
Kinetic Study of the Oxidative Addition Reaction between Methyl Iodide and [Rh(imino-β-diketonato)(CO)(PPh)3] Complexes, Utilizing UV–Vis, IR Spectrophotometry, NMR Spectroscopy and DFT Calculations


Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis of Rhodium(I) Complexes
2.2. DFT Study of Rhodium(I) and Rhodium(III) Complexes
2.2.1. Rhodium(I)
2.2.2. Rhodium(III)
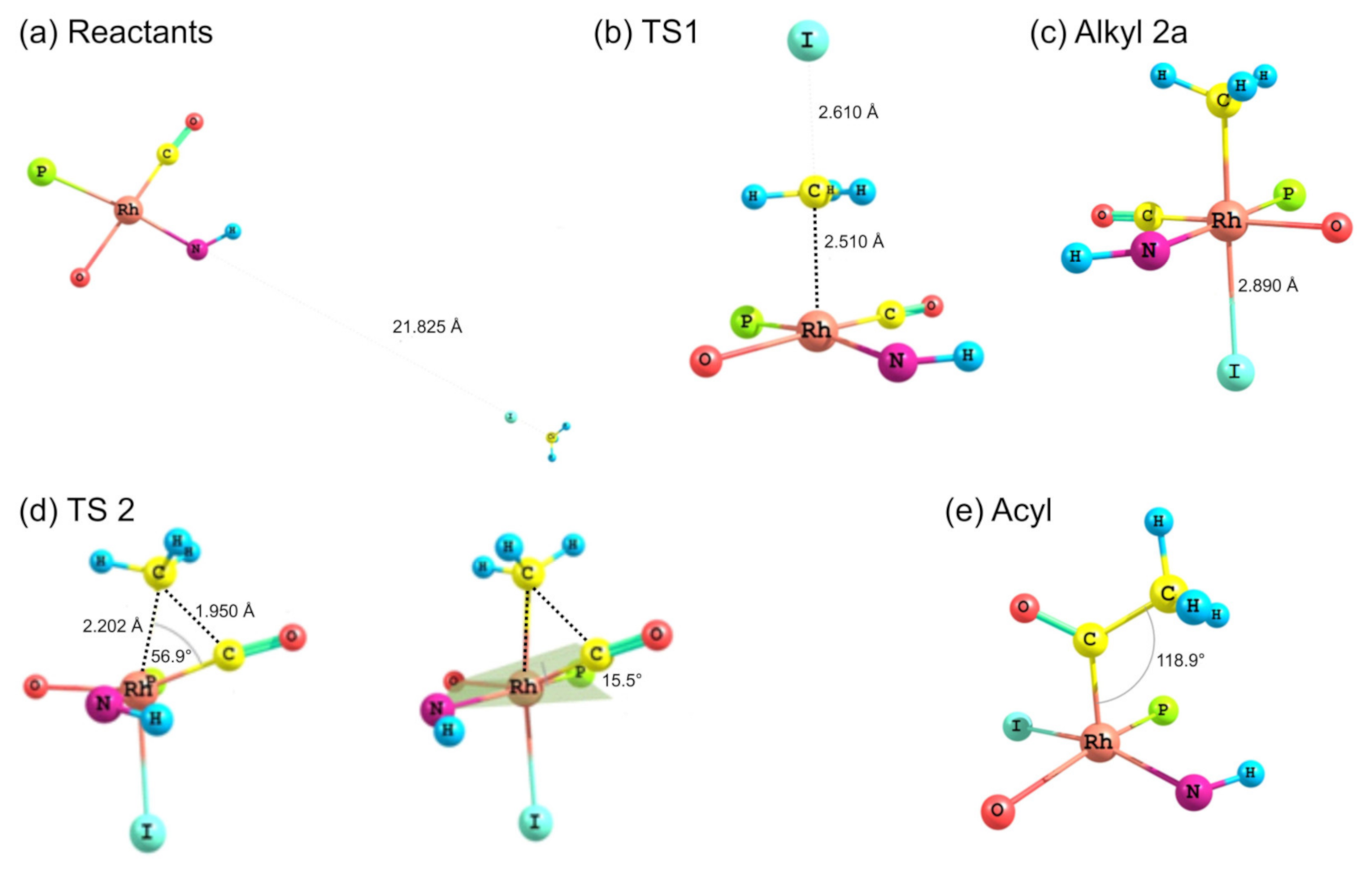
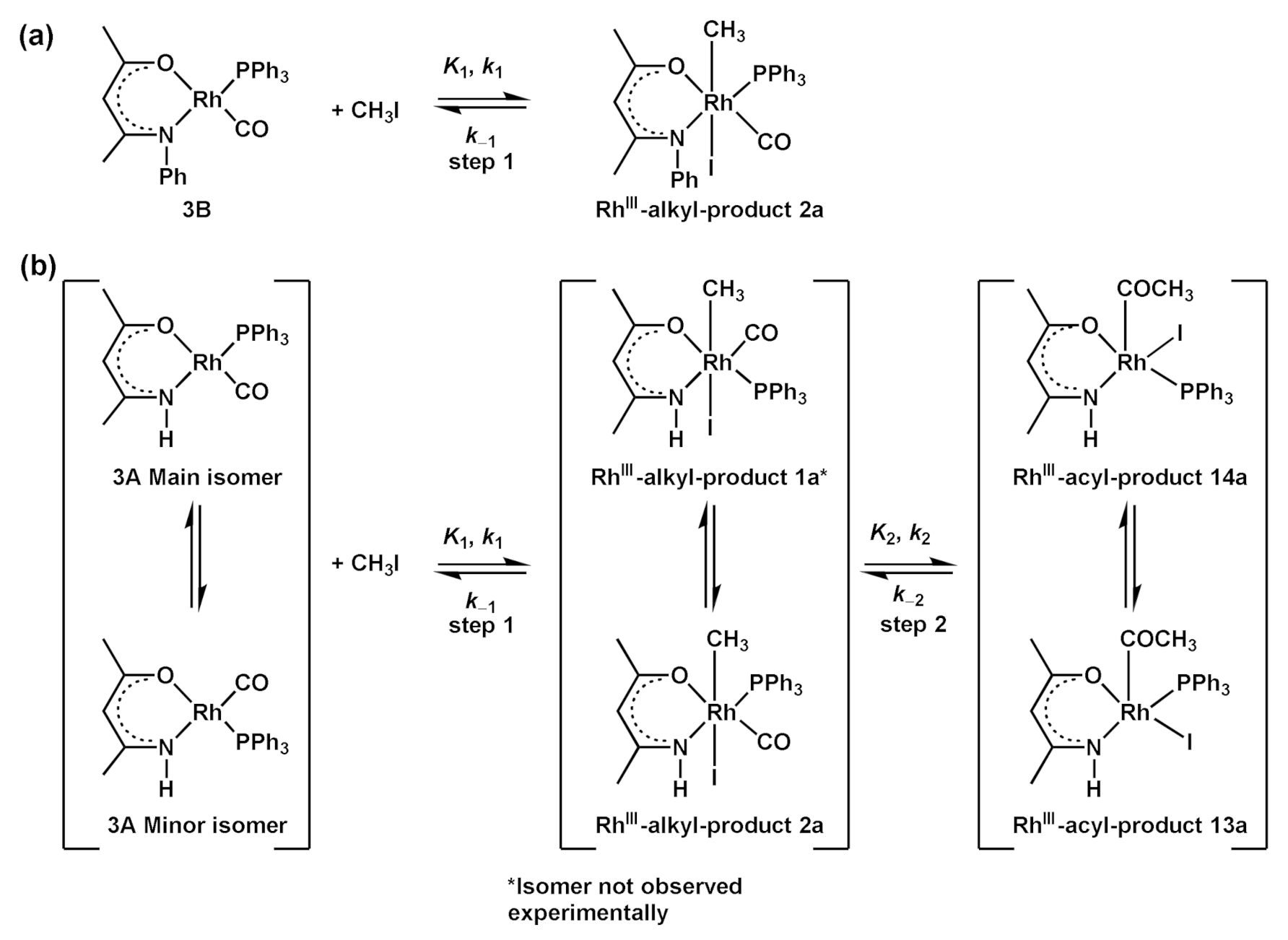
- Products of [Rh(CH3COCHCNHCH3)(CO)(PPh3)] 3A + CH3I reaction (Table 1): Evaluating the energies of the different Rh(III)-alkyl isomers in Table 1, it can be seen that the most preferred orientation of the product geometry is with the Oβ-ketoiminato atom O atom of the ligand trans to CO (O-trans-CO) and the CH3 and I atoms bonded at the apical positions (top and bottom, with respect to the square planar geometry), since it has the lowest relative energy (2a, reaction product of CH3I and 3A isomer O-trans-CO). The Rh(III)-alkyl isomer with the Nβ-ketoiminato atom of the ligand trans to CO, and the CH3 and I atoms bonded at the apical positions (1a, reaction product of CH3I and 3A isomer N-trans-CO) is only slightly higher in energy (0.07 eV). The energetically-preferred Rh(III)-alkyl isomer to form is, thus, 2a, while 1a may form in very small amounts. Evaluation of the energies of the Rh(III)-acyl isomers in Table 1, indicates that the two Rh(III)-acyl isomers 13a and 14a with the COCH3 ligand in the apical position have the lowest energy. Furthermore, the energy of Rh(III)-acyl isomers 13a and 14a are ca. 0.3 eV lower than the energy of the lowest energy Rh(III)-alkyl isomers 1a and 2a. This indicates that the Rh(III)-alkyl product of oxidative addition could convert, via CO insertion, to the lower more stable Rh(III)-acyl product as the final reaction product. Rh(III)-acyl 13a has slightly higher energy (0.04 eV) than 14a, therefore, the amount of 13a will be less than 14a.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Structures | Relative Energy (eV) | Product | Structures | Relative Energy (eV) | Product | Structures | Relative Energy (eV) |
|---|---|---|---|---|---|---|---|---|
| 1a |  | 0.34 (0.29) 0.32 0.49 | 7a |  | 0.41 (0.35) 0.38 0.52 | 13a |  | 0.04 (0.04) 0.00 0.01 |
| 1b |  | 0.34 (0.29) | 7b |  | 0.41 (0.35) | 13b |  | 0.04 (0.04) |
| 2a |  | 0.26 (0.21) 0.22 0.38 | 8a |  | 0.64 (0.58) 0.55 0.71 | 14a |  | 0.00 (0.03) 0.01 0.00 |
