
Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. MTT Assay
2.4. Immunoblot Analysis
2.5. Reactive Oxygen Species (ROS) Production Measurement
2.6. Mitochondrial Membrane Potential (MMP) Measurement
2.7. Animals and Treatment
2.8. Statistical Analysis
3. Results
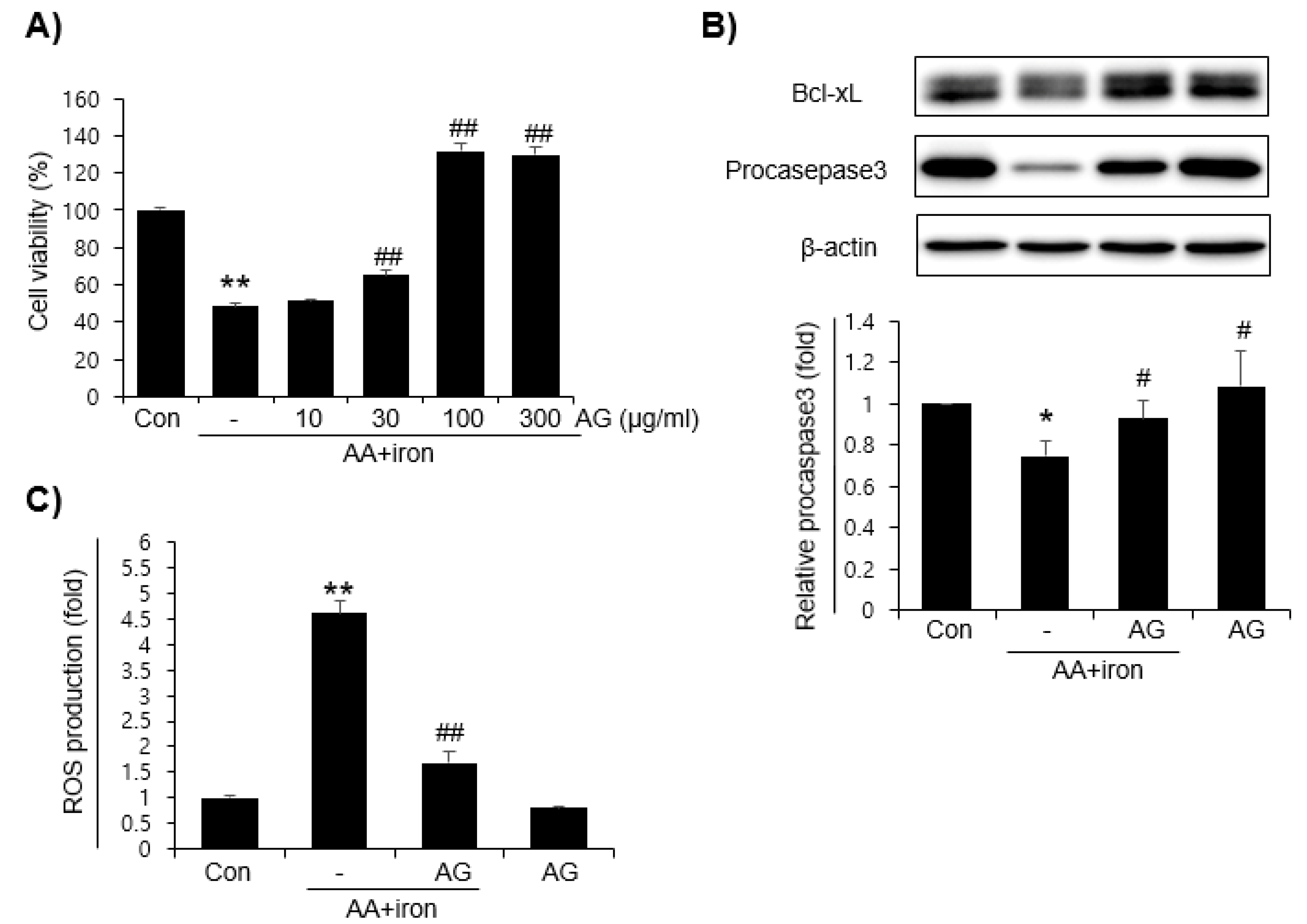
3.1. Effects of AG on AA+iron-Induced Oxidative Stress
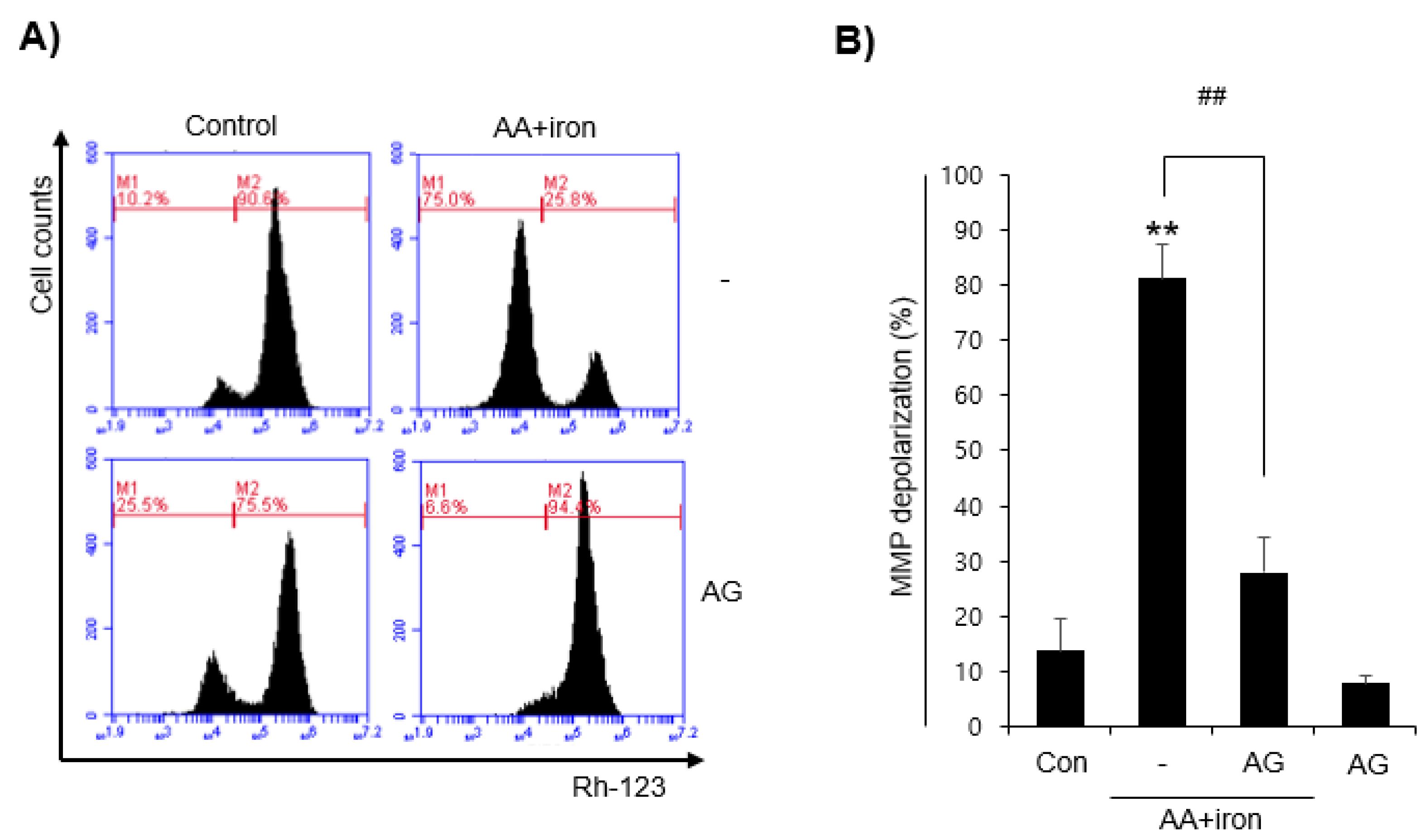
3.2. Effect of AG on Mitochondrial Damage
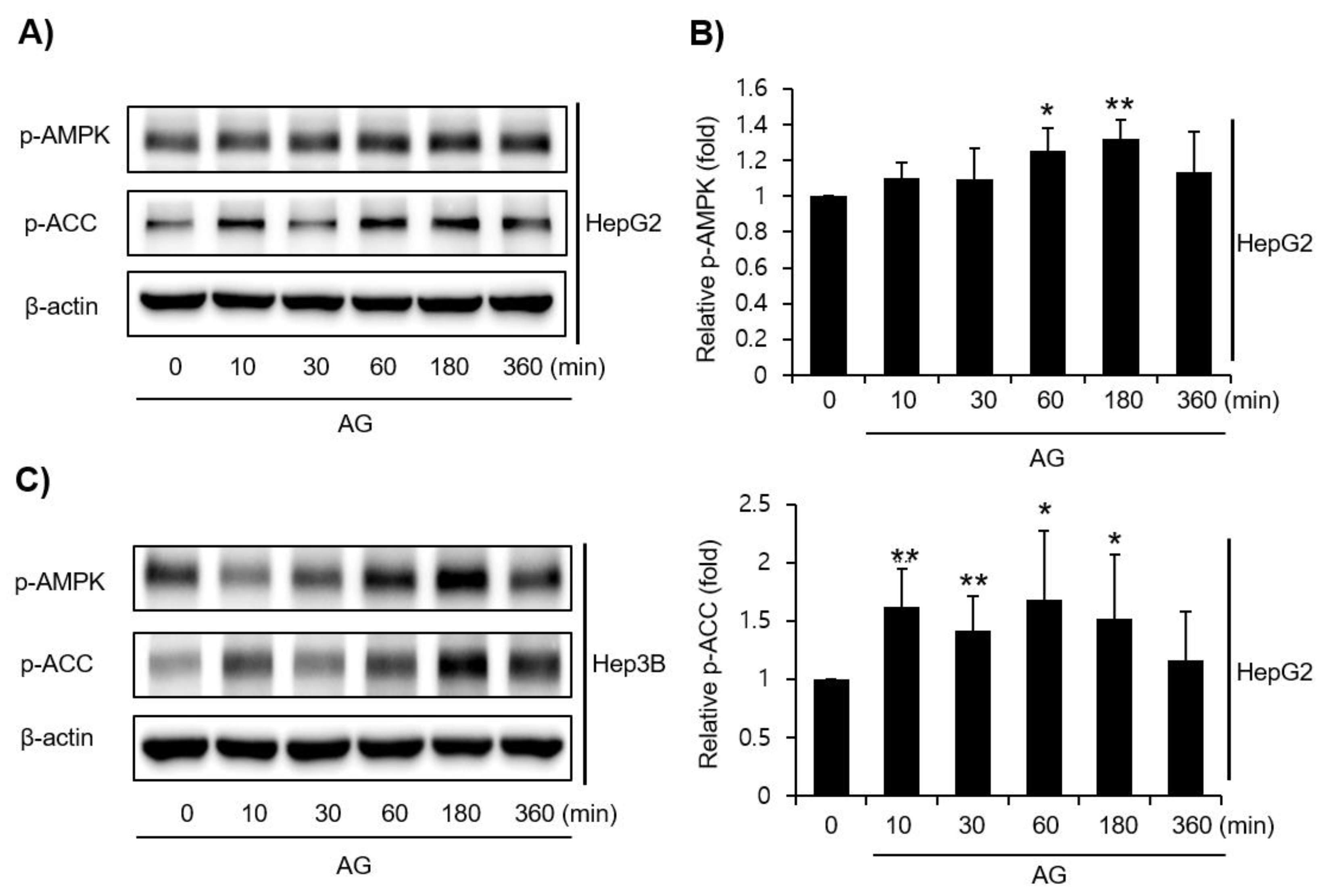
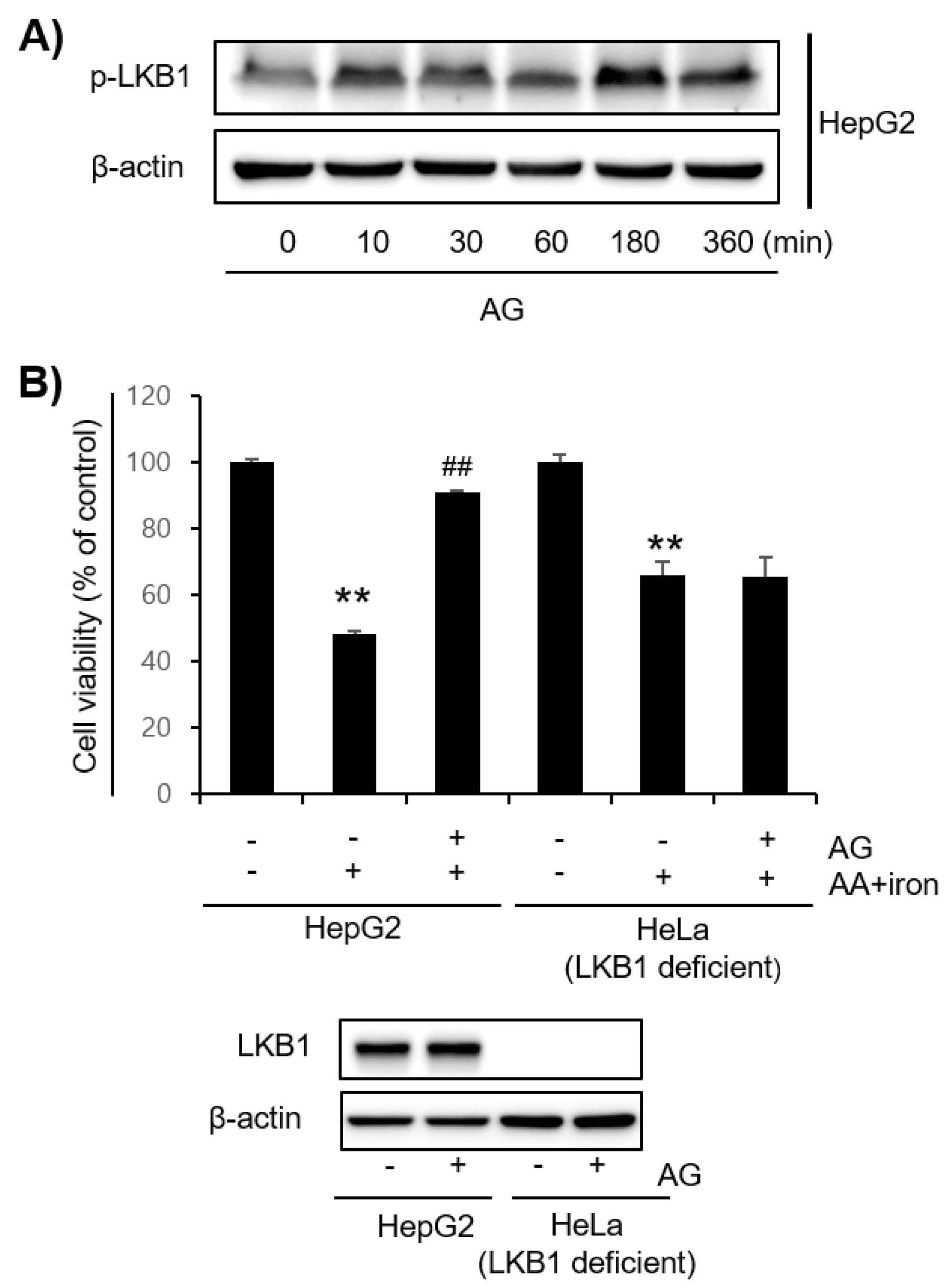
3.3. Effect of AG on LKB1-AMPK Pathway Activation
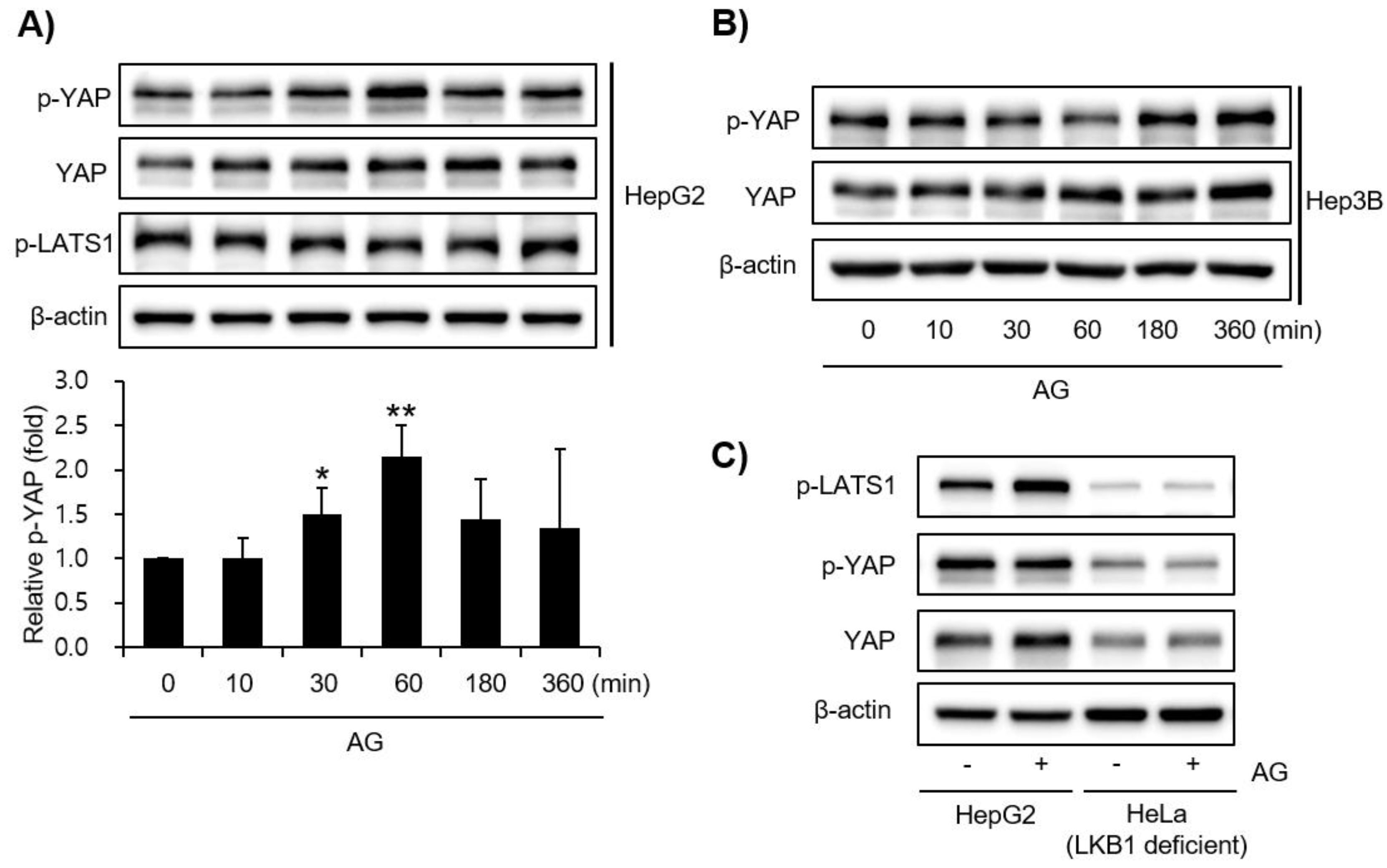
3.4. Effect of AG on YAP Signaling
3.5. Effect of Decursin on Oxidative Stress as an Active Component in AG
3.6. Effect of AG on CCl₄-Induced Liver Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Ota, T. Molecular mechanisms of nonalcoholic fatty liver disease (NAFLD)/Nonalcoholic Steatohepatitis (NASH). Adv. Exp. Med. Biol. 2021, 1261, 223–229. [Google Scholar] [PubMed]
- Raszeja-Wyszomirska, J.; Lawniczak, M.; Marlicz, W.; Miezyńska-Kurtycz, J.; Milkiewicz, P. Non-alcoholic fatty liver disease--new view. Pol. Merkur. Lekarski 2008, 24, 568–571. [Google Scholar] [PubMed]
- Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic acid as an early indicator of inflammation during non-alcoholic fatty liver disease development. Biomolecules 2020, 10, 1133. [Google Scholar] [CrossRef]
