
Determination of Flavonoids in Selected Scleranthus Species and Their Anti-Collagenase and Antioxidant Potential

 ,
,  ,
,  ,
,  ,
, 

Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Identification of Flavone C-Glycosides (6–9)
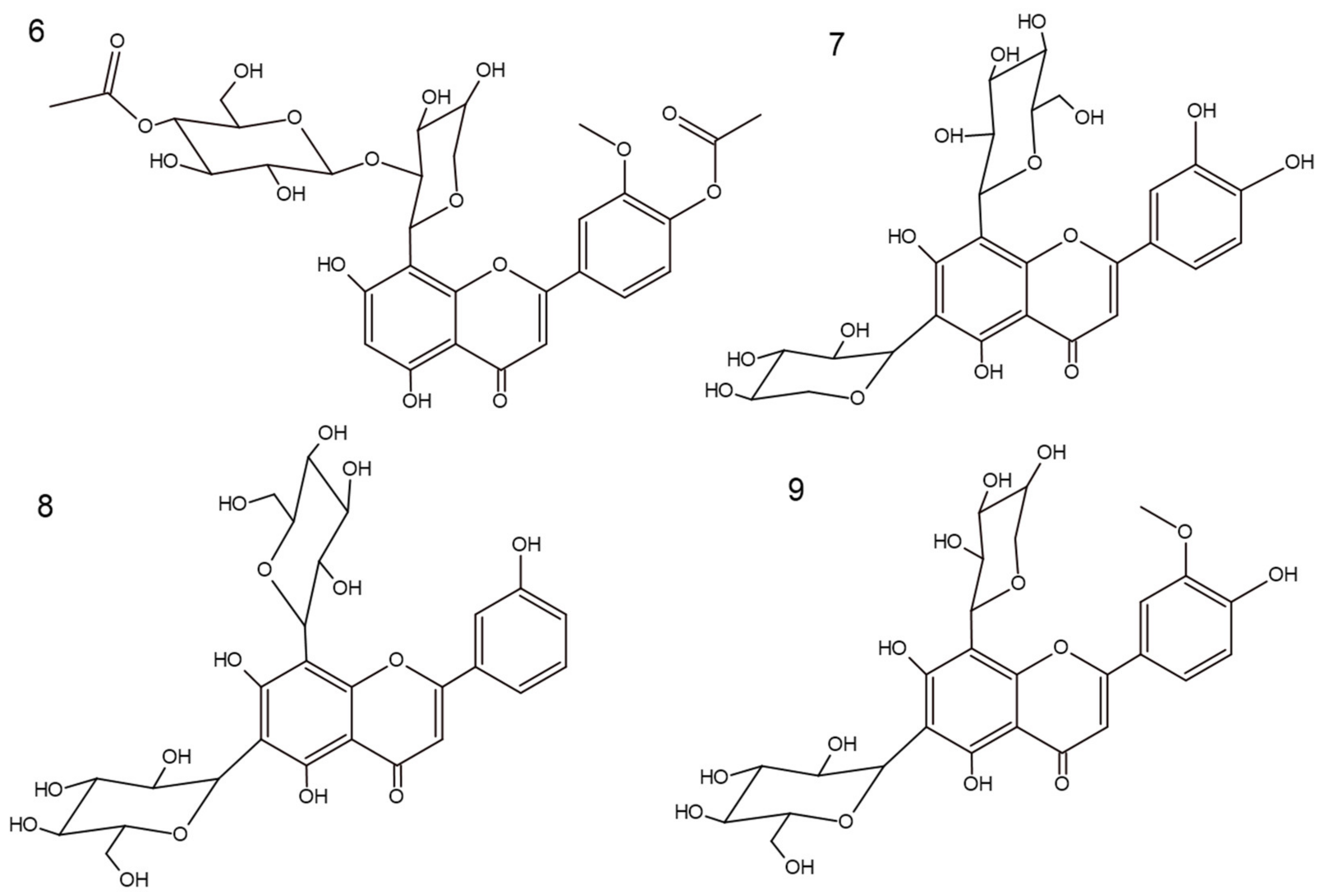
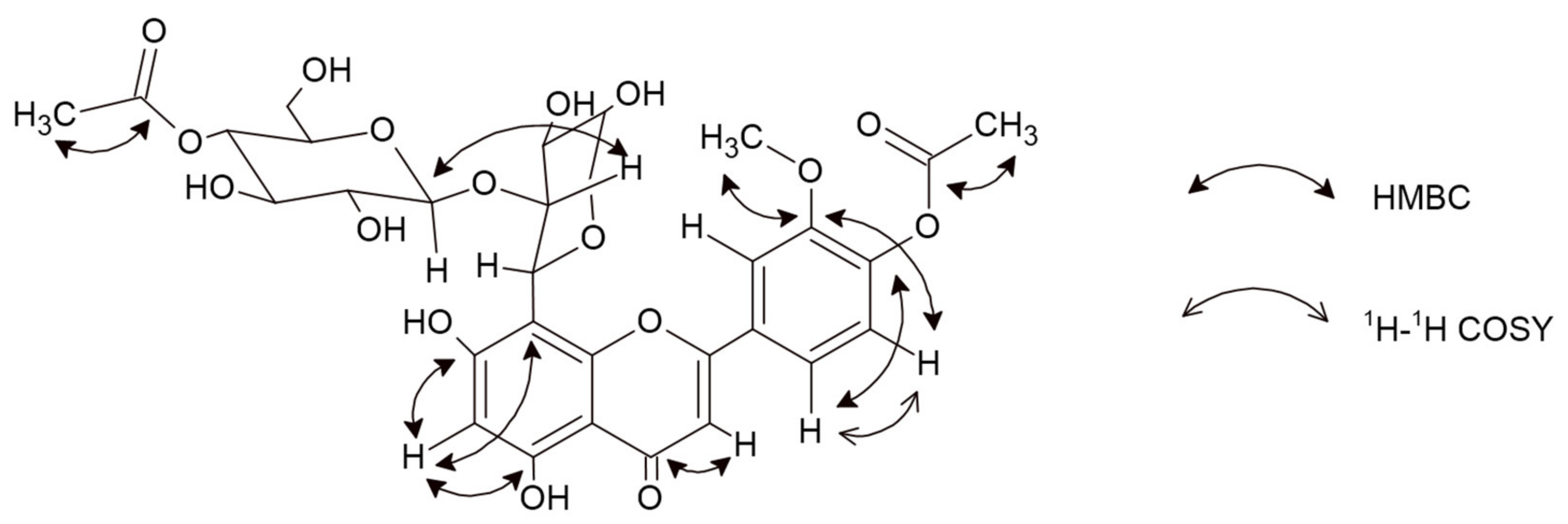
2.1.1. 5,7-Dihydroxy-3′-methoxy-4′-acetoxyflavone-8-C-β-d-arabinopyranoside-2″-O-(4‴-acetoxy)-glucoside (6)
2.1.2. 5,7,3′,4′-Tetrahydroxyflavone-6-C-xyloside-8-C-β-d-glucoside (Lucenin-1) (7)
2.1.3. 5,7,3′-Trihydroxyflavone-6-C-glucoside-8-C-β-d-glucoside (Vicenin-2) (8)
2.1.4. 5,7,4′-Trihydroxy-3′-methoxyflavone-6-C-β-d-glucopyranoside-8-C-α-arabinopyranoside (Chrysoeriol-6-C-β-d-glucopyranoside-8-C-α-arabinoside) (9)
2.2. Phytochemical Analysis of Extracts SP1–SP3, SA1–SA3 and Fractions SP4–SP6, SA4–SA6
2.3. Antioxidant Activities of SP1–SP6, SA1–SA6, and Compounds 1–9
2.4. In Vitro Anti-Collagenase Inhibition
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Preparation of Extracts SP1–SP3, SA1–SA3 and Fractions SP4–SP6, SA4–SA6
3.4. Phytochemical Analysis of Extracts SP1–SP3, SA1–SA3 and Fractions SP4–SP6, SA4–SA6
3.4.1. Determination of Total Phenol Content
3.4.2. Determination of Total Tannin Content
3.4.3. Determination of Phenolic Acid Content
3.4.4. Qualitative HPLC-MSn Analysis
3.4.5. Quantitative Analysis
3.5. Isolation of Flavone C-Glycosides (6–9)
3.6. Identification of Compounds 6–9
3.7. Antioxidant Activity
3.7.1. DPPH Assay
3.7.2. ABTS Assay
3.7.3. FRAP Assay
3.7.4. CUPRAC Assay
3.8. Anti-Collagenase Assay
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| NMR | Nuclear magnetic resonance |
| COSY | Correlation spectroscopy |
| HSQC | Heteronuclear single quantum coherence |
| HMBC | Heteronuclear multiple bond correlation |
| UV | Ultraviolet radiation |
| UV–vis | Ultraviolet–visible spectroscopy |
| ESI | Electrospray ionization |
| IC50 | Median inhibitory concentration |
| MS | Mass spectrometer |
| LC-MS | Liquid chromatography mass spectrometry |
| HPLC | High-performance liquid chromatography |
| HRESIMS | High-resolution electrospray ionization mass spectrometry |
| TLC | Thin-layer chromatography |
| TPC | Total phenolic content |
| TFC | Total flavonoid content |
| TPAC | Total phenolic acids content |
| TTC | Total tannins content |