| 2b |  | 0.26 (0.19) | 8b |  | 0.64 (0.58) | 14b |  | 0.00 (0.00) |
| 3a |  | 0.43 (0.34) 0.32 0.46 | 9a |  | 0.44 (0.37) 0.42 0.56 | 15a |  | - |
| 3b |  | 0.43 (0.35) | 9b |  | 0.44 (0.37) | 15b |  | - |
| 4a |  | 0.72 (0.64) 0.59 0.77 | 10a |  | 0.35 (0.28) 0.30 0.44 | 16a |  | 0.79 (0.72) 0.68 0.68 |
| 4b |  | 0.72 (0.63) | 10b |  | 0.35 (0.28) | 16b |  | 0.79 (0.70) |
| 5a |  | 0.46 (0.37) 0.22 0.38 | 11a |  | 0.87 (0.74) 0.51 0.69 | 17a |  | - |
| 5b |  | 0.46 (0.37) | 11b |  | 0.87 (0.74) | 17b |  | - |
| 6a |  | 0.66 (0.57) 0.52 0.70 | 12a |  | 0.68 (0.59) 0.48 0.63 | 18a |  | - |
| 6b |  | 0.66 (0.57) | 12b |  | 0.68 (0.62) | 18b |  | - |
| Product | Structures | Relative Energy (eV) | Product | Structures | Relative Energy (eV) | Product | Structures | Relative Energy (eV) |
|---|---|---|---|---|---|---|---|---|
| 1a |  | 0.49 (0.50) 0.33 0.48 | 7a |  | 0.15 (0.17) 0.07 0.19 | 13a |  | 0.00 (0.08) 0.00 0.00 |
| 1b |  | 0.49 (0.51) | 7b |  | 0.15 (0.16) | 13b |  | 0.00 (0.08) |
| 2a |  | 0.02 (0.00) 0.00 0.15 | 8a |  | 0.64 (0.64) 0.47 0.60 | 14a |  | 0.28 (0.33) 0.20 0.19 |
| 2b |  | 0.02 (0.03) | 8b |  | 0.64 (0.64) | 14b |  | 0.28 (0.31) |
| 3a |  | 0.57 (0.58) 0.41 0.59 | 9a |  | 0.34 (0.32) 0.27 0.41 | 15a |  | 0.37 (0.35) 0.25 0.24 |
| 3b |  | 0.57 (0.56) | 9b |  | 0.34 (0.34) | 15b |  | 0.37 (0.35) |
| 4a |  | 0.85 (0.85) 0.66 0.84 | 10a |  | 0.26 (0.27) 0.18 0.30 | 16a |  | 0.71 (-) 0.52 0.49 |
| 4b |  | 0.85 (0.84) | 10b |  | 0.26 (0.27) | 16b |  | 0.71 (-) |
| 5a |  | 0.44 (0.43) 0.22 0.38 | 11a |  | 0.48 (0.51) 0.31 0.46 | 17a |  | - |
| 5b |  | 0.44 (0.41) | 11b |  | 0.48 (0.51) | 17b |  | - |
| 6a |  | 0.15 (0.15) 0.03 0.16 | 12a |  | 0.66 (0.61) 0.43 0.59 | 18a |  | 0.36 (0.40) 0.32 0.29 |
| 6b |  | 0.15 (0.41) | 12b |  | 0.64 (0.66) | 18b |  | 0.36 (0.38) |
- Products of [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B + CH3I reaction (Table 2): From the energies presented in Table 2, it is clear that Rh(III)-alkyl 2a and Rh(III)-acyl 13a are equi-energetic—in other words, there is no driving force for the Rh(III)-alkyl 2a to convert via CO insertion to the Rh(III)-acyl 13a. The Rh(III)-alkyl 2a product, with the O atom of the ligand trans to the CO, and the CH3 and I atoms, bonded at the apical positions (top and bottom with respect to the square planar geometry), are predicted to be the final reaction product of the [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B + CH3I reaction.
