, 4.99, 4.08, 4.06 and 5.21 Å), Val47 (Alkyl 5.38 and 4.86 Å), Trp79 (π-alkyl 5.40 Å), Trp111 (π-alkyl 4.50, 5.41, 5.36 and 4.84 Å), Phe122 (π-alkyl 5.32, 4.91, 4.76 and 4.37 Å), Tyr209 (π-alkyl 4.88 Å), Trp219 (π-alkyl 5.45 Å), Cys298 (Alkyl 4.40 Å), Leu300 (Alkyl 4.65 Å) |
| Ginsenoside Rf | −9.44 | Val47 (1.82 Å), Gln49 (2.64 Å), Glu120 (2.15 Å), Phe121 (2.85 Å), Phe122 (2.20 and 2.89 Å), Ser302 (2.48 Å) | Phe122 (π-alkyl 3.02, 4.60 and 5.20 Å), Trp219 (π-alkyl 4.00, 5.07 and 5.47 Å, π-lone pair 2.65 Å), Leu301 (Alkyl 4.72 Å) |
| Ginsenoside Rh1 | −17.64 | Val47 (1.78 Å), Gln49 (2.82 Å), Glu120 (2.19 Å), Phe121 (2.82 Å), Phe122 (2.05 and 2.87 Å) | Val47 (Alkyl 5.49 Å), Phe122 (π-alkyl 2.86, 4.19, 4.71 and 4.97 Å), Trp219 (π-alkyl 3.99, 4.27, 5.30 and 5.33 Å), Ala299 (Alkyl 3.48 Å), Leu301(Alkyl 4.46 Å) |
| Ginsenoside Rh2 | −17.16 | Tyr48 (2.88 Å), Glu120 (2.52 Å), Phe122 (2.09 and 2.18 Å) | Trp20 (π-alkyl 4.06, 4.08, 4.71, 5.21 Å), Val47 (Alkyl 4.86 and 5.38 Å), Trp79 (π-alkyl 5.40 Å), Trp111 (π-alkyl 4.50, 4.84, 5.36 and 5.41 Å), Phe122 (π-alkyl 5.32, 4.91, 4.37, 4.76 Å), Tyr209 (π-alkyl 4.86 Å), Trp219 (π-alkyl 5.45 Å), Cys298 (Alkyl 4.40 Å), Leu300 (Alkyl 4.65 Å) |
| Protopanaxadiol | −13.74 | Val47 (1.83 Å), Gln49 (3.28 Å), Leu301 (2.75 Å) | Trp20 (π-alkyl 5.49 Å), Phe122 (π-alkyl 3.30, 4.04, 4.67, 5.34 Å), Trp219 (π-alkyl 4.94, 4.98 and 5.00 Å), Leu300 (Alkyl 5.22 Å), Leu301 (Alkyl 4.67 Å) |
| Protopanaxatriol | −9.86 | Val47 (1.67 Å), Gln49 (3.16 Å), Leu301 (2.93 Å) | Trp20 (π-alkyl 5.40 Å), Phe122 (π-alkyl 3.24 and 5.31 Å), Trp219 (π-alkyl 4.08, 4.53, 5.34 and 4.95 Å), Ala299 (Alkyl 3.93 Å), Leu301 (Alkyl 3.90 Å) |
| Quercetin | −31.20 | Thr19 (2.81 Å), Asp43 (2.32 Å), Tyr48 (2.15 Å), Trp111 (2.65 Å), Ser210 (2.27, 2.62 and 2.74 Å), Ile260 (2.09 Å) | His110 (π-π T shaped 4.87 Å), Tyr209 (π-π T shaped 5.38 Å)Ile260 (π-sigma 3.98 Å), Cys298 (π-sulfur 4.36 and 5.78 Å) |
| Compounds | Docked Energy (kcal/mol) | Hydrogen Bond Interactions (No. of H-Bond) | Hydrophobic Interactions |
|---|---|---|---|
| (20R) ginsenoside Rg2 | −4.09 | Gln49 (2.11 Å), Pro218 (1.77 Å), Ser302 (3.17 Å) | Phe122 (π-alkyl 3.96, 4.11 and 5.17 Å), Trp219 (π-alkyl 5.15, 5.01, 3.36 and 5.38 Å), Ala299 (Alkyl 4.23 Å), Leu300 (Alkyl 4.60 Å) |
| (20R) ginsenoside Rg3 | −5.96 | Trp20 (2.44 Å), Val47 (3.02 Å) Tyr48 (2.15 Å) | Phe122 (π-alkyl 4.54 Å), Leu124 (Alkyl 5.11 Å), Pro218 (Alkyl 4.60 Å), Trp219 (π-alkyl 5.15 and 4.69 Å), Leu300 (Alkyl 5.38 Å), Leu301 (Alkyl 5.27 Å) |