| ABTS | 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) |
| FRAP | Ferrous reducing antioxidant potential |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| CUPRAC | CUPric reducing antioxidant capacity |
| FALGPA | N-[3-(2-furyl)acryloyl]-leu-gly-pro-ala |
| EGCG | Epigallocatechin gallate |
| TOF | Time-of-flight |
| CD3OD | Deuterated methanol |
| DMSO | Dimethyl sulfoxide |
| CC | Column chromatography |
| MeOH | Methanol |
| Et2O | Diethyl ether |
| EtOAc | Ethyl acetate |
| n-BuOH | n-butanol |
| UPW | Ultra-pure water |
| ACN | Acetonitrile |
| mp | Melting point |
| SD | Standard deviation |
| ANOVA | Analysis of variance |
| ECM | Extracellular matrix |
| MMP | Matrix metalloproteinases |
| hex | Hexose moiety |
| pent | Pentose moiety |
| LOD | Limits of detection |
| LOQ | Limits of detection quantification |
| SDa | Standard deviation of the response |
References
- Smissen, R.D.; Garnock-Jones, P.J. Relationships, classification and evolution of Scleranthus (Caryophyllaceae) as inferred from analysis of morphological characters. Bot. J. Linn. Soc. 2002, 140, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Sell, P.D. Scleranthus (Caryophyllaceae). In Flora Europaea, 1st ed.; Tutin, T.G., Heywood, V.H., Burges, N.A., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: London, UK, 1964; pp. 148–149. [Google Scholar]
- Zibareva, L.; Volodin, V.; Saatov, Z.; Savchenko, T.; Whiting, P.; Lafont, R.; Dinan, L. Distribution of phytoecdysteroids in the Caryophyllaceae. Phytochemistry 2003, 64, 499–517. [Google Scholar] [CrossRef]
- Zdraveva, P.; Gevrenova, R.; Dimitrova, B. Phenolic compounds of Scleranthus annuus L. (Caryophyllaceae). In Proceedings of the 3rd Conference on Medicinal and Aromatic Plants of Southeast European Countries, Nitra, Slovakia, 5–8 September 2004. [Google Scholar]
- Jakimiuk, K.; Strawa, J.W.; Granica, S.; Tomczyk, M. New Flavone C-glycosides from Scleranthus perennis and their anti-collagenase activity. Molecules 2021, 26, 5631. [Google Scholar] [CrossRef] [PubMed]
- Bo, Y.S.; Hyun, P.K. Inhibition of collagenase by naturally-occurring flavonoids. Arch. Pharm. Res. 2005, 28, 1152–1155. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Phenolics and polyphenolics in foods, beverages and spices: Antioxidant activity and health effects—A review. J. Funct. Foods 2015, 18, 820–897. [Google Scholar] [CrossRef]