- The [Rh(N,O-BID)(CO)(PPh3)] + CH3I oxidative addition reaction is theoretically only possible if the activation barrier of the transition state is low enough to form the product—in this case, a Rh(III)-alkyl product. Similarly the CO insertion reaction is theoretically only possible if there is a driving force to lower energy (to form the product) and the activation barrier of the transition state is low enough to form the Rh(III)-acyl product. A DFT study of the transition states will be presented in Section 2.3.4.
2.3. Kinetics of Iodomethane Oxidative Addition to Rhodium(I)
2.3.1. IR Study
2.3.2. UV–Vis Study
2.3.3. NMR Study
2.3.4. DFT Study of Reaction Mechanism
2.3.5. Summary of [Rh(L,L′-BID)(CO)(PPh3)] + CH3I Reactions
3. Materials and Methods
3.1. Instrumentation
3.2. Synthesis
3.2.1. [Rh(N,O-BID)(CO)2]
3.2.2. [Rh(N,O-BID)(CO)(PPh3)]
3.3. Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Trzeciak, A.M.; Ziółkowski, J.J. Perspectives of rhodium organometallic catalysis. Fundamental and applied aspects of hydroformylation. Coord. Chem. Rev. 1999, 190–192, 883–900. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Yan, L.; Ding, Y. A mini review on strategies for heterogenization of rhodium-based hydroformylation catalysts. Front. Chem. Sci. Eng. 2018, 12, 113–123. [Google Scholar] [CrossRef]
- Crabtree, R.H. Rhodium and Iridium in Organometallic Catalysis. Platin. Met. Rev. 2006, 50, 171–176. [Google Scholar] [CrossRef]
- Heidingsfeldova, M.; Capca, M. Rhodium Complexes as Catalysts for Hydrosilylation Crosslinking of Silicone Rubber. J. Appl. Polym. Sci. 1985, 30, 1837–1846. [Google Scholar] [CrossRef]
- Chernyshov, B.V.I.; Kisil, I.M. Platinum metals catalvtic systems in nitric acid production system optimisation in non-concentrated acid plants. Platin. Met. Rev. 1993, 37, 136–143. [Google Scholar]
- Halligudi, S.B.; Bajaj, H.C.; Bhatt, K.N.; Krishnaratnam, M. Hydrogenation of benzene to cyclohexane catalyzed by rhodium (I) complex supported on montmorillonite clay. React. Kinet. Mech. Catal. 1992, 48, 547–552. [Google Scholar] [CrossRef]
- Mieczy, E.; Trzeciak, A.M.; Ziolkowski, J.J. Hydroformylation and isomerization of hex-1-ene catalyzed by [Rh(acac)(CO)(PPh3)]: Effect of modifying ligands. J. Mol. Catal. 1992, 73, 1–8. [Google Scholar]