- Engin, A. Non-alcoholic fatty liver disease. Adv. Exp. Med. Biol. 2017, 960, 443–467. [Google Scholar]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [Green Version]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Mrad, M.B.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef]
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postepy Hig. Med. Dosw. 2016, 70, 1150–1165. [Google Scholar] [CrossRef]
- Dunn, J.D.; Alvarez, L.A.; Zhang, X. Thierry Soldati, reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Lee, S.M.; Shin, S.M.; Hwang, S.J.; Brooks, J.S.; Kang, H.E.; Lee, M.G.; Kim, S.C.; Kim, S.G. Efficacy of sauchinone as a novel AMPK-activating lignan for preventing iron-induced oxidative stress and liver injury. Free Radic. Biol. Med. 2009, 47, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Fasciolo, G.; Meo, S.D.; Venditti, P. Vitamin E supplementation and mitochondria in experimental and functional hyperthyroidism: A mini-review. Nutrients 2019, 11, 2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.H.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Kim, H.M.; Lee, K.H.; Kim, K.Y.; Huang, D.S.; Kim, J.H.; Seong, R.S. Quantitative analysis of marker compounds in Angelica gigas, Angelica sinensis, and Angelica acutiloba by HPLC/DAD. Chem. Pharm. Bull. 2015, 63, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Mahat, B.; Chae, J.W.; Baek, I.H.; Song, G.Y.; Song, J.S.; Cho, S.K.; Kwon, K.I. Physicochemical characterization and toxicity of decursin and their derivatives from Angelica gigas. Biol. Pharm. Bull. 2012, 35, 1084–1090. [Google Scholar] [CrossRef] [Green Version]
- Bae, U.J.; Choi, E.K.; Oh, M.R.; Jung, S.J.; Park, J.; Jung, T.S.; Park, T.S.; Chae, S.W.; Park, B.H. Angelica gigas Ameliorates Hyperglycemia and Hepatic Steatosis in C57BL/KsJ-db/db Mice via Activation of AMP-Activated Protein Kinase Signaling Pathway. Am. J. Chin. Med. 2016, 44, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.J.; Kwon, M.R.; Chung, M.S. Pilot-scale subcritical-water extraction of nodakenin and decursin from Angelica gigas Nakai. Food Sci. Biotechnol. 2019, 29, 631–639. [Google Scholar] [CrossRef]
- Jang, E.J.; Kim, S.C.; Lee, J.H.; Lee, J.R.; Kim, I.K.; Baek, S.Y.; Kim, Y.W. Fucoxanthin, the constituent of Laminaria japonica, triggers AMPK-mediated cytoprotection and autophagy in hepatocytes under oxidative stress. BMC Complement. Altern. Med. 2018, 18, 97. [Google Scholar] [CrossRef]
- Lee, E.H.; Baek, S.Y.; Park, J.Y.; Kim, Y.W. Emodin in Rheum undulatum inhibits oxidative stress in the liver via AMPK with Hippo/Yap signalling pathway. Pharm. Biol. 2020, 58, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.Y.; Lee, J.H.; Jegal, K.H.; Cho, I.J.; Kim, Y.W.; Kim, S.C. Oxyresveratrol abrogates oxidative stress by activating ERK-Nrf2 pathway in the liver. Chem. Biol. Interact. 