- Mainka, M.; Czerwińska, M.E.; Osińska, E.; Ziaja, M.; Bazylko, A. Screening of antioxidative properties and inhibition of inflammation-linked enzymes by aqueous and ethanolic extracts of plants traditionally used in wound healing in Poland. Antioxidants 2021, 10, 698. [Google Scholar] [CrossRef]
- Kurtz, A.; Oh, S.J. Age related changes of the extracellular matrix and stem cell maintenance. Prev. Med. 2012, 54, S50–S56. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [Green Version]
- Jakimiuk, K.; Gesek, J.; Atanasov, A.G.; Tomczyk, M. Flavonoids as inhibitors of human neutrophil elastase. J. Enzyme Inhib. Med. Chem. 2021, 36, 1016–1028. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Maity, N.; Nema, N.K.; Sarkar, B.K. Bioactive compounds from natural resources against skin aging. Phytomedicine 2011, 19, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Yayli, N.; Seymen, H.; Baltaci, C. Flavone C-glycosides from Scleranthus uncinatus. Phytochemistry 2001, 58, 607–610. [Google Scholar] [CrossRef]
- Markham, K.R.; Chari, V.M. Carbon-13 NMR spectroscopy of flavonoids. In The Flavonoids; Harborne, J.B., Mabry, T.J., Eds.; Springer: Boston, MA, USA, 1970. [Google Scholar]
- Agrawal, P.K. Carbon-13 NMR of Flavonoids; Elsevier Science: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Kachlicki, P.; Piasecka, A.; Stobiecki, M.; Marczak, Ł. Structural characterization of flavonoid glycoconjugates and their derivatives with mass spectrometric techniques. Molecules 2016, 21, 1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, I.; Brazier-Hicks, M.; Stobiecki, M.; Frański, R.; Edwards, R. Selective disruption of wheat secondary metabolism by herbicide safeners. Phytochemistry 2006, 67, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.L.; Li, S.M.; Yu, S.L.; Yang, Y.X.; Zhang, Y.J.; Han, S.F.; Peng, X. Antioxidant capacity and quantification analysis of chemical constituents isolated from Tetrastigma hemsleyanum leaves. Acta Pol. Pharm. 2021, 78, 361–371. [Google Scholar] [CrossRef]
- Ferreres, F.; Gil-Izquierdo, A.; Vinholes, J.; Grosso, C.; Valentão, P.; Andrade, P.B. Approach to the study of C-glycosyl flavones acylated with aliphatic and aromatic acids from Spergularia rubra by high-performance liquid chromatography-photodiode array detection/electrospray ionization multi-stage mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 700–712. [Google Scholar] [CrossRef]