- Roth, B.J.F. The Production of Acetic Acid. Rhodium catalyzed carbonylation of methanol. Platin. Met. Rev. 1975, 19, 12–14. [Google Scholar]
- Jones, J.H. The CativaTM Process for the Manufacture of Acetic Acid: Iridium catalyst improves productivity in an established industrial process. Platin. Met. Rev. 2000, 44, 94–105. [Google Scholar]
- Maitlis, P.M.; Haynes, A.; Sunley, G.J.; Howard, M.J. Methanol carbonylation revisited: Thirty years on. J. Chem. Soc. Dalton Trans. 1996, 11, 2187–2196. [Google Scholar] [CrossRef]
- Stuurman, N.F.; Buitendach, B.E.; Twigge, L.; Swarts, P.J.; Conradie, J. Rhodium(triphenylphosphine)carbonyl-2,4-dioxo-3-pentyl-4-decanyloxybenzoate: Synthesis, electrochemistry and oxidative addition kinetics. New J. Chem. 2018, 42, 4121–4132. [Google Scholar] [CrossRef]
- Brink, A.; Roodt, A.; Steyl, G.; Visser, H.G. Steric vs. electronic anomaly observed from iodomethane oxidative addition to tertiary phosphine modified rhodium(i) acetylacetonato complexes following progressive phenyl replacement by cyclohexyl [PR3 = PPh3, PPh2Cy, PPhCy2 and PCy3]. Dalton Trans. 2010, 39, 5572–5578. [Google Scholar] [CrossRef] [PubMed]
- Ruballo, A.; Selina, V..; Cherkasova, T.G.; Varshavskii, Y.S. IR-spectroscopic study of dicarbonyl complexes of rhodium (I) with β-aminovinyl ketones by Ruballo Selina Cherkasova Varshavskii. Koord. Khimiya 1992, 17, 530–536. [Google Scholar]
- Galding, M.R.; Cherkasova, T.G.; Osetrova, L.V.; Varshavsky, Y.S. 31P NMR Spectra of beta-Aminovinylketonato Rhodium (I) Carbonyl-Phosphine Complexes Rh(AVK)(PPh3) (CO). Rhodium Express 1993, 1, 14–17. [Google Scholar]
- Ferreira, H.; Conradie, M.M.; van Rooyen, P.H.; Conradie, J. Packing polymorphism of dicarbonyl-[2-(phenylamino)pent-3-en-4-onato]rhodium(I). J. Organomet. Chem. 2017, 851, 235–247. [Google Scholar] [CrossRef]
- Bonati, F.; Wilkinson, G. 600. Dicarbonyl-β-diketonato- and related complexes of rhodium(I). J. Chem. Soc. 1964, 3156–3160. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. A kinetic study of the oxidative addition of methyl iodide to [Rh((C4H3S)COCHCOCF3)(CO)(PPh3)] utilizing UV/vis and IR spectrophotometry and 1H, 19F and 31P NMR spectroscopy. Synthesis of [Rh((C4H3S)COCHCOCF3)(CO)(PPh3)(CH3)(I)]. Inorg. Chim. Acta 2008, 361, 208–218. [Google Scholar] [CrossRef]