2016, 245, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Baek, S.Y.; Park, J.Y.; Kim, Y.W. Rifampicin activates AMPK and alleviates oxidative stress in the liver as mediated with Nrf2 signaling. Chem. Biol. Interact. 2020, 315, 108889. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ko, S.M.; Choi, E.J.; Ham, S.H.; Kwon, Y.D.; Lee, Y.B.; Cho, H.Y. Simultaneous determination of decursin, decursinol angelate, nodakenin, and decursinol of angelica gigas nakai in human plasma by UHPLC-MS/MS: Application to pharmacokinetic study. Molecules 2018, 23, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Balboa, M.A.; Balsinde, J. Oxidative stress and arachidonic acid mobilization. Biochim. Biophys. Acta 2006, 1761, 385–391. [Google Scholar] [CrossRef]
- Turlin, B.; Deugnier, Y. Iron overload disorders. Clin. Liver Dis. 2002, 6, 481–496. [Google Scholar] [CrossRef]
- Fleming, R.E.; Bacon, B.R. Orchestration of iron homeostasis. N. Engl. J. Med. 2005, 352, 1741–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, J.W.; Searle, J. Hepatic iron deposition in human disease and animal models. Biometals 1996, 9, 205–209. [Google Scholar] [CrossRef]
- Mattera, R.; Stone, G.P.; Bahhur, N.; Kuryshev, Y.A. Increased release of arachidonic acid and eicosanoids in iron-overloaded cardiomyocytes. Circulation 2001, 103, 2395–2401. [Google Scholar] [CrossRef]