- Tsiokanos, E.; Tsafantakis, N.; Termentzi, A.; Aligiannis, N.; Skaltsounis, L.A.; Fokialakis, N. Phytochemical characteristics of bergamot oranges from the Ionian islands of Greece: A multi-analytical approach with emphasis in the distribution of neohesperidose flavanones. Food Chem. 2021, 343, 128400. [Google Scholar] [CrossRef]
- Chopin, J.; Dellamonica, G. The Flavonoids; Chapman and Hall: London, UK, 1988. [Google Scholar]
- Olennikov, D.N.; Kashchenko, N.I. Glycosylflavones from Silene armeria and S. compacta. Chem. Nat. Compd. 2019, 55, 934–936. [Google Scholar] [CrossRef]
- Sut, S.; Dall’Acqua, S.; Uysal, S.; Zengin, G.; Aktumsek, A.; Picot-Allain, C.; Mahomoodally, F. LC-MS, NMR fingerprint of Potentilla argentea and Potentilla recta extracts and their in vitro biopharmaceutical assessment. Ind. Crops Prod. 2019, 131, 125–133. [Google Scholar] [CrossRef]
- Ciganović, P.; Jakimiuk, K.; Tomczyk, M.; Zovko Končić, M. Glycerolic licorice extracts as active cosmeceutical ingredients: Extraction optimization, chemical characterization, and biological activity. Antioxidants 2019, 8, 445. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and flavonols’ antiradical structure–activity relationship—A quantum chemical study. Antioxidants 2020, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Pientaweeratch, S.; Panapisal, V.; Tansirikongkol, A. Antioxidant, anti-collagenase and anti-elastase activities of Phyllanthus emblica, Manilkara zapota and silymarin: An in vitro comparative study for anti-aging applications. Pharm. Biol. 2016, 54, 1865–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strawa, J.; Wajs-Bonikowska, A.; Jakimiuk, K.; Waluk, M.; Poslednik, M.; Nazaruk, J.; Tomczyk, M. Phytochemical examination of woolly burdock Arctium tomentosum leaves and flower heads. Chem. Nat. Compd. 2020, 56, 345–347. [Google Scholar] [CrossRef]
- Rutkowski, L. Klucz do Oznaczania Roślin Naczyniowych Polski Niżowej; Wydawnictwo Naukowe PWN: Warsaw, Poland, 2006. [Google Scholar]
- European Pharmacopoeia, 10th ed.; European Directorate for the Quality of Medicines and HealthCare of the Council of Europe (EDQM): Strasbourg, France, 2019; p. 1588.