- Stuurman, N.F.; Conradie, J. Iodomethane oxidative addition and CO migratory insertion in monocarbonylphosphine complexes of the type [Rh((C6H5)COCHCO((CH2)nCH3))(CO)(PPh3)]: Steric and electronic effects. J. Organomet. Chem. 2009, 694, 259–268. [Google Scholar] [CrossRef]
- Varshavsky, Y.S.; Galding, M.R.; Cherkasova, T.G.; Podkorytov, I.S.; Nikol’skii, A.B.; Trzeciak, A.M.; Olejnik, Z.; Lis, T.; Ziółkowski, J.J. 31P-NMR and X-ray studies of new rhodium(I) β-ketoiminato complexes Rh(R1C(O)CHC(NH)R2)(CO)(PZ3) where PZ3=PPh3, PCy3, P(OPh)3 or P(NC4H4)3. J. Organomet. Chem. 2001, 628, 195–210. [Google Scholar] [CrossRef]
- Damoense, L.; Purcell, W.; Roodt, A.; Leipoldt, J.G. The Crystal Structure of (2-Aminovinyl-4-pentanonato-KO, KN)-carbonyl-triphenylphosphinerhodium (I). Rhodium Express 1994, 5, 10–13. [Google Scholar]
- Venter, G.J.S.; Steyl, G.; Roodt, A. Crystal structure of carbonyl-(4-(2,6-dichlorophenylamino)pent-3-en-2-onato-κ2N,O)-(triphenylphosphine-κP)-rhodium(I) acetone solvate, C31.5H28Cl2NO2.5PRh. Z. Krist.—New Cryst. Struct. 2013, 228, 410–412. [Google Scholar] [CrossRef][Green Version]
- Venter, G.J.S. The crystal structure of carbonyl-[4-(2,4-dichlorophenylamino)pent-3-en-2-onato-κ2N,O]-(triphenylphosphine-κP)rhodium(I), RhC30H25Cl2NO2P. Z. Krist.—New Cryst. Struct. 2017, 232, 901–903. [Google Scholar] [CrossRef]
- Venter, G.J.S.; Steyl, G.; Roodt, A. Carbonyl[4-(2,3-dimethylphenylamino)pent-3-en-2-onato-κ2N,O](triphenylphosphine-κP)rhodium(I). Acta Crystallogr. Sect. E Struct. Reports Online 2009, 65, m1321–m1322. [Google Scholar] [CrossRef]
- Venter, G.J.S.; Steyl, G.; Roodt, A. Carbonyl[4-(2,6-dimethylphenylamino)pent-3-en-2-onato-κ2N,O](triphenylphosphine-κP)rhodium(I) acetone hemisolvate. Acta Crystallogr. Sect. E Struct. Reports Online 2009, 65, m1606–m1607. [Google Scholar] [CrossRef]
- Conradie, J. Density functional theory calculations of Rh-β-diketonato complexes. Dalton Trans. 2015, 44, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Conradie, M.M.; Conradie, J. Methyl iodide oxidative addition to [Rh(acac)(CO)(PPh3)]: An experimental and theoretical study of the stereochemistry of the products and the reaction mechanism. Dalton Trans. 2011, 40, 8226–8237. [Google Scholar] [CrossRef]
- Conradie, J.; Lamprecht, G.J.; Otto, S.; Swarts, J.C. Synthesis and characterisation of ferrocene-containing β-diketonato complexes of rhodium(I) and rhodium(III). Inorg. Chim. Acta 2002, 328, 191–203. [Google Scholar] [CrossRef]