- Tadolini, B.; Hakim, G. The mechanism of iron (III) stimulation of lipid peroxidation. Free Radic. Res. 1996, 25, 221–227. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Synergistic toxicity of iron and arachidonic acid in HepG2 cells overexpressing CYP2E1. Mol. Pharmacol. 2001, 60, 742–752. [Google Scholar] [PubMed]
- Shin, S.M.; Kim, S.G. Inhibition of arachidonic acid and iron-induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1,2-dithiole-3-thione congeners. Mol. Pharmacol. 2009, 75, 242–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Saltiel, A.R. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J. Biol. Chem. 2020, 295, 12279–12289. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Colombo, S.L.; Moncada, S. AMPK alpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem. Pharmacol. 2013, 86, 200–209. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef]
- Li, Y.; Ding, H.; Dong, J.; Rahman, S.U.; Feng, S.; Wang, X.; Wu, J.; Wang, Z.; Liu, G.; Li, X.; et al. Glucagon attenuates lipid accumulation in cow hepatocytes through AMPK signaling pathway activation. J. Cell Physiol. 2019, 234, 6054–6066. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, N.; Varela-Rey, M.; Fernández-Ramos, D.; Woodhoo, A.; Vázquez-Chantada, M.; Embade, N.; Espinosa-Hevia, L.; Bustamante, F.J.; Parada, L.A.; Rodriguez, M.S.; et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010, 52, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, U.J.; Bae, S.J.; Song, Y.R.; Kim, Y.W. A Critical YAP in Malignancy of HCC Is Regulated by Evodiamine. Int. J. Mol. Sci. 2022, 23, 1855. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, Y.W.; Shin, S.M.; Kim, C.W.; Yu, I.J.; Kim, S.G. Synergistic hepatotoxicity of N, N-dimethylformamide with carbon tetrachloride in association with endoplasmic reticulum stress. Chem. Biol. Interact. 2010, 184, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, D.; Lin, L.; Jiang, R.; Dai, J.; Tang, L.; Yang, Y.; Ge, P.; Wang, B.; Zhang, L. Potential roles of AMP-activated protein kinase in liver regeneration in mice with acute liver injury. Mol. Med. Rep. 2018, 17, 5390–5395. [Google Scholar] [CrossRef] [Green Version]
- Clawson, G.A. Mechanisms of carbon tetrachloride hepatotoxicity. Pathol. Immunopathol. Res. 1989, 8, 104–112. [Google Scholar] [CrossRef]
- Brattin, W.J.; Glende, E.A., Jr.; Recknagel, R.O. Pathological mechanisms in carbon tetrachloride hepatotoxicity. J. Free Radic. Biol. Med. 1985, 1, 27–38. [Google Scholar] [CrossRef]
- Naji, K.M.; Al-Shaibani, E.S.; Alhadi, F.A.; Al-Soudi, S.A.; D’Souza, M.R. Hepatoprotective and antioxidant effects of single clove garlic against CCl4-induced hepatic damage in rabbits. BMC Complement. Altern. Med. 2017, 17, 411. [Google Scholar] [CrossRef]