- Krošlák, E.; Maliar, T.; Nemeček, P.; Viskupičová, J.; Maliarová, M.; Havrlentová, M.; Kraic, J. Antioxidant and proteinase inhibitory activities of selected poppy (Papaver somniferum L.) genotypes. Chem. Biodivers. 2017, 14, e1800374. [Google Scholar] [CrossRef]
- Okuda, M.; Kaihara, S.; Murakami, T.; Koide, D.; Ohe, K. Wiley StatsRef: Statistics Reference Online; John Wiley Sons, Ltd.: Chichester, UK, 2014; Volume 18, pp. 71–77. [Google Scholar]
- Siciliano, T.; De Tommasi, N.; Morelli, I.; Braca, A. Study of flavonoids of Sechium edule (Jacq) Swartz (Cucurbitaceae) different edible organs by liquid chromatography photodiode array mass spectrometry. J. Agric. Food Chem. 2004, 52, 6510–6515. [Google Scholar] [CrossRef]
- Yayli, N.; Baltaci, C.; Genç, H.; Terzioǧlu, S. Phenolic and flavone C-glycosides from Scleranthus uncinatus. Pharm. Biol. 2002, 40, 369–373. [Google Scholar] [CrossRef]
- Jakimiuk, K.; Wink, M.; Tomczyk, M. Flavonoids of the Caryophyllaceae. Phytochem. Rev. 2021, 20, 179–218. [Google Scholar] [CrossRef]

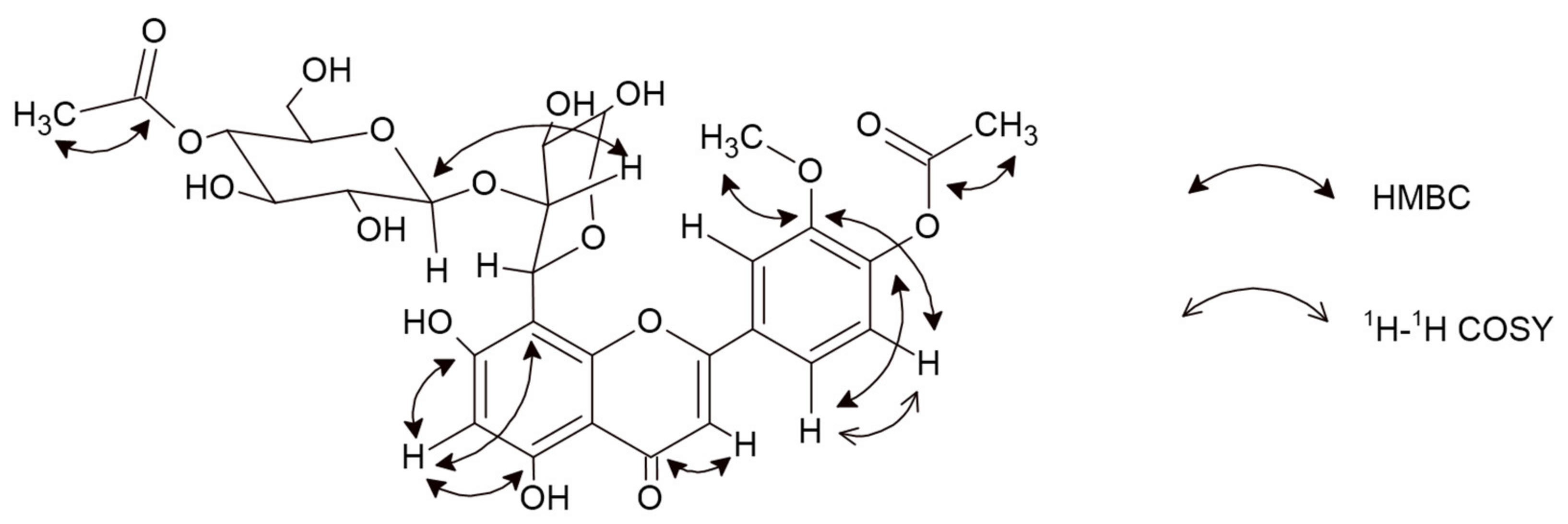

{kind=link}
{kind=link}
| Analyte | Rt (Min) | UV Spectra (Λ Max Nm) | -ESI–MS (m/z) | Fragmentation A | Predicted Compounds |
|---|---|---|---|---|---|
| I | 7.6 | 268, 302 | 459 | 293 | Apiopaeonoside (5) I |
| II | 7.9 | 260, 300sh, 325 | 385 | 191 | Quinic acid derivative |
| III | 11.35 | 270, 345 | 579 | 459, 399, 369, 313 | Flavone C-hex-C-pent derivative |
| IV | 12.8 | 257, 273, 348 | 579 | 459, 399, 369 | Lucenin-1 (7) I |
| V | 17.51 | 270, 348 | 579 | 459 | Flavone 6,8-di-C-glycoside derivative |
| VI | 18.01 | 254, 270, 348 | 593 | 473 | vicenin-2 (8) I |
| VII | 19.45 | 254, 270, 348 | 593 | 473, 383, 312 | Flavone 6,8-di-C-glycoside derivative |
| VIII | 21.86 | 255, 273, 348 | 593 | 473, 383, 312 | Chrysoeriol-6-C-β-d-glucopyranoside-8-C-α-arabinoside (9) I |
| IX | 33.18 | 275, 348 | 593 | 473, 383 | Flavone 6,8-di-C-glycoside derivative |
| X | 35.23 | 270, 348 | 593 | 473, 383 | Flavone 6,8-di-C-glycoside derivative |
| XI | 40.13 | 265, 348 | 563 | 473, 383 | Flavone C-hex-C-pent derivative |
| XII | 41.64 | 270, 345 | 579 | 489, 399, 327 | Flavone C-hex-C-pent derivative |
