
Products of Photo- and Thermochemical Rearrangement of 19-Membered di-tert-Butyl-Azoxybenzocrown

 ,
,  ,
,
Abstract
:
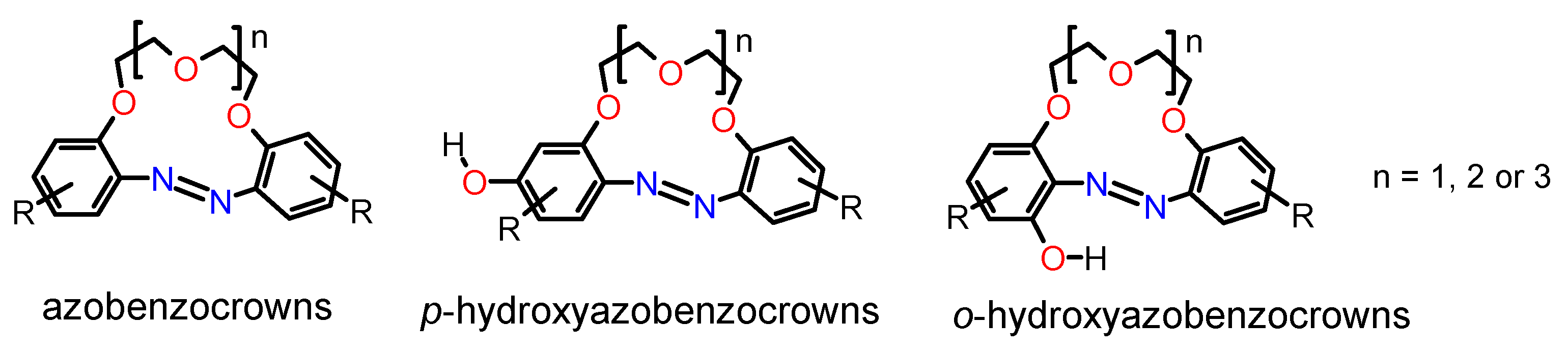
1. Introduction
2. Results and Discussion
2.1. Rearrangement of a 19-Membered Azoxybenzocrown with tert-Butyl Substituents in Para Positions to the Oligoether Linkage (Meta to Azoxy Group)
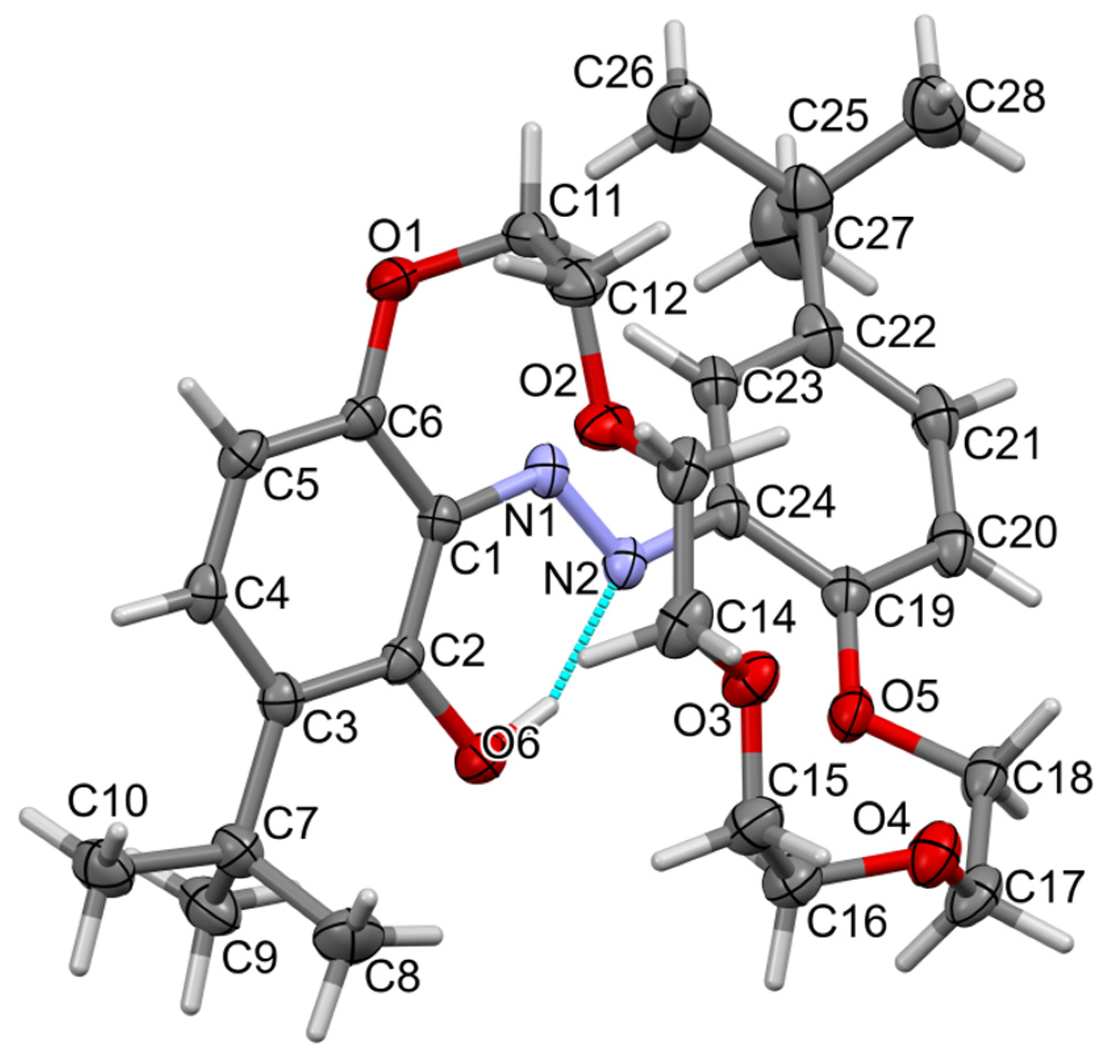
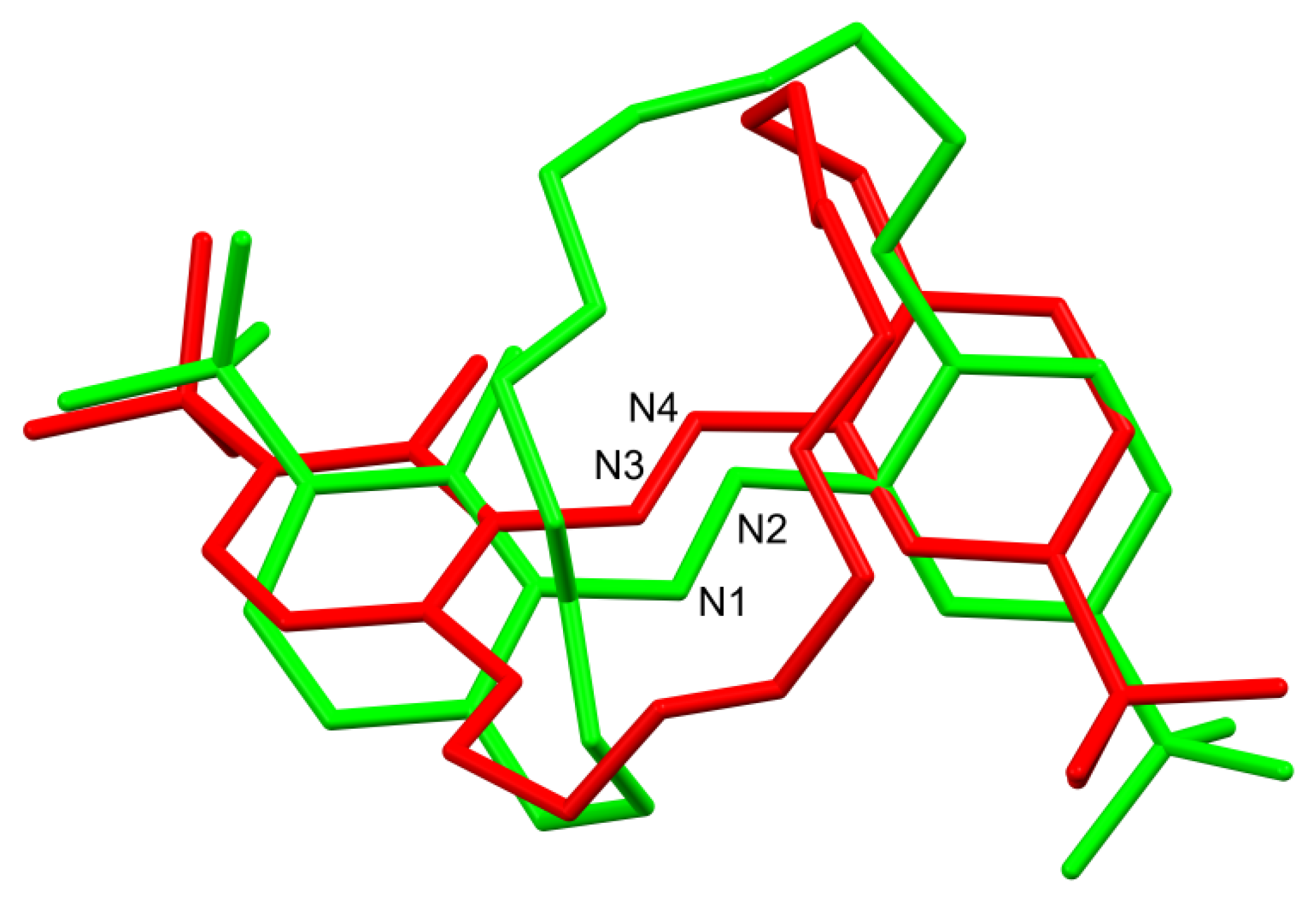
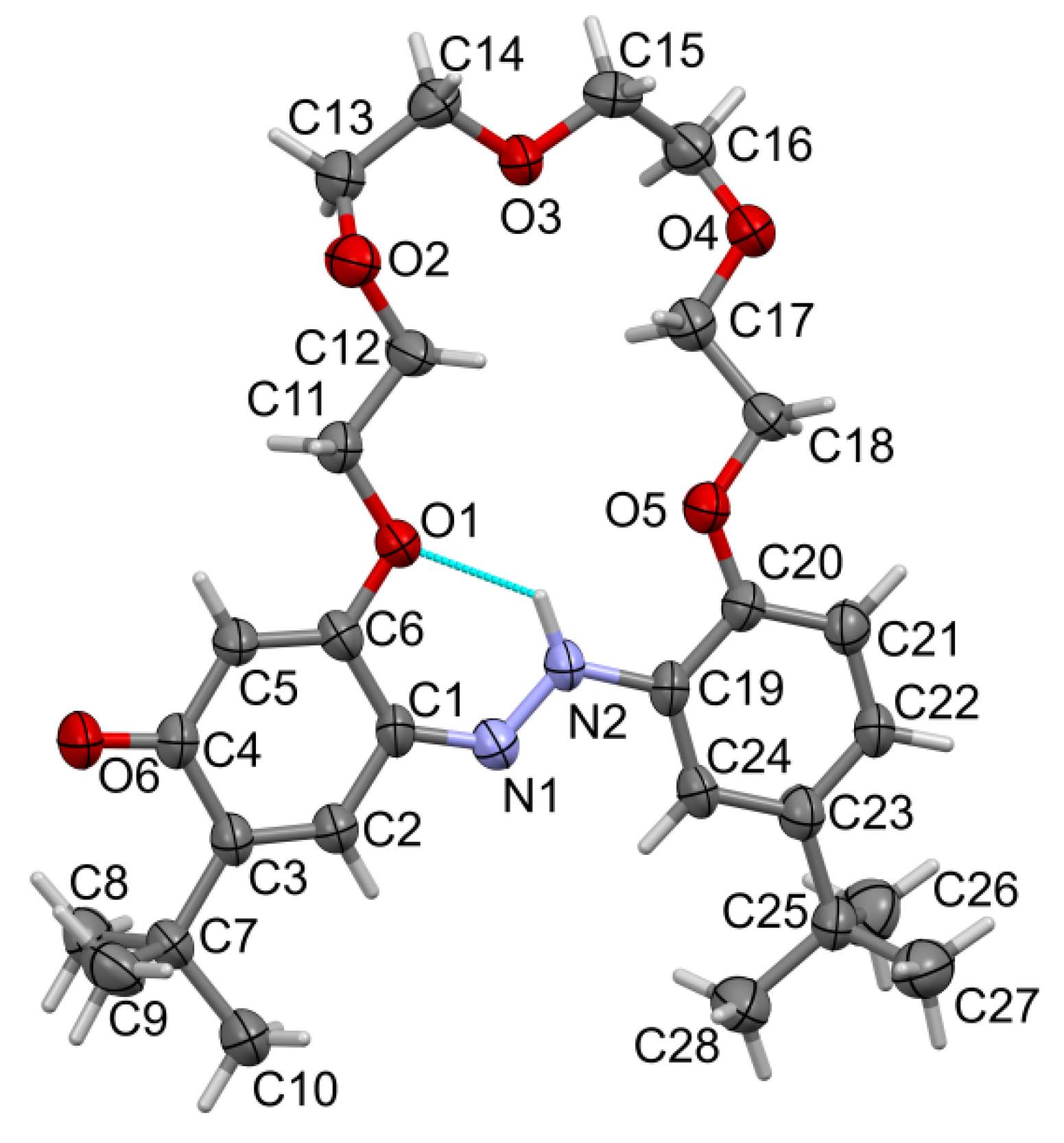
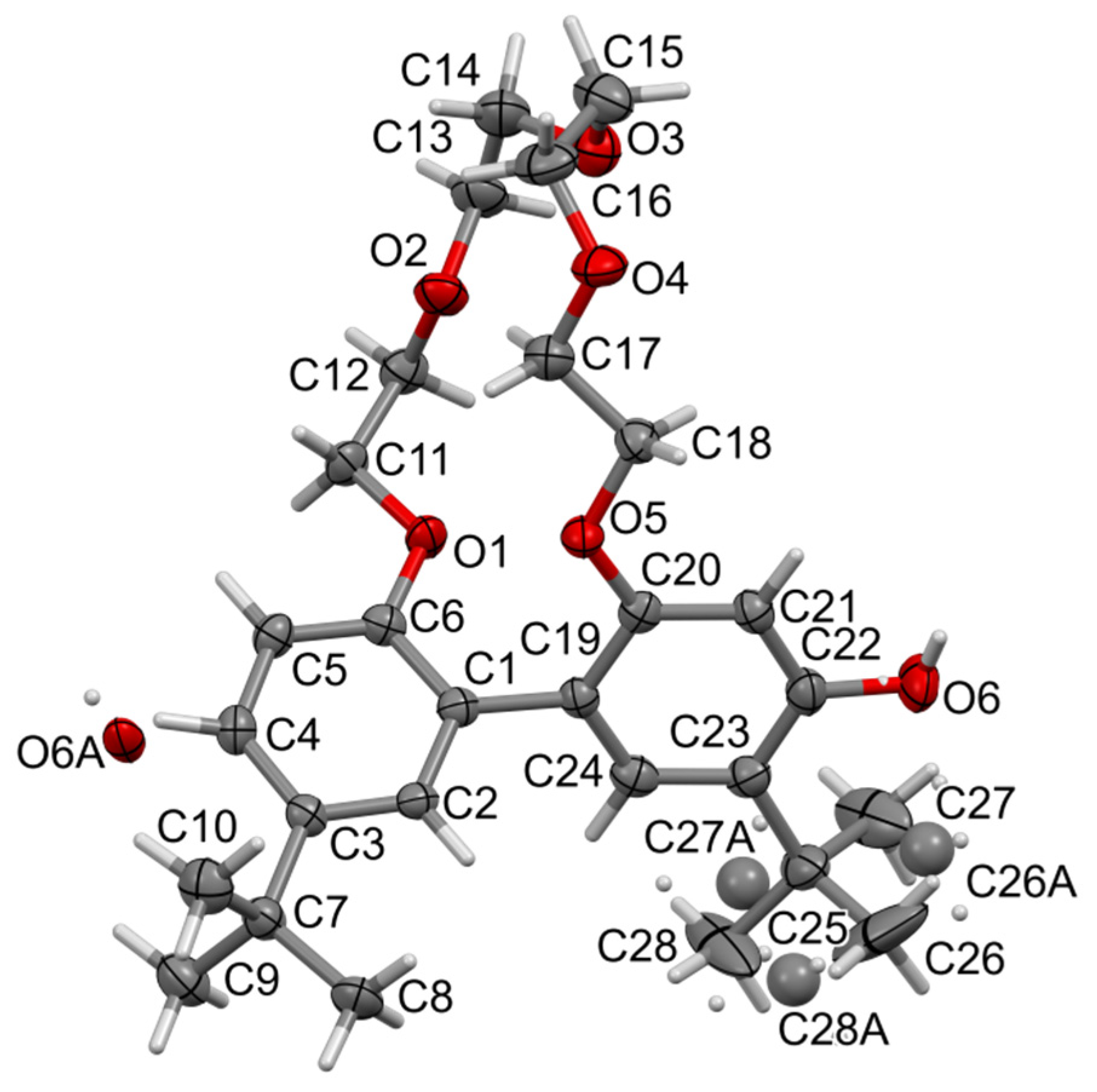
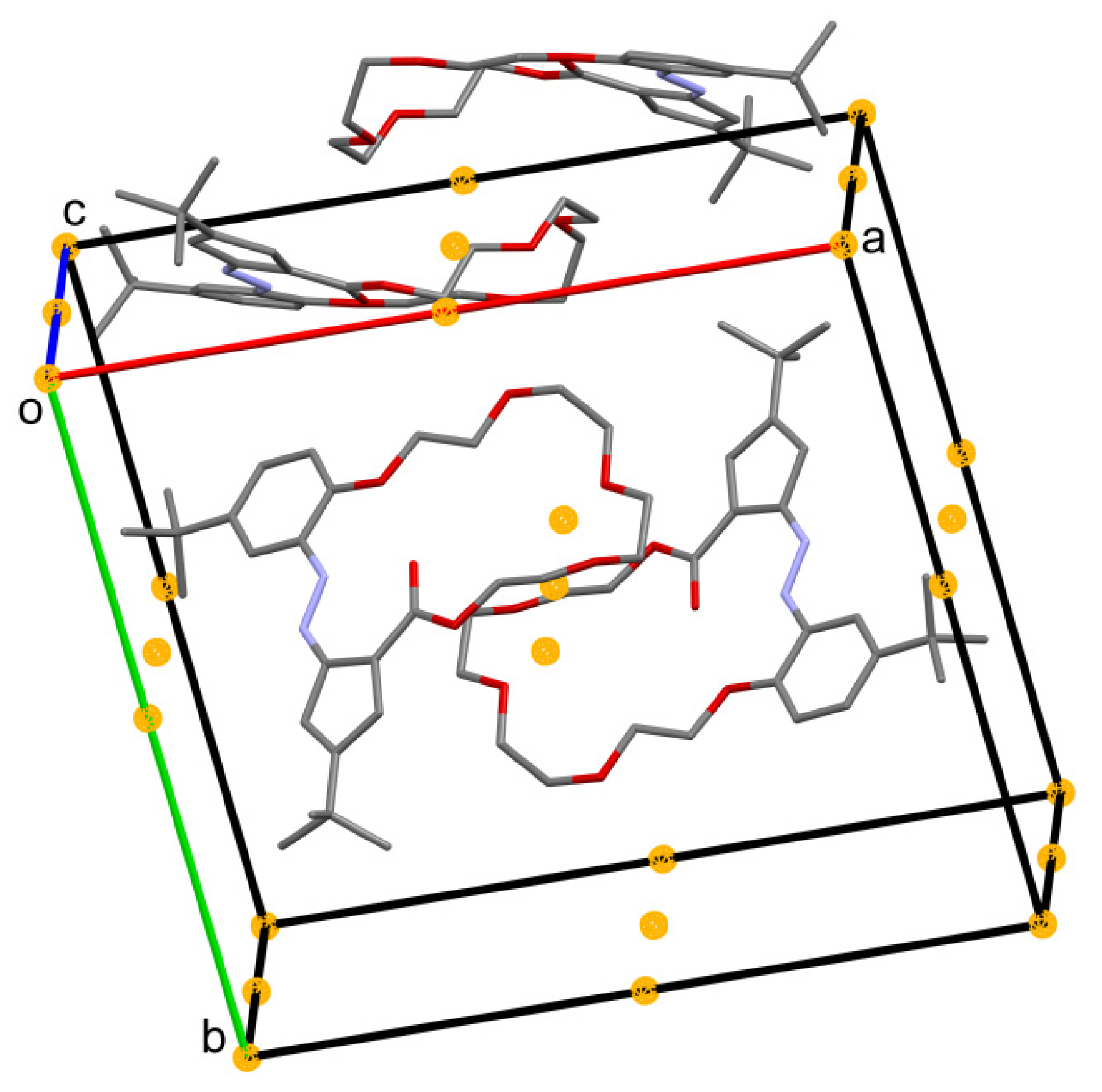
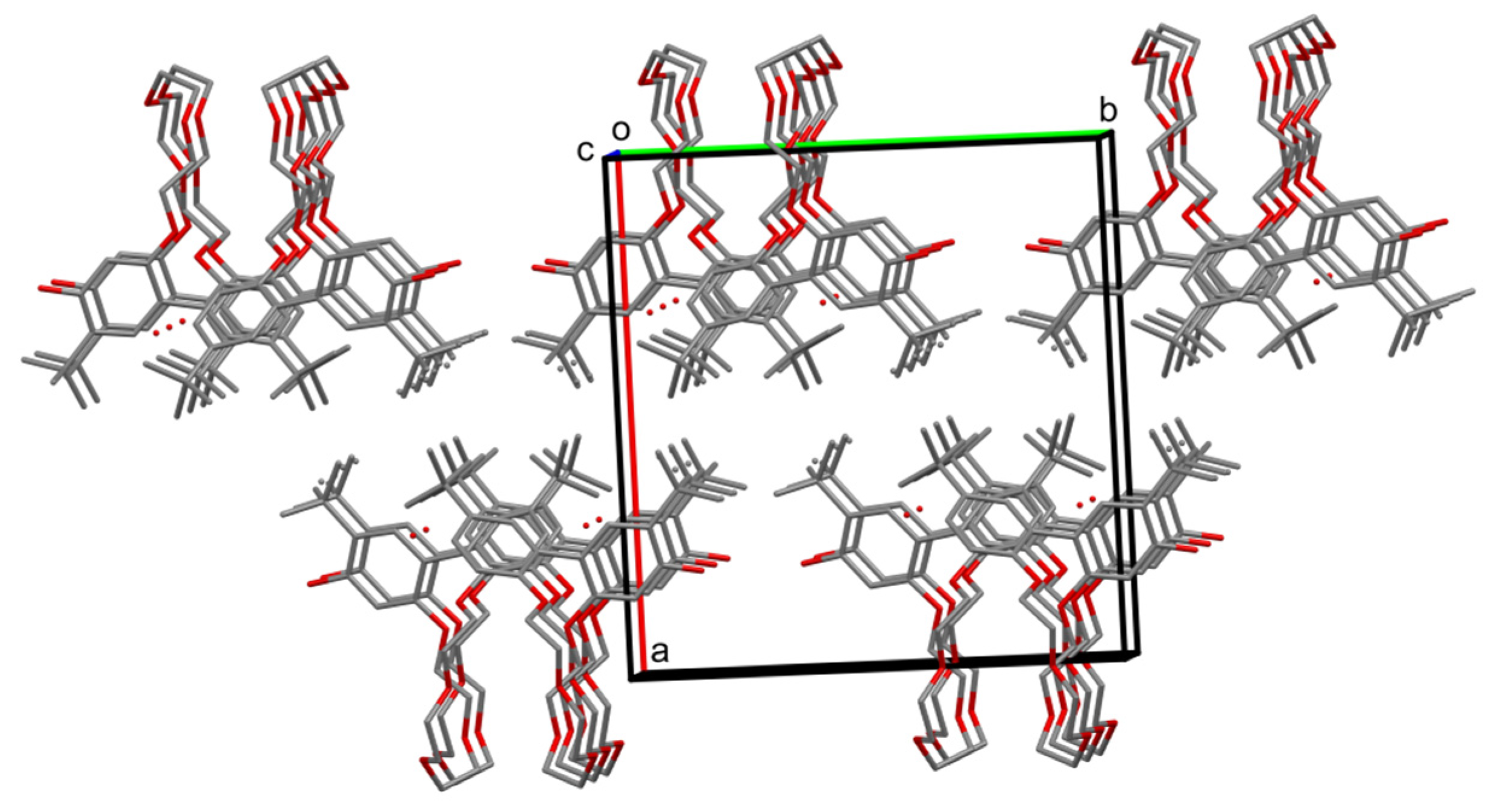
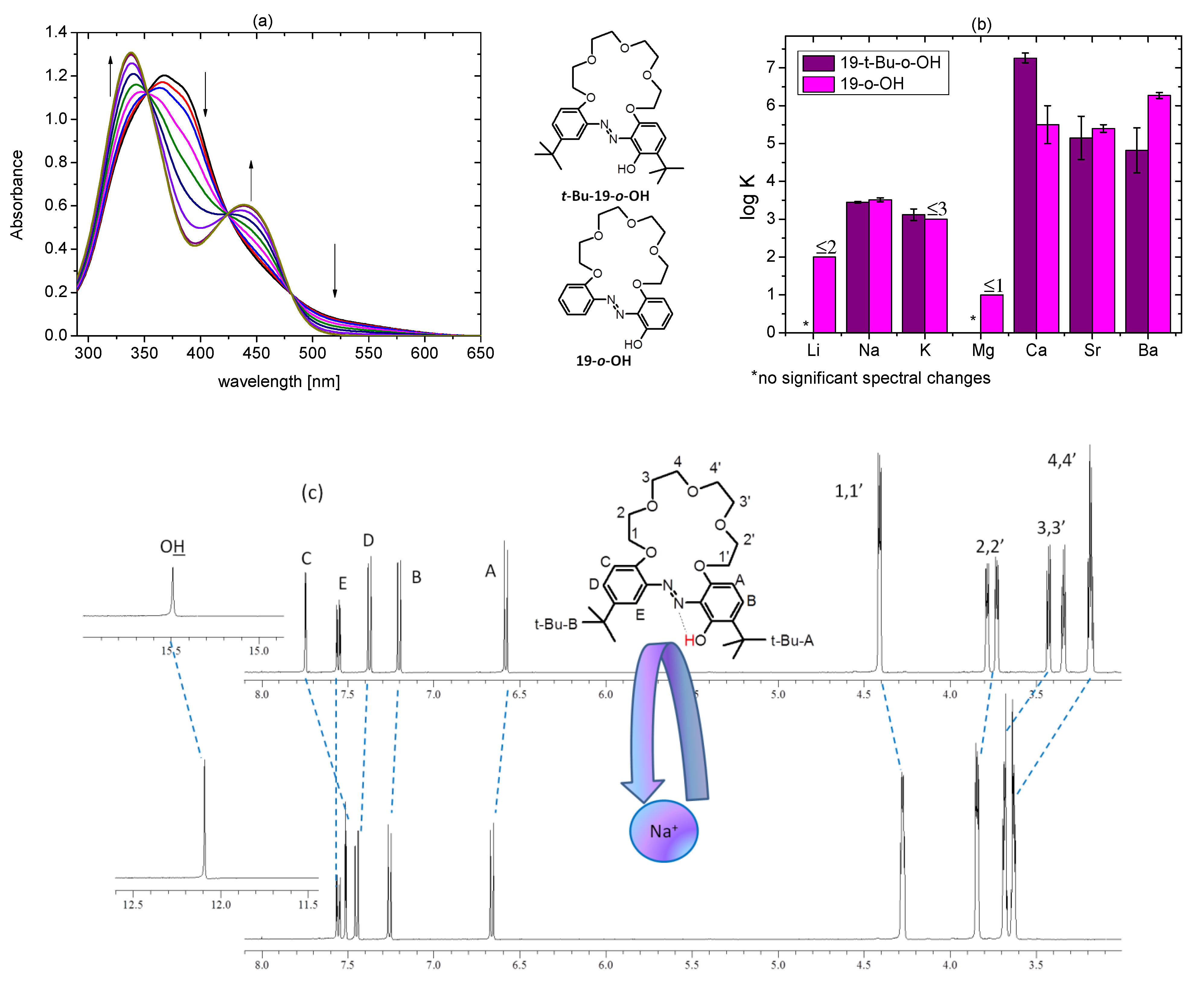
2.2. X-ray Structures of Newly Obtained Macrocycles
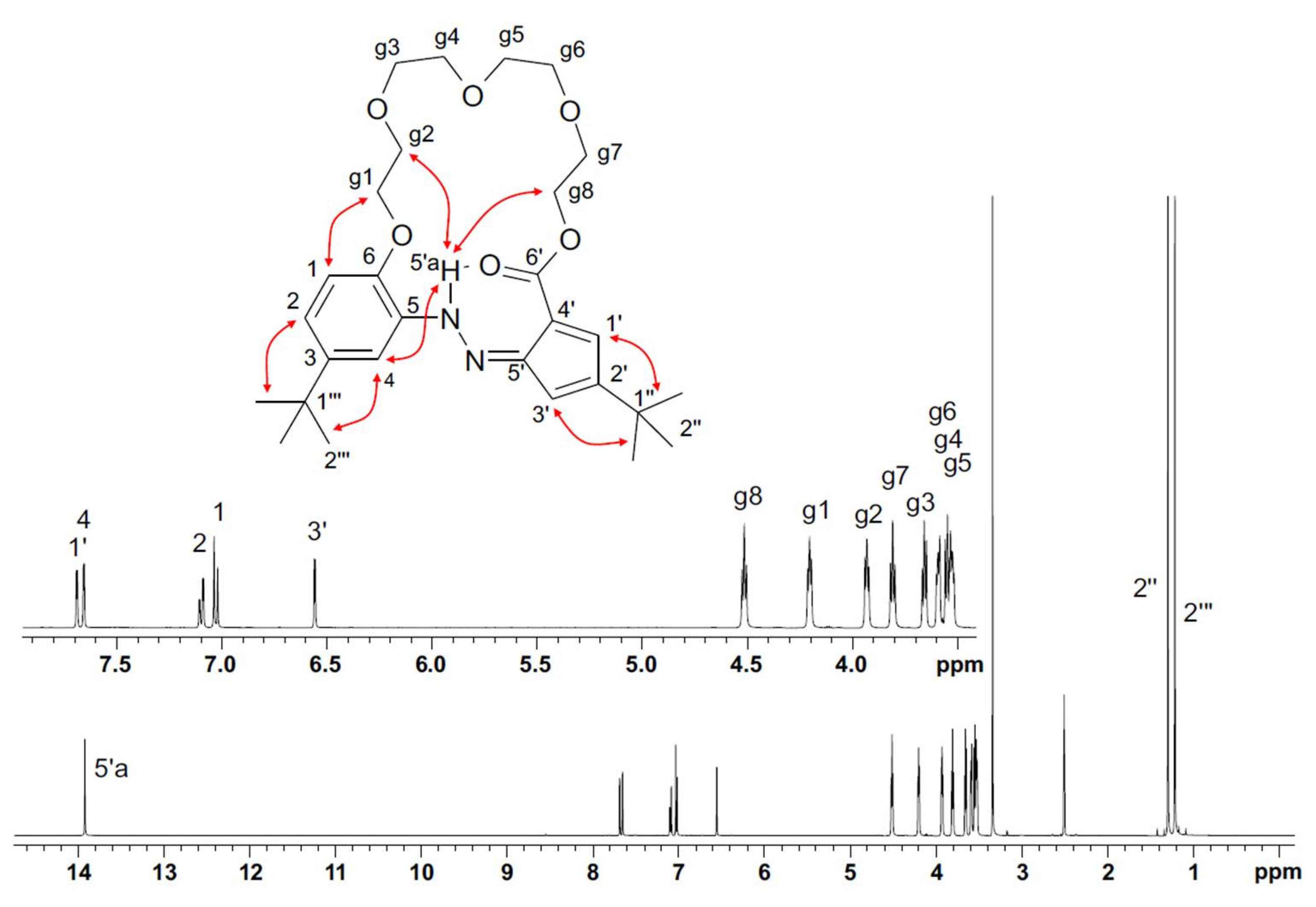
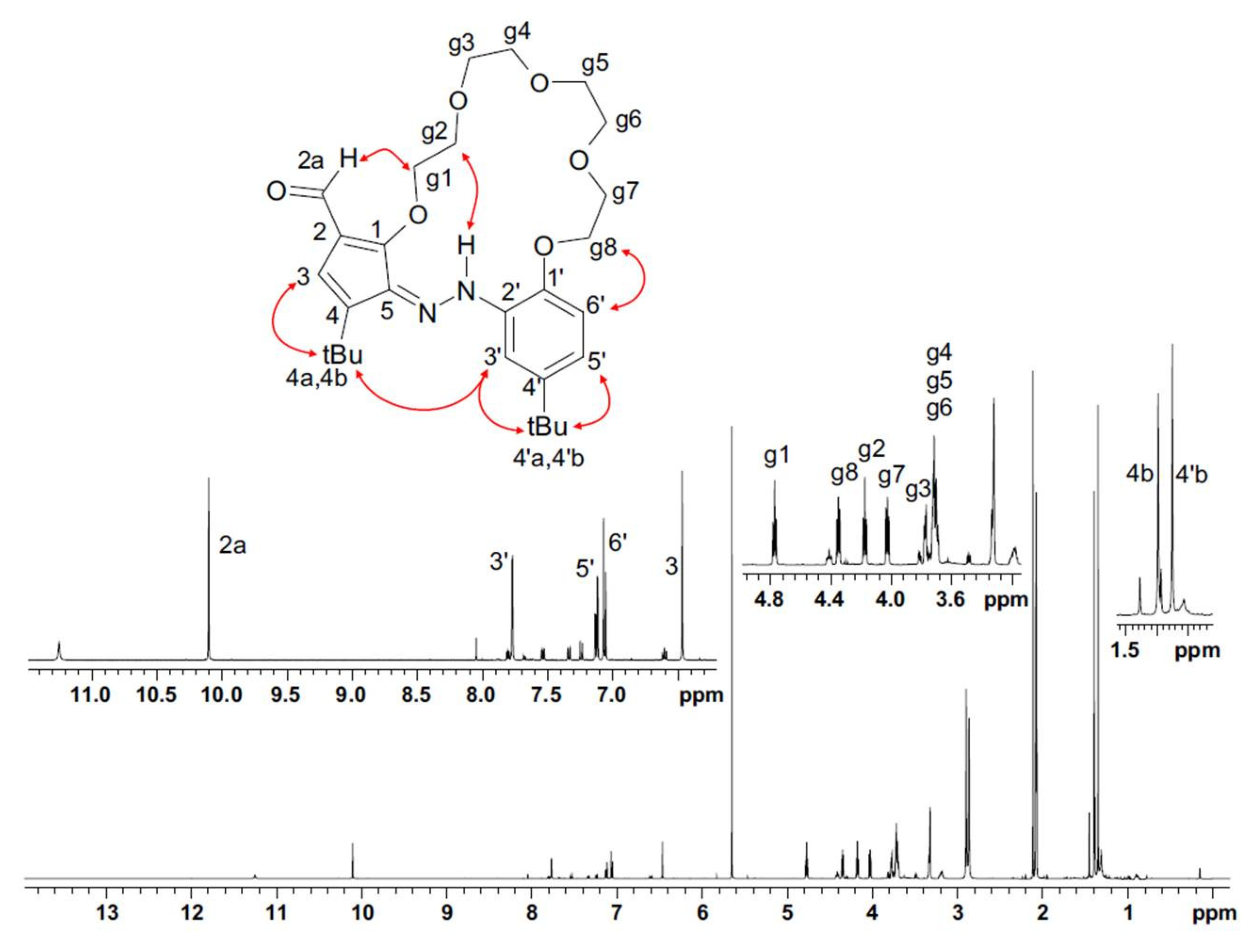
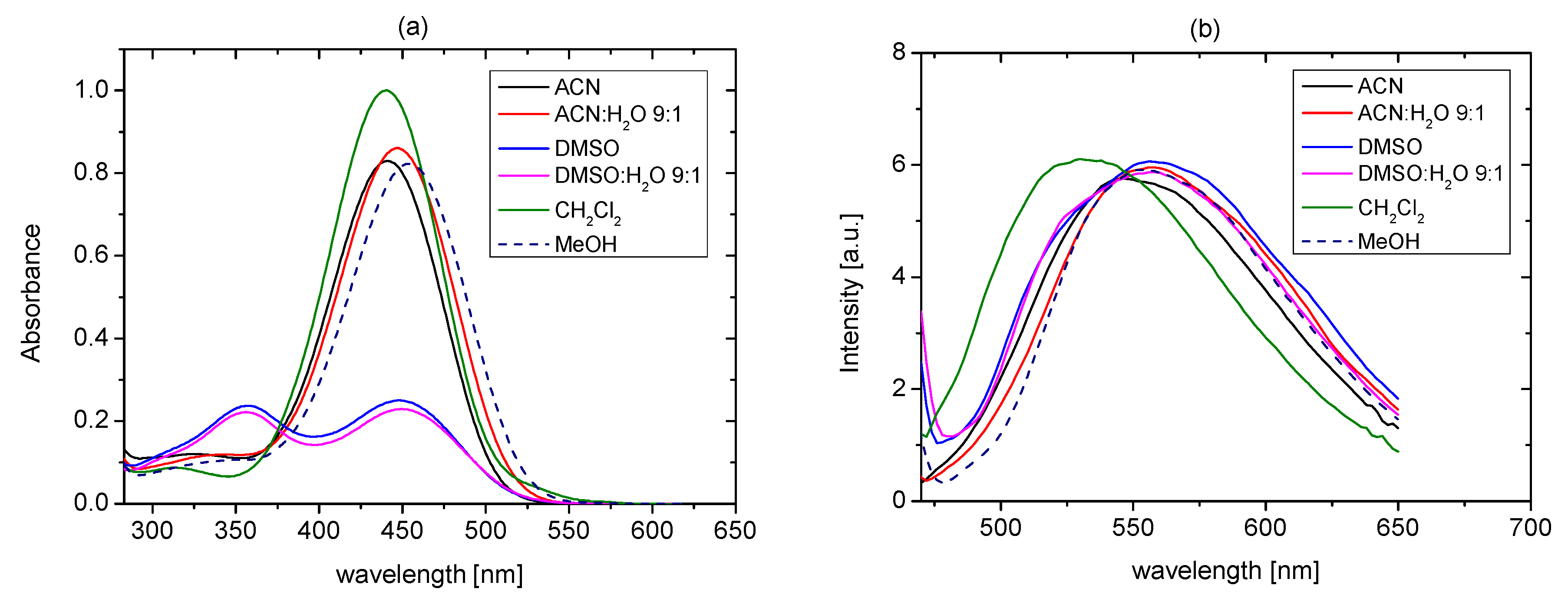
2.3. Studies of the Structures in Solution
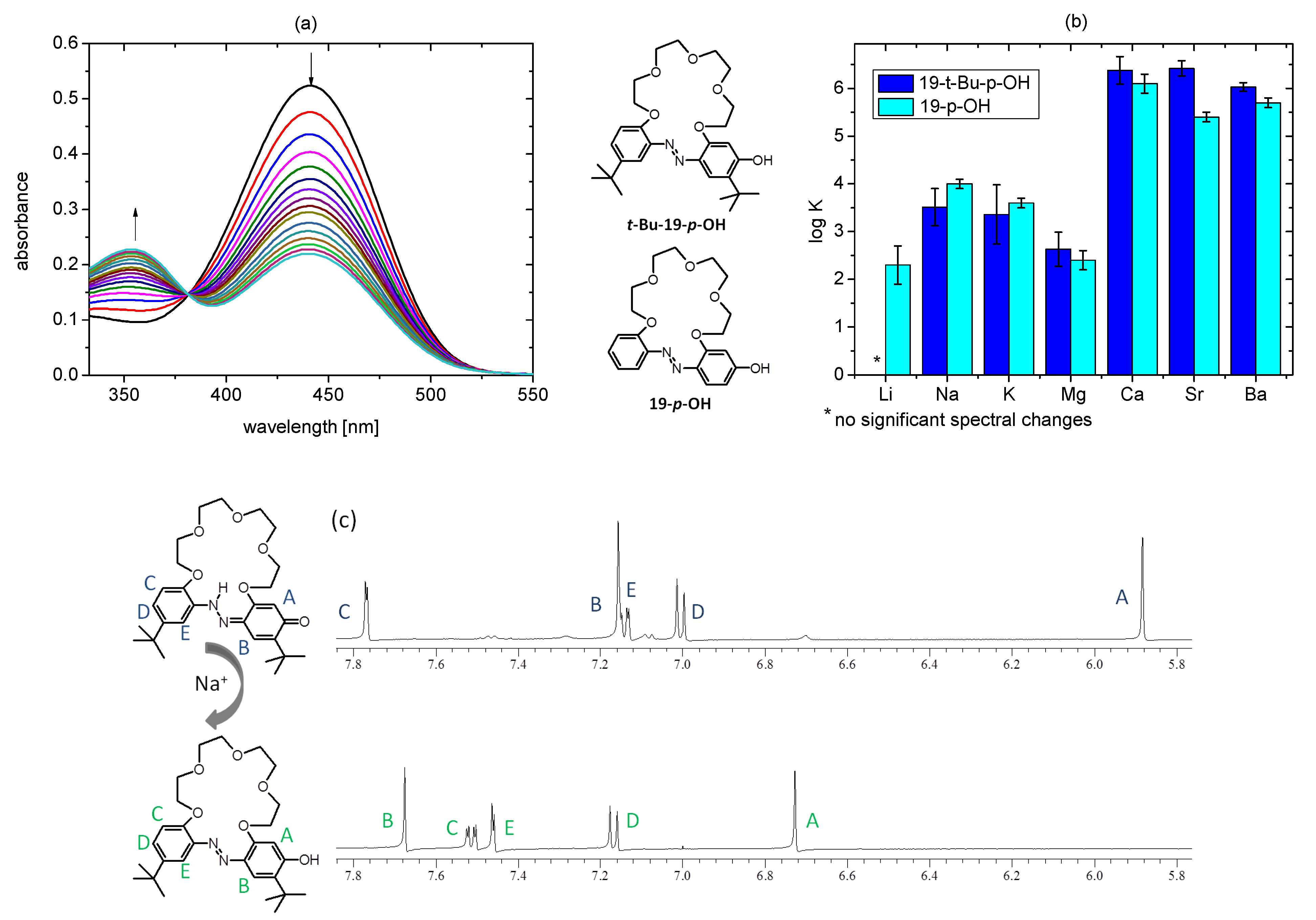
2.4. Tautomeric Equilibrium of Hydroxyazobenzocrowns
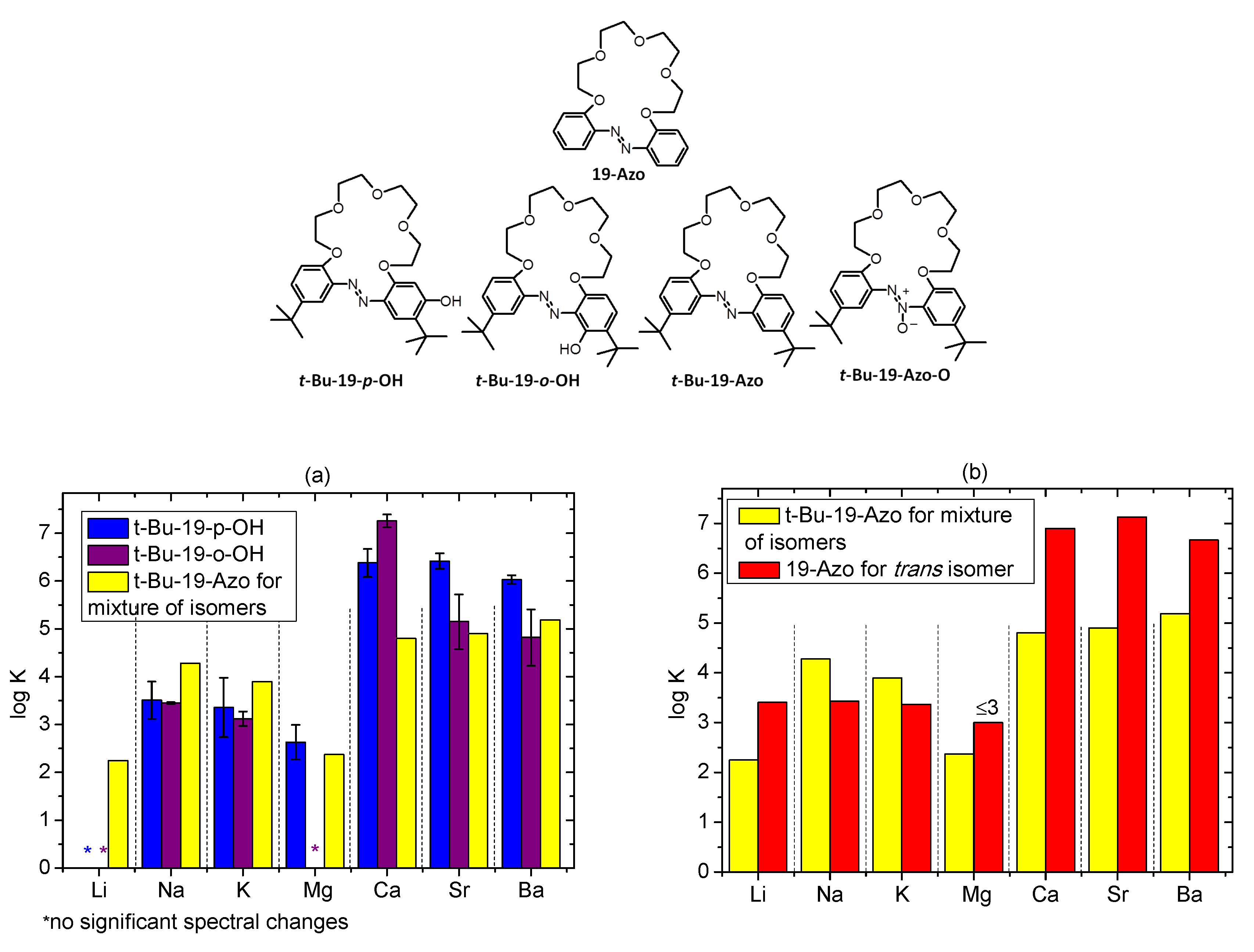
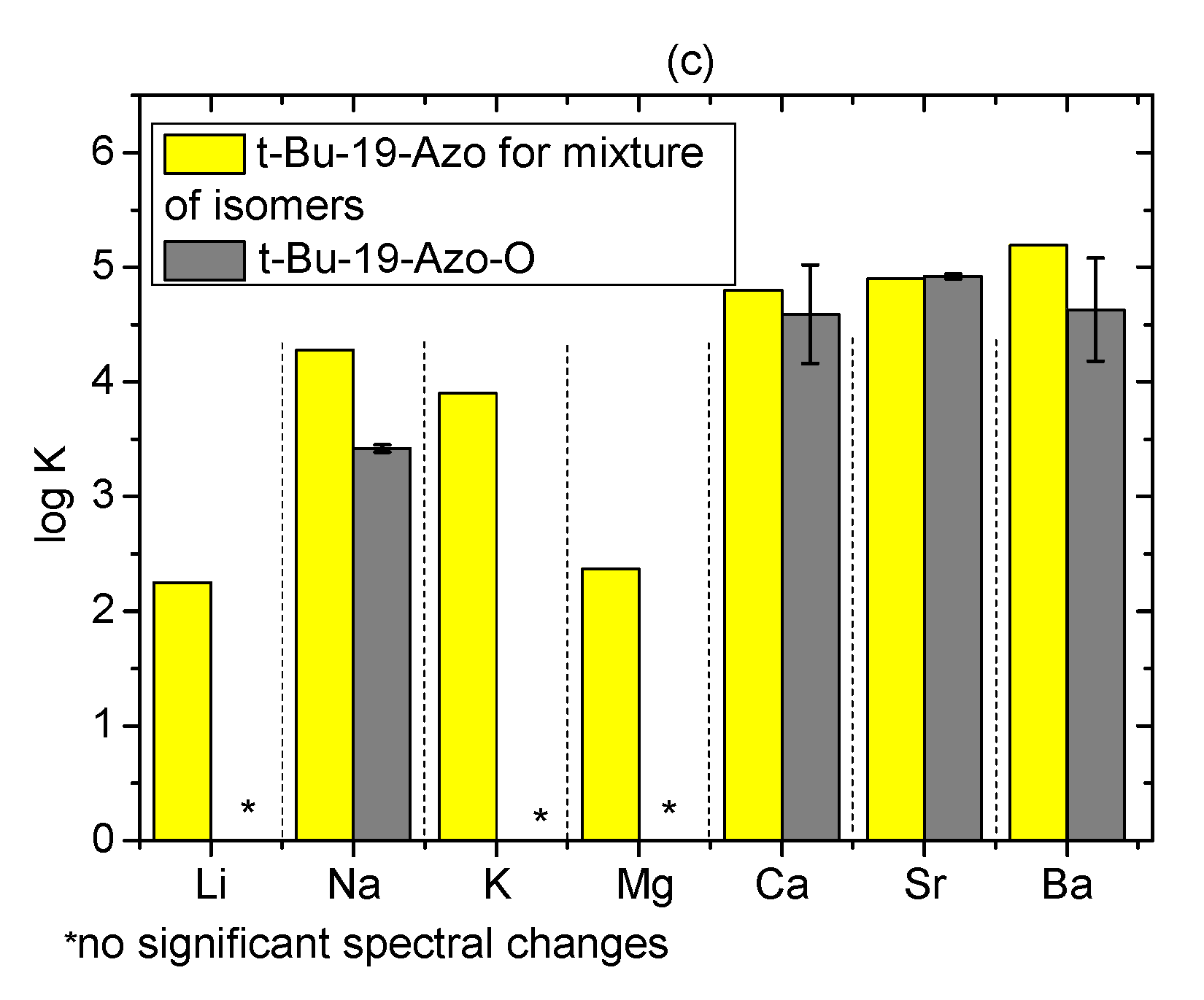
2.5. Metal Cations Complexation in Solution
3. Materials and Methods
3.1. General Information
3.2. Preparation
3.2.1. Preparation of Substrate for Rearrangements–di-tert-Butyl-19-Azoxybenzocrown (t-Bu-19-Azo-O)
3.2.2. Photochemical Rearrangement of t-Bu-19-Azo-O
3.2.3. Thermochemical Rearrangement of t-Bu-19-Azo-O
3.2.4. Spectral Characterization of the Products of Photochemical and Thermochemical Rearrangements of t-Bu-19-Azo-O
3.3. X-ray Crystal Structure Determination
3.4. Structure Determination of t-Bu-20-ester, t-Bu-19-al and t-Bu-19-p-OH in Solutions
- NMR of t-Bu-20-ester
- NMR of t-Bu-19-al
- NMR of t-Bu-19-p-OH