| (20S) ginsenoside Rg3 | −6.88 | Tyr48 (2.58 Å), Gln49 (1.72 Å), His110 (2.41 Å), Phe122 (1.67 Å) | Trp20 (π-alkyl 5.41, 4.98, 3.70, 4.53, 5.07 and 4.78 Å), Trp79 (π-alkyl 5.08 Å), Trp111 (π-alkyl 4.37, 5.11, 4.19 and 4.66 Å), Phe122 (π-alkyl 5.22, 4.60, 4.33 and 4.17 Å), Tyr209 (π-alkyl 4.93 Å), Pro218 (Alkyl 4.81Å), Trp219 (π-alkyl 4.82 Å), Ile260 (Alkyl 5.29 Å), Cys298 (Alkyl 4.61Å) |
| Compound K | −7.83 | Val47 (1.90 Å), Tyr48 (1.92 Å) Gln49 (3.31 and 3.14 Å), Trp111 (2.88 Å) | Trp20 (π-alkyl 4.50, 4.86, 4.66, 4.76, 4.55, 4.61 and 4.33 Å), Val47 (Alkyl 4.82 and 4.10 Å), Trp79 (π-alkyl 4.55 Å), Trp111 (π-alkyl 5.44, 5.00 and 4.69 Å), Phe121 (π-alkyl 4.18 Å), Phe122 (π-alkyl 5.38, 4.48 and 4.29 Å), Pro218 (Alkyl 5.46, 4.00 and 5.05 Å), Trp219 (π-alkyl 5.17 Å), Cys298 (Alkyl 4.06 Å) |
| Ginsenoside Rh1 | −7.47 | Val47 (2.70 Å), Tyr48 (2.27 Å) Gln49 (3.07 Å), Cys298 (2.27 and 1.67 Å) | Trp20 (π-alkyl 5.18 Å), Pro23 (Alkyl 4.34 and 4.73 Å), Pro24 (Alkyl 3.92 Å), Val47 (Alkyl 4.15 and 4.46 Å), Phe122 (π-alkyl 4.68, 4.50 and 4.72 Å), Pro218 (Alkyl 5.41, 5.00 and 4.39 Å), Trp219 (π-alkyl 5.23 Å) Leu300 (Alkyl 4.12 Å) |
| Ginsenoside Rh2 | −9.63 | Trp20 (2.66 Å), Val47 (4.76 and 3.05 Å), Tyr48 (2.10 Å) | Trp20 (π-alkyl 4.35, 4.69, 4.95 and 4.84 Å), Phe122 (π-alkyl 3.77 and 5.31Å), Pro218 (Alkyl 4.56, 3.45 and 5.28 Å), Trp219 (π-alkyl 4.63, 5.45 and 4.27 Å), Leu300 (Alkyl 4.60 Å), Leu301 (Alkyl 4.39 Å) |
| Protopanaxadiol | −8.62 | Gln49 (1.89 Å), Tyr209 (2.97 Å) | Trp20 (π-alkyl 5.17, 4.00, 4.66, 3.94 and 4.60 Å), Val47 (Alkyl 4.36 Å), Tyr48 (π-alkyl 4.94 Å), Trp79 (π-alkyl 4.23 Å), Trp111 (π-alkyl 5.07, 4.53, 5.10 and 5.40 Å), Phe122 (π-alkyl 4.65, 4.44, 5.40 and 4.76 Å), Tyr209 (π-alkyl 4.87 Å), Trp219 (π-alkyl 5.06 Å), Cys298 (Alkyl 5.39 Å) |
| Protopanaxatriol | −6.39 | Val47 (2.37 Å), Gln49 (2.48 Å) | Trp20 (π-alkyl 4.31 and 4.56 Å), Lys21 (Alkyl 5.20 Å), Pro24 (Alkyl 5.18 Å), Val47 (Alkyl 3.83 Å), Trp79 (π-alkyl 4.97 and 4.60 Å), Trp111 (π-alkyl 5.10, 5.36, 5.10 and 4.55 Å), Phe122 (π-alkyl 5.21, 4.49 and 4.54 Å), Pro218 (Alkyl 4.28 and 3.68 Å), Cys298 (Alkyl 4.45 Å) |
| Quercetin | −21.86 | Gln183 (2.04 Å), Asp216 (1.69 Å), Ile260 (2.22 Å), Lys262 (3.12 and 2.92 Å) | Trp20 (π-π T shaped 5.38 Å), Tyr48 (π-π T shaped 5.50 Å and π-donor 3.12 Å), Tyr209 (π-π stacked 4.36 Å), Ser210 (π-donor 3.94 Å), Lys262 (π-alkyl 4.24 and 4.87 Å), Cys298 (π-sulfur 5.65 Å) |