| XIII | 42.4 | 255, 270, 347 | 635 | 413, 308 | Flavone C-hex-C-pent derivative |
| XIV | 44.8 | 255, 270, 348 | 621 | 579, 531, 399, 327 | scleranthoside A (1) I |
| XV | 46.2 | 270, 345 | 677 | 413, 323 | Flavone-C-pent-O-hex derivative |
| XVI | 48.6 | 255, 270, 348 | 593 | 413, 323 | scleranthoside B (2) I |
| XVII | 56.69 | 255, 270, 349 | 677 | 413 | Flavone-C-pent-O-hex derivative |
| XVIII | 58.47 | 254, 268, 348 | 677 | 545, 455, 413, 322 | scleranthoside C (3) I |
| XIX | 64.11 | 268, 348 | 635 | 413 | Luteolin-8-C-pent-O-hex derivative |
| XX | 66.86 | 268, 348 | 635 | 413 | Luteolin-8-C-pent-O-hex derivative |
| XXI | 74.55 | 253, 270, 348 | 677 | 413, 323 | Luteolin-8-C-pent-O-hex derivative |
| XXII | 75.98 | 252, 268, 348 | 677 | 413 | scleranthoside D (4) I |
| XXIII | 90.87 | 270, 348 | 677 | 413 | Luteolin-8-C-pent-O-hex derivative |
| XXIV | 98.34 | 252, 268, 348 | 677 | 635, 545 455, 413 | scleranthoside D (6) I |
| Sample | TPC (Mg Peq/G Extract) A | TPAC (Mg Caeq/G Extract) B | TTC (Mg Peq/G Extract) A |
|---|---|---|---|
| SA1 SP1 | 103.16 ± 0.80 122.96 ± 1.96 | 7.20 ± 0.19 4.68 ± 0.18 | 8.64 ± 0.15 9.78 ± 0.98 |
| SA2 SP2 | 103.08 ± 1.84 107.76 ± 0.52 | 6.66 ± 0.18 6.03 ± 0.27 | 11.01 ± 0.98 20.16 ± 1.47 |
| SA3 SP3 | 95.72 ± 1.69 99.20 ± 0.40 | 6.12 ± 0.17 4.95 ± 0.12 | 7.35 ± 0.18 1.6 ± 0.15 |
| SA4 SP4 | 106.04 ± 2.71 165.16 ± 2.83 | 8.91 ± 0.27 5.58 ± 0.15 | 8.07 ± 1.35 12.57 ± 0.89 |
| SA5 SP5 | 217.20 ± 1.60 240.00 ± 2.01 | 11.34 ± 0.63 7.47 ± 2.80 | 6.3 ± 0.90 6.69 ± 0.97 |
| SA6 SP6 | 159.44 ± 1.40 161.32 ± 2.91 | 9.19 ± 0.54 5.67 ± 0.18 | 8.22 ± 0.45 1.74 ± 0.18 |
| Compound A | SA1 | SP1 | SA2 | SP2 | SA3 | SP3 | SA4 | SP4 | SA5 | SP5 | SA6 | SP6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BLQ | 0.12 ± 0.01 | 0.20 ± 0.01 | 0.23 ± 0.01 | ND | 0.11 ± 0.01 | BLQ | 0.17 ± 0.01 | 1.61 ± 0.01 | 1.61 ± 0.01 | 0.57 ± 0.03 | 0.52 ± 0.04 |
| 2 | 0.56 ± 0.01 | 1.66 ± 0.01 | 1.10 ± 0.07 | 2.20 ± 0.11 | 0.73 ± 0.01 | 2.09 ± 0.07 | 0.12 ± 0.01 | 1.26 ± 0.01 | 5.08 ± 0.02 | 12.85 ± 0.01 | 1.28 ± 0.01 | 5.22 ± 0.01 |
| 3 | 0.36 ± 0.01 | 4.90 ± 0.05 | 0.94 ± 0.03 | 6.29 ± 0.13 | 0.62 ± 0.01 | 6.38 ± 0.06 | 0.34 ± 0.01 | 5.55 ± 0.01 | 2.03 ± 0.2 | 59.10 ± 0.08 | 0.32 ± 0.01 | 1.05 ± 0.02 |