3.5. Studies of Crown–Metal Cations Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yue, L.; Yang, K.; Lou, X.Y.; Yang, Y.W.; Wang, R. Versatile roles of macrocycles in organic-inorganic hybrid materials for biomedical applications. Matter 2020, 3, 1557–1588. [Google Scholar] [CrossRef]
- Sabah, K.J.; Zahid, N.I.; Hashim, R. Synthesis of new chiral macrocycles-based glycolipids and its application in asymmetric Michael addition. Res. Chem. Intermed. 2021, 47, 2653–2667. [Google Scholar] [CrossRef]
- Shang, J.; Liu, Y.; Pan, T. Macrocycles in bioinspired catalysis: From molecules to materials. Front. Chem. 2021, 9, 635315. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Song, Z.; Yu, L.; Ye, S.; He, H. Fluorescent probes based on macrocyclic hosts: Construction, mechanism and analytical applications. Trends Anal. Chem. 2020, 133, 116086. [Google Scholar] [CrossRef]
- Tay, H.M.; Beer, P. Optical sensing of anions by macrocyclic and interlocked hosts. Org. Biomol. Chem. 2021, 19, 4652–4677. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, Z.; Yuan, L. Recent advances of photoresponsive supramolecular switches. Asian J. Chem. 2021, 10, 74–90. [Google Scholar] [CrossRef]
- Sokołowska, P.; Dąbrowa, K.; Jarosz, S. Visible-Light Responsive Sucrose-Containing Macrocyclic Host for Cations. Org. Lett. 2021, 23, 2687–2692. [Google Scholar] [CrossRef]
- Luboch, E.; Bilewicz, R.; Kowalczyk, M.; Wagner-Wysiecka, E.; Biernat, J.F. Azo Macrocyclic Compounds. In Advances in Supramolecular Chemistry; Gokel, G.W., Ed.; Cerberus Press: South Miami, FL, USA, 2003; Volume 9, pp. 71–162. [Google Scholar]
- Wagner-Wysiecka, E.; Łukasik, N.; Biernat, J.F.; Luboch, E. Azo group(s) in selected macrocyclic compounds. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 189–257. [Google Scholar] [CrossRef] [Green Version]