- Conradie, J.; Lamprecht, G.J.; Roodt, A.; Swarts, J.C. Kinetic study of the oxidative addition reaction between methyl iodide and [Rh(FcCOCHCOCF3)(CO)(PPh3)]: Structure of [Rh(FcCOCHCOCF3)(CO)(PPh3)(CH3)(I)]. Polyhedron 2007, 26, 5075–5087. [Google Scholar] [CrossRef]
- Wilson, J.M.; Sunley, G.J.; Adams, H.; Haynes, A. Oxidative addition of MeI to cationic Rh(I) carbonyl complexes with pyridyl bis(carbene) ligands. J. Organomet. Chem. 2005, 690, 6089–6095. [Google Scholar] [CrossRef]
- Van Zyl, G.J.; Lamprecht, G.J.; Leipoldt, J.G.; Swaddle, T.W. Kinetics and mechanism of the oxidative addition of iodomethane to β-diketonatobis(triphenylphosphite)rhodium(I) complexes. Inorg. Chim. Acta 1988, 143, 223–227. [Google Scholar] [CrossRef]
- Leipoldt, J.G.; Basson, S.S.; Botha, L.J. The oxidative addition of iodomethane to thioacetylacetonatocarbonylphosphine-rhodium(I) complexes. Inorg. Chim. Acta 1990, 168, 215–220. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. Stereochemistry of the reaction products of the oxidative addition reaction of methyl iodide to [Rh((C4H3S)COCHCOR)(CO)(PPh3)]: A NMR and computational study. R = CF3, C6H5, C4H3S. Inorg. Chim. Acta 2009, 362, 519–530. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. A density functional theory study of the oxidative addition of methyl iodide to square planar [Rh(acac)(P(OPh)3)2] complex and simplified model systems. J. Organomet. Chem. 2010, 695, 2126–2133. [Google Scholar] [CrossRef]
- Griffin, T.R.; Cook, D.B.; Haynes, A.; Pearson, J.M.; Monti, D.; Morris, G.E. Theoretical and Experimental Evidence for SN2 Transition States in Oxidative Addition of Methyl Iodide to cis-[M(CO)2I2]− (M = Rh, Ir). J. Am. Chem. Soc. 1996, 118, 3029–3030. [Google Scholar] [CrossRef]
- Feliz, M.; Freixa, Z.; van Leeuwen, P.W.N.M.; Bo, C. Revisiting the Methyl Iodide Oxidative Addition to Rhodium Complexes: A DFT Study of the Activation Parameters. Organometallics 2005, 24, 5718–5723. [Google Scholar] [CrossRef]
- Mateyise, N.G.S.; Conradie, J.; Conradie, M.M. Synthesis, characterization, electrochemistry, DFT and kinetic study of the oligothiophene-containing complex [Rh((C4H3S-C4H2S)COCHCOCF3)(CO)(PPh3)]. Polyhedron 2021, 199, 115095. [Google Scholar] [CrossRef]
- Basson, S.S.; Leipoldt, J.G.; Nel, J.T. The oxidative addition of methyl iodide to β-diketonecarbonyltriphenylphosphinerhodium(I) complexes. Inorg. Chim. Acta 1984, 84, 167–172. [Google Scholar] [CrossRef]