| 4 | BLQ | 1.98 ± 0.02 | BLQ | 0.71 ± 0.01 | ND | 2.07 ± 0.01 | 0.24 ± 0.06 | 3.81 ± 0.01 | 8.14 ± 0.14 | 18.97 ± 0.08 | ND | ND |
| 6 | 1.05 ± 0.01 | 1.06 ± 0.02 | 0.58 ± 0.01 | 0.91 ± 0.04 | 0.68 ± 0.01 | 0.73 ± 0.01 | 0.36 ± 0.01 | 0.93 ± 0.01 | 3.79 ± 0.01 | 6.93 ± 0.04 | BLQ | ND |
| 7 | 0.72 ± 0.02 | 0.11 ± 0.01 | 0.56 ± 0.04 | BLQ | 0.46 ± 0.03 | 0.10 ± 0.01 | ND | ND | 0.25 ± 0.01 | ND | 3.40 ± 0.01 | 1.44 ± 0.02 |
| 8 | 0.19 ± 0.01 | 0.16 ± 0.01 | 0.16 ± 0.01 | 0.15 ± 0.01 | 0.13 ± 0.01 | 0.15 ± 0.01 | ND | ND | 0.28 ± 0.11 | ND | 0.90 ± 0.01 | 1.37 ± 0.01 |
| 9 | 0.11 ± 0.01 | ND | BLQ | ND | BLQ | ND | ND | ND | 0.33 ± 0.12 | ND | 0.45 ± 0.05 | 0.50 ± 0.01 |
| Total | 13.74 ± 0.1 | 17.4 ± 0.27 | 13.49 ± 0.18 | 18.69 ± 0.21 | 11.27 ± 0.05 | 18.97 ± 0.32 | 4.62 ± 0.16 | 17.6 ± 0.26 | 71.8 ± 3.1 | 136.13 ± 0.39 | 20.52 ± 0.78 | 21.03 ± 0.36 |
| 5 | ND | 1.37 ± 0.04 | ND | 2.11 ± 0.04 | ND | 1.08 ± 0.02 | ND | ND | 0.98 ± 0.12 | 0.83 ± 0.11 | 0.74 ± 0.04 | 7.74 ± 0.13 |
| Sample | DPPH A (µM Teq) | ABTS A (µM Teq) | FRAP B (mM Fe2+/mL) | CUPRAC A (µM Teq) |
|---|---|---|---|---|
| SA1 SP1 | 193.16 ± 2.56 140.43 ± 2.76 | 215.74 ± 2.46 196.95 ± 1.23 | 2.02 ± 0.49 1.92 ± 0.43 | 7.14 ± 1.32 5.82 ± 0.32 |
| SA2 SP2 | 186.41 ± 1.93 64.07 ± 0.97 | 253.65 ± 3.72 67.94 ± 2.03 | 3.17 ± 0.66 1.57 ± 0.07 | 5.38 ± 0.76 9.34 ± 1.63 |
| SA3 SP3 | 118.49 ± 1.46 122.28 ± 1.83 | 425.69 ± 2.33 56.11 ± 2.13 | 3.30 ± 0.15 2.64 ± 0.33 | 13.29 ± 0.76 17.69 ± 1.96 |
| SA4 SP4 | 157.09 ± 0.97 273.53 ± 1.93 | 175.99 ± 2.03 443.96 ± 2.42 | 1.96 ± 0.53 4.65 ± 0.35 | 1.86 ± 0.32 5.33 ± 0.54 |
| SA5 SP5 | 354.32 ± 2.90 269.31 ± 0.97 | 577. 82 ± 3.51 311.45 ± 3.05 | 6.74 ± 0.98 6.73 ± 0.41 | 67.80 ± 1.32 97.26 ± 1.87 |
| SA6 SP6 | 386.59 ± 2.19 292.51 ± 2.03 | 459.29 ± 1.40 271.94 ± 1.68 | 7.50 ± 0.32 5. 78 ± 0.42 | 45.82 ± 1.01 70.44 ± 1.28 |
| 1 | 405.15 ± 3.12 | 765.16 ± 2.83 | 39.25 ± 1.42 | 581.32 ± 3.61 |
| 2 | 198.43 ± 2.40 | 188.57 ± 1.86 | 5.68 ± 0.68 | 114.56 ± 2.04 |
| 3 | 7.11 ± 0.59 | 198.57 ± 2.46 | 2.37 ± 0.36 | 67.30 ± 1.30 |
| 4 | 9.64 ± 0.82 | 302.05 ± 2.42 | 2.65 ± 0.47 | 44.22 ± 1.35 |
| 5 | 61.96 ± 1.90 | 444.23 ± 0.47 | 2.75 ± 0.11 | 48.46 ± 1.75 |