- Benkhaya, S.; M’rabet, S.; El Harfi, A. Classifications, properties, recent synthesis and applications of azo dyes. Heliyon 2020, 6, e03271. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Qi, D.; Li, J. Design, synthesis and applications of responsive macrocycles. Commun. Chem. 2020, 3, 189. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Liu, P.; Zhu, H.; Shi, B.; Hong, X.; Huang, F. Azobenzene-based macrocyclic arenes: Synthesis, crystal structures, and light-controlled molecular encapsulation and release. Angew. Chem. 2021, 8, 5830–5834. [Google Scholar] [CrossRef]
- Alehashem, M.S.; Ariffin, A.B.; Savage, P.B.; Dabdawb, W.A.Y.; Thomas, N.F. Treasures old and new: What we can learn regarding the macrocyclic problem from past and present efforts in natural product total synthesis. RSC Adv. 2020, 10, 10989–11012. [Google Scholar] [CrossRef]
- Shiga, M.; Nakamura, H.; Takagi, M.; Ueno, K. Synthesis of azobenzo-crown ethers and their complexation behavior with metal ions. Bull. Chem. Soc. Jpn. 1984, 57, 412–415. [Google Scholar] [CrossRef]
- Tahara, R.; Morozumi, T.; Nakamura, H.; Shimomura, M. Photoisomerization of azobenzocrown ethers. Effect of complexation of alkaline earth metal ions. J. Phys. Chem. B. 1997, 101, 7736–7743. [Google Scholar] [CrossRef]
- Simonov, Y.A.; Luboch, E.; Biernat, J.F.; Bolotina, N.V.; Zavodnik, V.E. Inclusion compounds of NaI with 13-membered azo- and azoxycrown ethers. J. Incl. Phenom. Macrocycl. Chem. 1997, 28, 17–32. [Google Scholar] [CrossRef]