- Basson, S.S.; Leipoldt, J.G.; Roodt, A.; Venter, J.A.; van der Walt, T.J. The oxidative addition of iodomethane to acetylacetonatocarbonylphosphinerhodium(I) complexes. Inorg. Chim. Acta 1986, 119, 35–38. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999, 101, 396–408. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687, Erratum in: Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972–9981. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tuñon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]












| Concentration CH3I (mol dm−3) | Reaction Step | Wavelength (cm−1) | kobs (s−1) |
|---|---|---|---|
| [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B + CH3I | |||
| 0.02720 | Rh(I) loss | 1972.62 | 0.00720(5) |
| Rh(III)-alkyl gain | 2059.72 | 0.007(3) | |
| 0.05383 | Rh(I) loss | 1972.62 | 0.0132(2) |
| Rh(III)-alkyl gain | 2059.72 | 0.0124(1) | |
| 0.12780 | Rh(I) loss | 1972.62 | 0.02338(4) |
| Rh(III)-alkyl gain | 2059.72 | 0.0242(2) | |
| [Rh(CH3COCHCNHCH3)(CO)(PPh3)] 3A + CH3I | |||
| 0.0236 | Rh(I) loss | 1967.66 | 0.00227(5) |
| Rh(III)-alkyl gain | 2061.30 | 0.0077(4) | |
| Rh(III)-alkyl loss | 2061.30 | 0.00103(2) | |
| Rh(III)-acyl gain | 1713.74 | 0.0014(5) | |
| 0.0537 | Rh(I) loss | 1967.66 | 0.0059(1) |
| Rh(III)-alkyl gain | 2061.30 | 0.015(1) | |
| Rh(III)-alkyl loss | 2061.30 | 0.00174(3) | |
| Rh(III)-acyl gain | 1713.74 | 0.00185(3) | |
| 0.1283 | Rh(I) loss | 1967.66 | 0.0158(2) |
| Rh(III)-alkyl gain | 2061.30 | 0.029(1) | |
| Rh(III)-alkyl loss | 2061.30 | 0.00191(3) | |
| Rh(III)-acyl gain | 1713.74 | 0.00181(2) | |
| Complex | Temperature (K) | k2 (mol−1 dm3 s−1) |
|---|---|---|
| [Rh(CH3COCHCNHCH3)(CO)(PPh3)] 3A | 298 | 0.144(1) |
| [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B | 288 | 0.125(3) |
| 293 | 0.188(4) | |
| 298 | 0.213(5) | |
| 303 | 0.29(2) | |
| 308 | 0.44(2) |
| Reaction Step | Resonance Signal | ppm | kobs (s−1) | Average kobs (s−1) |
|---|---|---|---|---|
| [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B + 0.0502 M CH3I | ||||
| Rh(I)-loss (one isomer) | Methine | 5.018 | 0.0112(6) | 0.011(1) |
| O-CH3 | 1.567 | 0.0111(7) | ||
| N-CH3 | 1.688 | 0.0111(6) | ||
| Rh(III)-alkyl gain (one isomer) | Methine | 5.004 | 0.014(1) | 0.014(1) |
| O-CH3 | 1.655 | 0.014(1) | ||
| N-CH3 | 1.869 | 0.014(1) | ||
| [Rh(CH3COCHCNHCH3)(CO)(PPh3)] 3A + 0.0301 M CH3I | ||||