| Zenarestat | −31.35 | Thr19 (2.69 Å), Trp20 (2.53 Å) Lys77 (2.72 Å), Ser159 (3.45 Å) Asn160 (3.04 Å), Cys298 (3.49 Å) | Trp20 (π-π T shaped 5.79, 5.32 and 5.78 Å, π-sigma 3.48 Å), Tyr48 (π-π T shaped 5.83 Å, and π-donor 3.97 and 3.49 Å), His110 (Fluorine 3.42 and 3.35 Å), Tyr209 (π-π stacked 4.29 Å), Cys298 (π-sulfur 5.85 and 5.94 Å) |
| Compounds | Sorbitol Content (mg)/Lens Wet Weight (g) | Inhibition (%) |
|---|---|---|
| Blank (glucose-free) | - | - |
| Control | 1.72 ± 0.03 | - |
| Protopanaxadiol | 0.06 ± 0.01 a | 96.51 ± 2.91 a |
| (20S) ginsenoside Rg3 | 0.37 ± 0.01 b | 78.48 ± 4.27 b |
| (20R) ginsenoside Rg3 | 0.89 ± 0.01 c | 48.25 ± 3.99 c |
| Ginsenoside Rh2 | 0.05 ± 0.01 a | 97.09 ± 5.41 a |
| Compound K | 0.46 ± 0.01 b | 73.25 ± 8.11 b |
| Protopanaxatriol | 0.09 ± 0.01 a | 94.76 ± 3.46 a |
| Ginsenoside Rf | 0.28 ± 0.01 a | 83.72 ± 4.88 a |
| (20S) ginsenoside Rg2 | 0.73 ± 0.01 c | 57.55 ± 5.11 c |
| (20R) ginsenoside Rg2 | 0.83 ± 0.01 c | 51.74 ± 2.99 c |
| Ginsenoside Rh1 | 0.10 ± 0.01 a | 94.18 ± 4.71 a |
| Quercetin a | 0.22 ± 0.01 a | 87.20 ± 3.91 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.Y.; Zaib, S.; Jannat, S.; Khan, I.; Rahman, M.M.; Park, S.K.; Chang, M.S. Inhibition of Aldose Reductase by Ginsenoside Derivatives via a Specific Structure Activity Relationship with Kinetics Mechanism and Molecular Docking Study. Molecules 2022, 27, 2134. https://doi.org/10.3390/molecules27072134
Ali MY, Zaib S, Jannat S, Khan I, Rahman MM, Park SK, Chang MS. Inhibition of Aldose Reductase by Ginsenoside Derivatives via a Specific Structure Activity Relationship with Kinetics Mechanism and Molecular Docking Study. Molecules. 2022; 27(7):2134. https://doi.org/10.3390/molecules27072134
Chicago/Turabian StyleAli, Md Yousof, Sumera Zaib, Susoma Jannat, Imtiaz Khan, M. Mizanur Rahman, Seong Kyu Park, and Mun Seog Chang. 2022. "Inhibition of Aldose Reductase by Ginsenoside Derivatives via a Specific Structure Activity Relationship with Kinetics Mechanism and Molecular Docking Study" Molecules 27, no. 7: 2134. https://doi.org/10.3390/molecules27072134
APA StyleAli, M. Y., Zaib, S., Jannat, S., Khan, I., Rahman, M. M., Park, S. K., & Chang, M. S. (2022). Inhibition of Aldose Reductase by Ginsenoside Derivatives via a Specific Structure Activity Relationship with Kinetics Mechanism and Molecular Docking Study. Molecules, 27(7), 2134. https://doi.org/10.3390/molecules27072134