- Skwierawska, A.; Luboch, E.; Biernat, J.F.; Kravtsov, V.C.; Simonov, Y.A.; Dvorkin, A.A.; Bel’skii, V.K. Stereochemistry of 16-membered azo- and azoxycrown ethers. Structures of their sandwich potassium iodide complexes. J. Incl. Phenom. Macrocycl. Chem. 1998, 31, 71–86. [Google Scholar] [CrossRef]
- Luboch, E.; Biernat, J.F.; Kravtsov, V.C.; Simonov, Y.A. 13-Membered azocrown ether. Structure of the lithium bromide complex and membrane properties. J. Incl. Phenom. Macrocycl. Chem. 1998, 31, 109–118. [Google Scholar] [CrossRef]
- Luboch, E.; Biernat, J.F.; Kravtsov, V.C.; Simonov, Y.A.; Belskii, V.K. Structures of NaI complexes of 16-membered azo- and azoxycrown ethers. Correlation of crystal structure and carrier-doped membrane electrode selectivity. Supramol. Chem. 1999, 11, 109–118. [Google Scholar] [CrossRef]
- Luboch, E.; Wagner-Wysiecka, E.; Poleska-Muchlado, Z.; Kravtsov, V.C. Synthesis and properties of azobenzocrown ethers with pi-electron donor, or pi-electron donor and pi-electron acceptor group(s) on benzene rings. Tetrahedron 2005, 61, 10738–10747. [Google Scholar] [CrossRef]
- Luboch, E. The Wallach rearrangement as a method for the synthesis of functionalized azobenzocrown ethers. Pol. J. Chem. 2008, 82, 1315–1318. [Google Scholar]
- Luboch, E.; Wagner-Wysiecka, E.; Rzymowski, T. 4-Hexylresorcinol-derived hydroxyazobenzocrown ethers as chromoionophores. Tetrahedron 2009, 65, 10671–10678. [Google Scholar] [CrossRef]
- Szarmach, M.; Wagner-Wysiecka, E.; Fonari, M.S.; Luboch, E. Bis(azobenzocrown ether)s—Synthesis and ionophoric properties. Tetrahedron 2012, 68, 507–515. [Google Scholar] [CrossRef]
- Szarmach, M.; Wagner-Wysiecka, E.; Luboch, E. Rearrangement of azoxybenzocrowns into chromophoric hydroxyazobenzocrowns and the use of hydroxyazobenzocrowns for the synthesis of ionophoric biscrown compounds. Tetrahedron 2013, 69, 10893–10905. [Google Scholar] [CrossRef]