| Rh(I)-loss (two isomers) | Methine | 5.002 and 4.990 | 0.0040(2) | 0.0043(2) |
| O-CH3 | 1.583 (a) and 1.838 (b) | 0.0047(1) | ||
| N-CH3 | 2.056 (a) and 1.965 (b) | 0.0042(2) | ||
| Rh(III)-alkyl gain (one isomer) | Methine | 4.881 | - a | - a |
| O-CH3 | 2.017; 1.862 | - a | ||
| N-CH3 | - a | |||
| Rh(III)-alkyl loss (one isomer) | Methine | 4.881 | 0.00171(4) | 0.00170(4) |
| O-CH3 | 2.017; 1.862 | 0.00170(4) | ||
| N-CH3 | 0.00168(4) | |||
| Rh(III)-acyl gain (isomer A) b | Methine | 4.976 and 5.014 | 0.00157(2) | 0.00152(1) |
| O-CH3 | 2.157 (a) and 2.133 (b) | 0.00150(1) | ||
| N-CH3 | 2.861 (a) and 2.555 (b) | 0.00150(1) | ||
| Complex | Energy (eV) |
|---|---|
| [Rh(CH3COCHCNPhCH3)(CO)(PPh3)] 3B | |
| Reactants: 3B and CH3I | 0.54 |
| TS1 | 0.83 (imaginary frequency 270.0i cm−1) |
| Alkyl: [Rh(III)-alkyl 2a] | 0.00 |
| [Rh(CH3COCHCNHCH3)(CO)(PPh3)] 3A | |
| Reactants: 3A and CH3I | 0.82 |
| TS1 | 1.08 (imaginary frequency 266.0i cm−1) |
| Alkyl: [Rh(III)-alkyl product 2a] | 0.26 |
| TS2 | 1.17 (imaginary frequency 419.0i cm−1) |
| Acyl: [Rh(III)-acyl product 14a] | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, H.; Conradie, M.M.; Conradie, J. Kinetic Study of the Oxidative Addition Reaction between Methyl Iodide and [Rh(imino-β-diketonato)(CO)(PPh)3] Complexes, Utilizing UV–Vis, IR Spectrophotometry, NMR Spectroscopy and DFT Calculations. Molecules 2022, 27, 1931. https://doi.org/10.3390/molecules27061931
Ferreira H, Conradie MM, Conradie J. Kinetic Study of the Oxidative Addition Reaction between Methyl Iodide and [Rh(imino-β-diketonato)(CO)(PPh)3] Complexes, Utilizing UV–Vis, IR Spectrophotometry, NMR Spectroscopy and DFT Calculations. Molecules. 2022; 27(6):1931. https://doi.org/10.3390/molecules27061931
Chicago/Turabian StyleFerreira, Hendrik, Marrigje Marianne Conradie, and Jeanet Conradie. 2022. "Kinetic Study of the Oxidative Addition Reaction between Methyl Iodide and [Rh(imino-β-diketonato)(CO)(PPh)3] Complexes, Utilizing UV–Vis, IR Spectrophotometry, NMR Spectroscopy and DFT Calculations" Molecules 27, no. 6: 1931. https://doi.org/10.3390/molecules27061931
APA StyleFerreira, H., Conradie, M. M., & Conradie, J. (2022). Kinetic Study of the Oxidative Addition Reaction between Methyl Iodide and [Rh(imino-β-diketonato)(CO)(PPh)3] Complexes, Utilizing UV–Vis, IR Spectrophotometry, NMR Spectroscopy and DFT Calculations. Molecules, 27(6), 1931. https://doi.org/10.3390/molecules27061931