
Discrimination of Excited States of Acetylacetone through Theoretical Molecular-Frame Photoelectron Angular Distributions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cross-Sections in the Molecular Frame: MFPADs
2.2. LCAO B-Spline Code
2.3. Computational Details
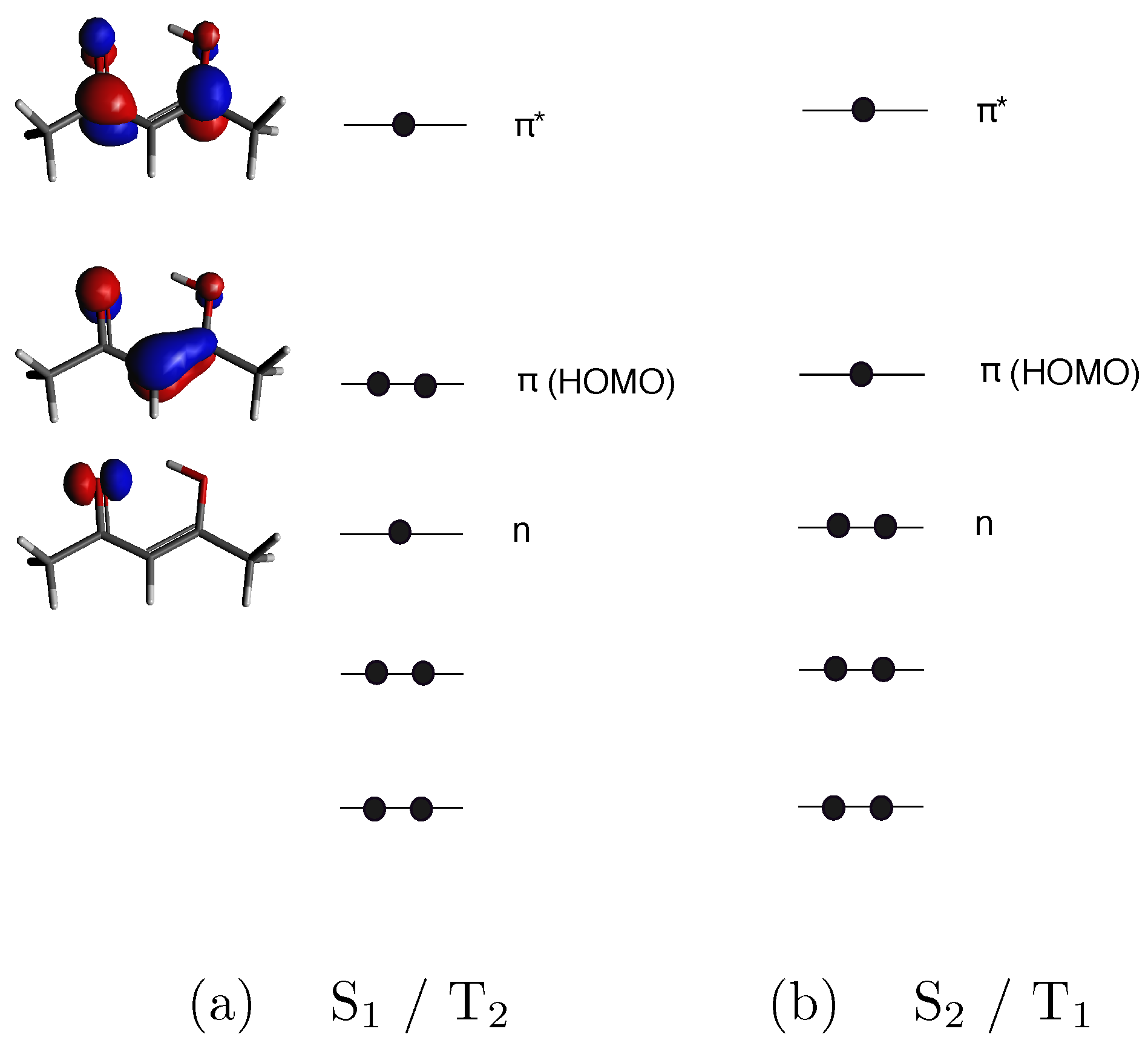
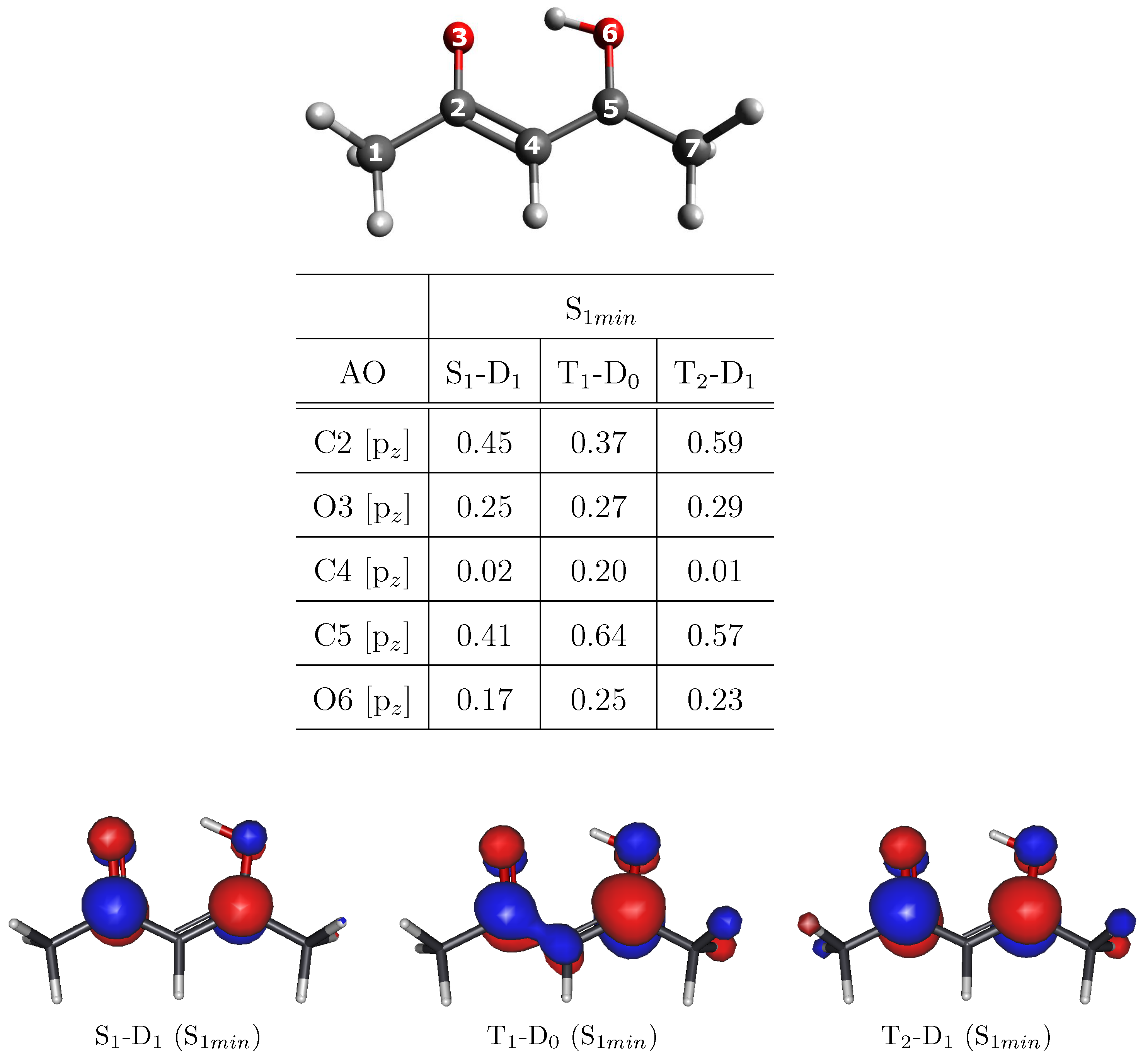