- Wagner-Wysiecka, E.; Szarmach, M.; Chojnacki, J.; Łukasik, N.; Luboch, E. Cation sensing by diphenyl-azobenzocrowns. J. Photochem. Photobiol. A—Chem. 2017, 333, 220–232. [Google Scholar] [CrossRef]
- Wagner-Wysiecka, E.; Szulc, P.; Luboch, E.; Chojnacki, J.; Szwarc-Karabyka, K.; Łukasik, N.; Murawski, M.; Kosno, M. Photochemical rearrangement of a 19-membered azoxybenzocrown: Products and their properties. ChemPlusChem 2020, 85, 2067–2083. [Google Scholar] [CrossRef]
- Wallach, O.; Belli, L. Ueber die Umwandlung von Azoxybenzol in Oxyazobenzol. Ber. Dtsch. Chem. Ges. 1880, 13, 525–527. [Google Scholar] [CrossRef] [Green Version]
- Badger, G.M.; Buttery, R.G. Aromatic azo-compounds. Part VI. The action of light on azoxy-compounds. J. Chem. Soc. 1954, 2243–2245. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Agadzhanyan, T.E.; Maimind, V.I.; Kudryavtsev, R.V. Study of the mechanism of the para and ortho rearrangements of substituted azoxy compounds. Russ. Chem. Bull. 1963, 12, 1216–1219. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Schroder, A.; Mohanty, D.K. Azoxyaromatic polyethers. Polymer 1997, 38, 239–243. [Google Scholar] [CrossRef]
- Yamamoto, J.; Hamada, R.; Tsuboi, T. Photochemical reaction of azoxybenzenes in the presence of acetic acid. Nippon Kagaku Kaishi 2002, 3, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, J.; Sakamoto, T.; Kusunoki, K.; Umezu, M.; Matsuura, T. The Wallach Rearrangement of a few azoxy compounds. Nippon Kagaku Kaishi 1977, 1, 66–71. [Google Scholar] [CrossRef]