| 6 | 6.69 ± 0.97 | 169.27 ± 1.68 | 3.05 ± 0.25 | 18.57 ± 0.76 |
| 7 | 409.16 ± 2.03 | 796.61 ± 1.23 | 45.30 ± 2.49 | 116.16 ± 0.76 |
| 8 | 176.71 ± 0.97 | 369.24 ± 3.05 | 5.16 ± 0.96 | 27.15 ± 1.32 |
| 9 | 407.05 ± 1.83 | 586.69 ± 2.91 | 9.07 ± 0.45 | 18.20 ± 0.76 |
| Compound | IC50 (µM) |
|---|---|
| 6 | 39.59 ± 1.21 |
| 7 | 73.86 ± 1.03 |
| 8 | 53.37 ± 0.88 |
| 9 | 71.06 ± 0.97 |
| EGCG A | 34.82 ± 0.53 |
| Sample | Percentage of Inhibition (%) A |
|---|---|
| SA1 SP1 | 32.04 ± 0.45 22.06 ± 0.34 |
| SA2 SP2 | 10.90 ± 0.11 6.46 ± 0.09 |
| SA3 SP3 | 12.12 ± 0.41 11.13 ± 0.14 |
| SA4 SP4 | 19.42 ± 0.22 6.69 ± 0.10 |
| SA5 SP5 | 16.59 ± 0.19 15.40 ±0.32 |
| SA6 SP6 | 7.11 ± 0.17 2.39 ± 0.07 |
| Compound | Regression Equation A | R2 | Linear Range (µg/mL) | LOD (µg/mL) | LOQ (µg/mL) | Accuracy (%) | Precision (%) B |
|---|---|---|---|---|---|---|---|
| 3 | y = 2374x + 63.119 | 0.9999 | 0.05–10 | 0.03 | 0.09 | 98.43 ± 3.70 | 1.3/1.95 |
| 5 | y = 4563.2x − 28.098 | 0.9999 | 0.01–5 | 0.003 | 0.01 | 101.28 ± 3.95 | 0.97/1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakimiuk, K.; Strawa, J.W.; Granica, S.; Locatelli, M.; Tartaglia, A.; Tomczyk, M. Determination of Flavonoids in Selected Scleranthus Species and Their Anti-Collagenase and Antioxidant Potential. Molecules 2022, 27, 2015. https://doi.org/10.3390/molecules27062015
Jakimiuk K, Strawa JW, Granica S, Locatelli M, Tartaglia A, Tomczyk M. Determination of Flavonoids in Selected Scleranthus Species and Their Anti-Collagenase and Antioxidant Potential. Molecules. 2022; 27(6):2015. https://doi.org/10.3390/molecules27062015
Chicago/Turabian StyleJakimiuk, Katarzyna, Jakub W. Strawa, Sebastian Granica, Marcello Locatelli, Angela Tartaglia, and Michał Tomczyk. 2022. "Determination of Flavonoids in Selected Scleranthus Species and Their Anti-Collagenase and Antioxidant Potential" Molecules 27, no. 6: 2015. https://doi.org/10.3390/molecules27062015
APA StyleJakimiuk, K., Strawa, J. W., Granica, S., Locatelli, M., Tartaglia, A., & Tomczyk, M. (2022). Determination of Flavonoids in Selected Scleranthus Species and Their Anti-Collagenase and Antioxidant Potential. Molecules, 27(6), 2015. https://doi.org/10.3390/molecules27062015