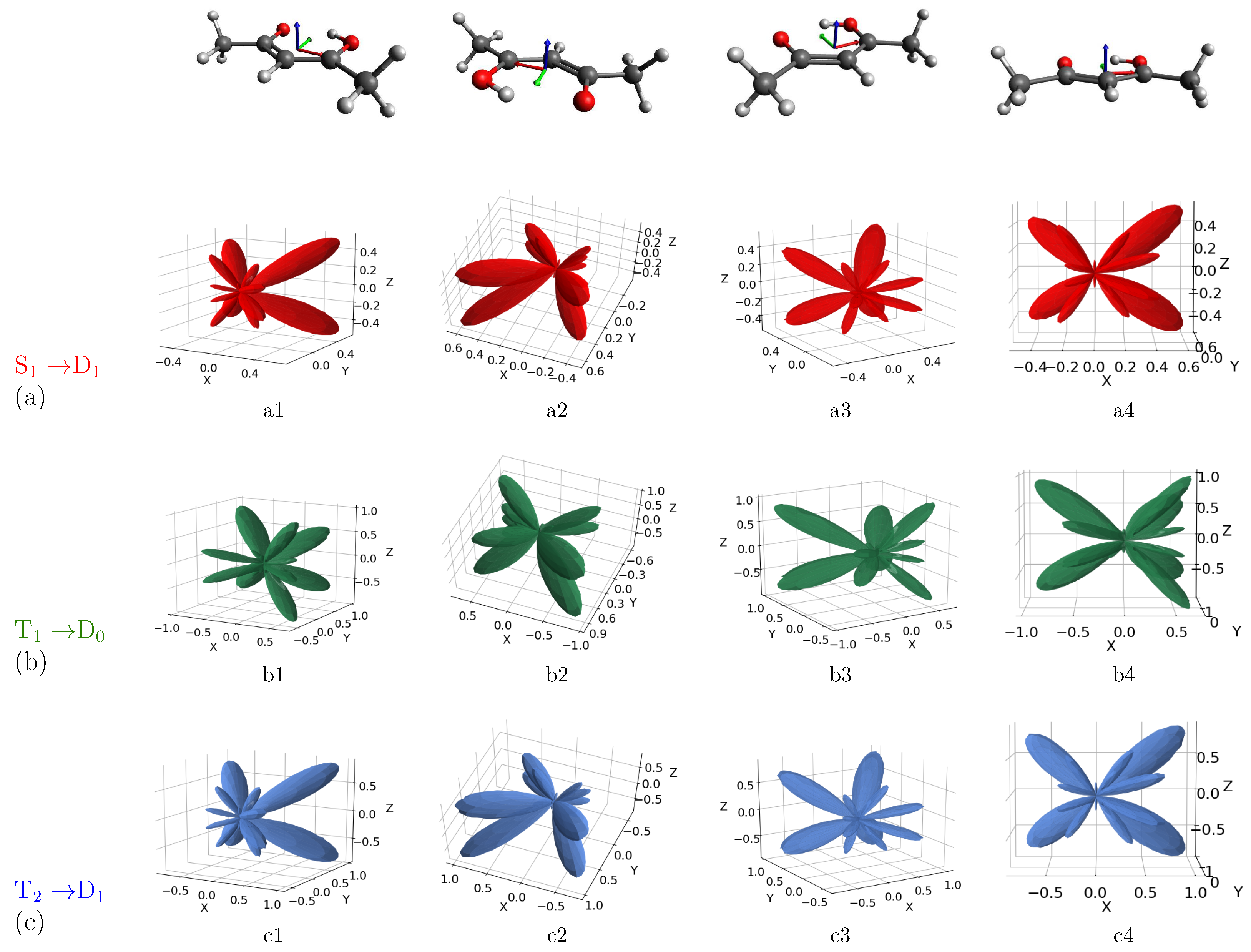
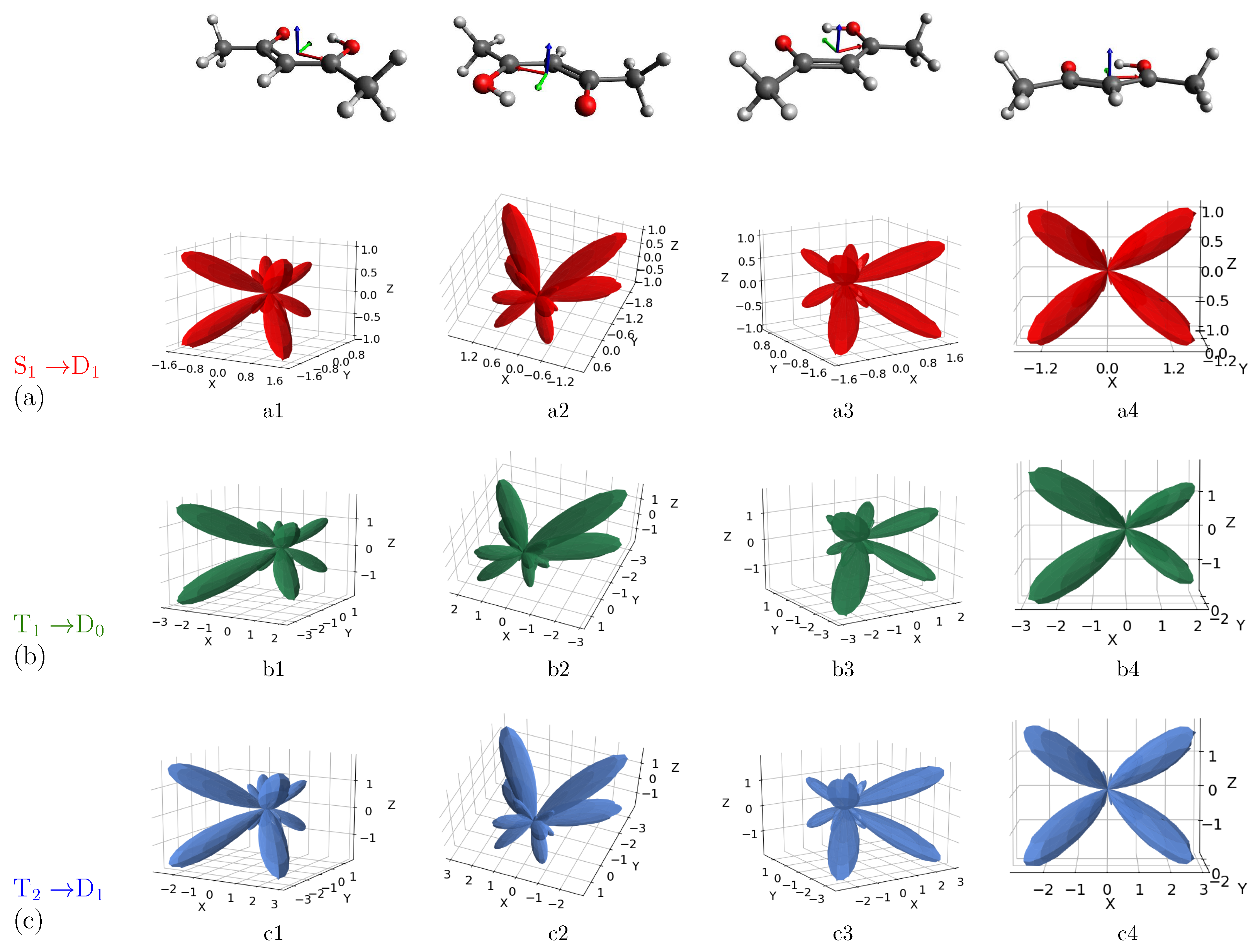