- Bunce, N.J. On the involvement of diazonium ions in the photorearrangement of azoxybenzene. Can. J. Chem. 1977, 55, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Skwierawska, A.M.; Biernat, J.F.; Kravtsov, V.C. Synthesis and electrode properties of 19-membered azo- and azoxycrown ethers. Structure of dibenzo-19-azocrown-7. Tetrahedron 2006, 62, 149–154. [Google Scholar] [CrossRef]
- Coppens, P. The evaluation of absorption and extinction in single-crystal structure analysis. In Crystallographic Computing; Ahmed, F.R., Ed.; Munksgaard: Copenhagen, Denmark, 1970; pp. 255–270. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Kyvala, M.; Lukes, I. Program package “OPIUM”. Available online: https://web.natur.cuni.cz/~kyvala/opium.html (accessed on 28 February 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input | Solvent | Maximum Solution Temperature [°C] | Yield [%] | The Total Yield of Four Macrocyclic, Colored Products | |||
|---|---|---|---|---|---|---|---|
| t-Bu-20-ester | t-Bu-19-al | t-Bu-19-o-OH | t-Bu-19-p-OH | ||||
| A. | Toluene | 74 | 3.5 | 4.0 | 45.4 | 40.5 | 93.4 |
| B. | Xylene | 68 | 4.3 | 8.0 | 45.0 | 40.0 | 97.3 |
| C. | DMF | 73 | 0.6 | - | 53.8 | 16.9 | 71.3 |
| D. | 2-propanol | 64 | 7.1 | 5.7 | 10.0 | 64.3 | 87.1 |
| E. | s-butanol | 68 | 8.0 | 7.0 | 5.0 | 70.0 | 90.0 |
| F. | n-butanol | 75 | 2.5 | 2.3 | 11.3 | 48.8 | 64.9 |
| G. | 2-propanol/ acetic acid (0.2 mL) | 73 | - | - | 73.0 | 13.0 | 86.0 |
| H. | toluene/ acetic acid (0.2 mL) | 70 | - | - | 66.7 | 6.2 | 72.9 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| t-Bu-19-o-OH | ||||
| O6—H6···N2 | 0.93 (2) | 1.63 (2) | 2.500 (2) | 155 (4) |
| O12—H12···N4 | 0.95 (2) | 1.66 (3) | 2.526 (2) | 150 (4) |