- Sahreen, S.; Khan, M.R.; Khan, R.A. Hepatoprotective effects of methanol extract of Carissa opaca leaves on CCl4-induced damage in rat. BMC Complement. Altern. Med. 2011, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Boll, M.; Weber, L.W.; Becker, E.; Stampfl, A. Mechanism of carbon tetrachloride-induced hepatotoxicity. Hepatocellular damage by reactive carbon tetrachloride metabolites. Z. Nat. C. J. Biosci. 2001, 56, 649–659. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.-R.; Jang, B.; Lee, S.-M.; Bae, S.-J.; Bak, S.-B.; Kim, Y.-W. Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling. Molecules 2022, 27, 1858. https://doi.org/10.3390/molecules27061858
Song Y-R, Jang B, Lee S-M, Bae S-J, Bak S-B, Kim Y-W. Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling. Molecules. 2022; 27(6):1858. https://doi.org/10.3390/molecules27061858
Chicago/Turabian StyleSong, Yu-Rim, Boyun Jang, Sung-Min Lee, Su-Jin Bae, Seon-Been Bak, and Young-Woo Kim. 2022. "Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling" Molecules 27, no. 6: 1858. https://doi.org/10.3390/molecules27061858
APA StyleSong, Y.-R., Jang, B., Lee, S.-M., Bae, S.-J., Bak, S.-B., & Kim, Y.-W. (2022). Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling. Molecules, 27(6), 1858. https://doi.org/10.3390/molecules27061858