| t-Bu-19-p-OH | ||||
| N2—H2···O1 | 0.89 (2) | 2.04 (5) | 2.730 (6) | 134 (6) |
| N4—H4···O7 | 0.89 (2) | 2.04 (5) | 2.695 (6) | 130 (5) |
| t-Bu-20-ester | ||||
| N2—H2···O6 | 0.89 (2) | 1.85 (2) | 2.716 (3) | 164 (4) |
| t-Bu-17-p-OH | ||||
| O6—H6···O2 i | 0.84 (2) | 1.89 (4) | 2.657 (7) | 150 (6) |
| Isomer | “Free” t-Bu-19-Azo | Equimolar Amount of NaClO4 | 10-Fold Excess of NaClO4 |
|---|---|---|---|
| Z | 65 | 63 | 31 |
| E | 35 | 37 | 69 |
| t-Bu-19-o-OH | t-Bu-19-p-OH | t-Bu-20-ester | t-Bu-17-p-OH | |
|---|---|---|---|---|
| CCDC | 2112902 | 2105497 | 2105498 | 2130657 |
| Crystal data | ||||
| Chemical formula | C28H40N2O6 | C28H40N2O6 | C28H40N2O6 | C28H40O6 |
| Mr | 500.62 | 500.62 | 500.62 | 472.60 |
| Crystal system, space group | Monoclinic, P21/n | Monoclinic, P21/c | Monoclinic, P21/c | |
| Temperature (K) | 120 | 120 | 120 | 120 |
| a, b, c (Å) | 13.432 (2), 15.7712 (18), 26.177 (3) | 9.7213 (10), 16.1636 (13), 19.3472 (16) | 19.5034 (7), 17.5123 (11), 7.891 (2) | 17.208 (2), 15.999 (3), 10.021 (5) |
| a, b, γ (°) | 90, 97.077 (11), 90 | 67.341 (6), 77.329 (7), 74.107 (7) | 90, 99.288 (9), 90 | 90, 100.597 (19), 90 |
| V (Å3) | 5503.0 (13) | 2675.9 (4) | 2659.8 (8) | 2711.8 (13) |
| Z | 8 | 4 | 4 | 4 |
| Radiation type | Cu Ka | Mo Ka | Mo Ka | Mo Ka |
| m (mm−1) | 0.69 | 0.09 | 0.09 | 0.08 |
| Crystal size (mm) | 0.34 × 0.18 × 0.05 | 0.09 × 0.05 × 0.03 | 0.34 × 0.23 × 0.05 | 0.12 × 0.06 × 0.02 |
| Data collection | ||||
| Diffractometer | STOE IPDS 2T | STOE IPDS 2T | STOE IPDS 2T | STOE IPDS 2T |
| Absorption correction | Integration * | – | – | – |
| Tmin, Tmax | 0.782, 0.979 | – | – | – |
| No. of measured, independent and observed [I > 2s(I)] reflections | 22476, 9605, 7767 | 19172, 9458, 3910 | 7178, 7178, 5882 | 16293, 4920, 2659 |
| Rint | 0.022 | 0.129 | ? | 0.089 |
| (sin θ/λ)max (Å−1) | 0.600 | 0.597 | 0.687 | 0.601 |
| Refinement | ||||
| R[F2 > 2s(F2)], wR(F2), S | 0.049, 0.132, 1.04 | 0.098, 0.315, 0.99 | 0.077, 0.201, 1.15 | 0.062, 0.154, 1.01 |
| No. of reflections | 9605 | 9458 | 7178 | 4920 |
| No. of parameters | 669 | 669 | 336 | 351 |
| No. of restraints | 2 | 2 | 1 | 4 |
| H atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.80, −0.31 | 0.35, −0.31 | 0.39, −0.39 | 0.22, −0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner-Wysiecka, E.; Szulc, P.; Luboch, E.; Chojnacki, J.; Sowiński, P.; Szwarc-Karabyka, K. Products of Photo- and Thermochemical Rearrangement of 19-Membered di-tert-Butyl-Azoxybenzocrown. Molecules 2022, 27, 1835. https://doi.org/10.3390/molecules27061835
Wagner-Wysiecka E, Szulc P, Luboch E, Chojnacki J, Sowiński P, Szwarc-Karabyka K. Products of Photo- and Thermochemical Rearrangement of 19-Membered di-tert-Butyl-Azoxybenzocrown. Molecules. 2022; 27(6):1835. https://doi.org/10.3390/molecules27061835
Chicago/Turabian StyleWagner-Wysiecka, Ewa, Paulina Szulc, Elżbieta Luboch, Jarosław Chojnacki, Paweł Sowiński, and Katarzyna Szwarc-Karabyka. 2022. "Products of Photo- and Thermochemical Rearrangement of 19-Membered di-tert-Butyl-Azoxybenzocrown" Molecules 27, no. 6: 1835. https://doi.org/10.3390/molecules27061835
APA StyleWagner-Wysiecka, E., Szulc, P., Luboch, E., Chojnacki, J., Sowiński, P., & Szwarc-Karabyka, K. (2022). Products of Photo- and Thermochemical Rearrangement of 19-Membered di-tert-Butyl-Azoxybenzocrown. Molecules, 27(6), 1835. https://doi.org/10.3390/molecules27061835