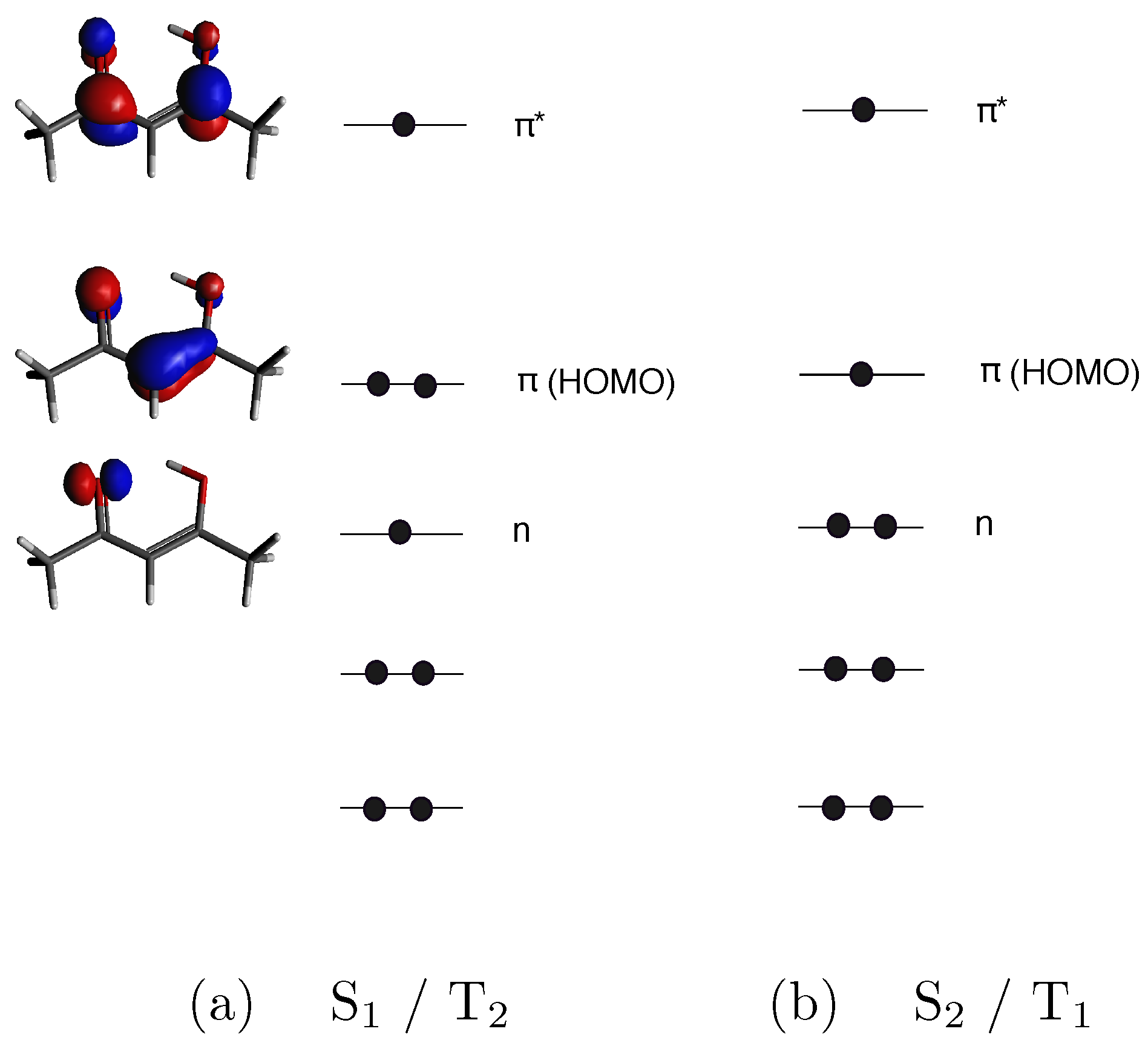
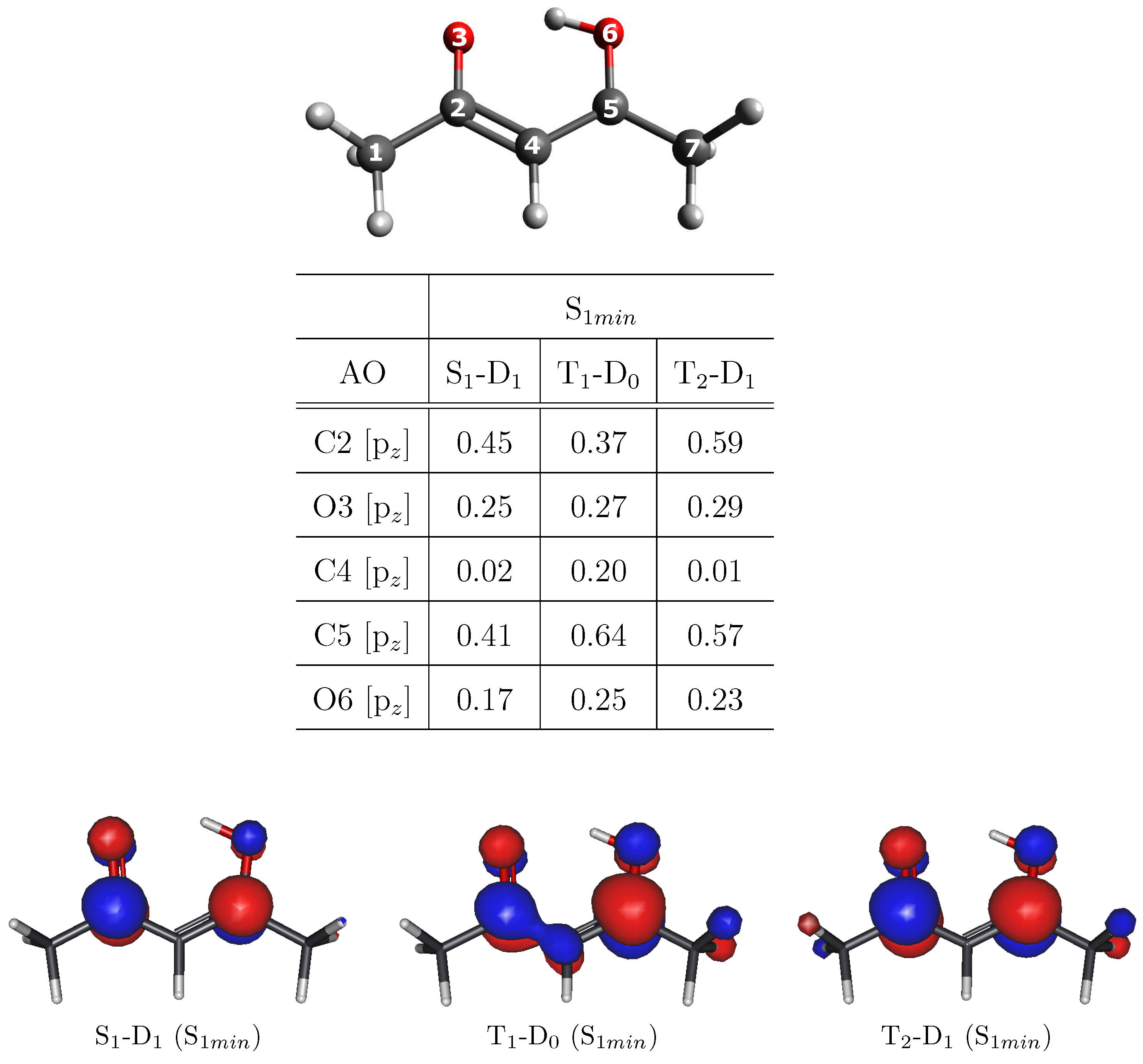
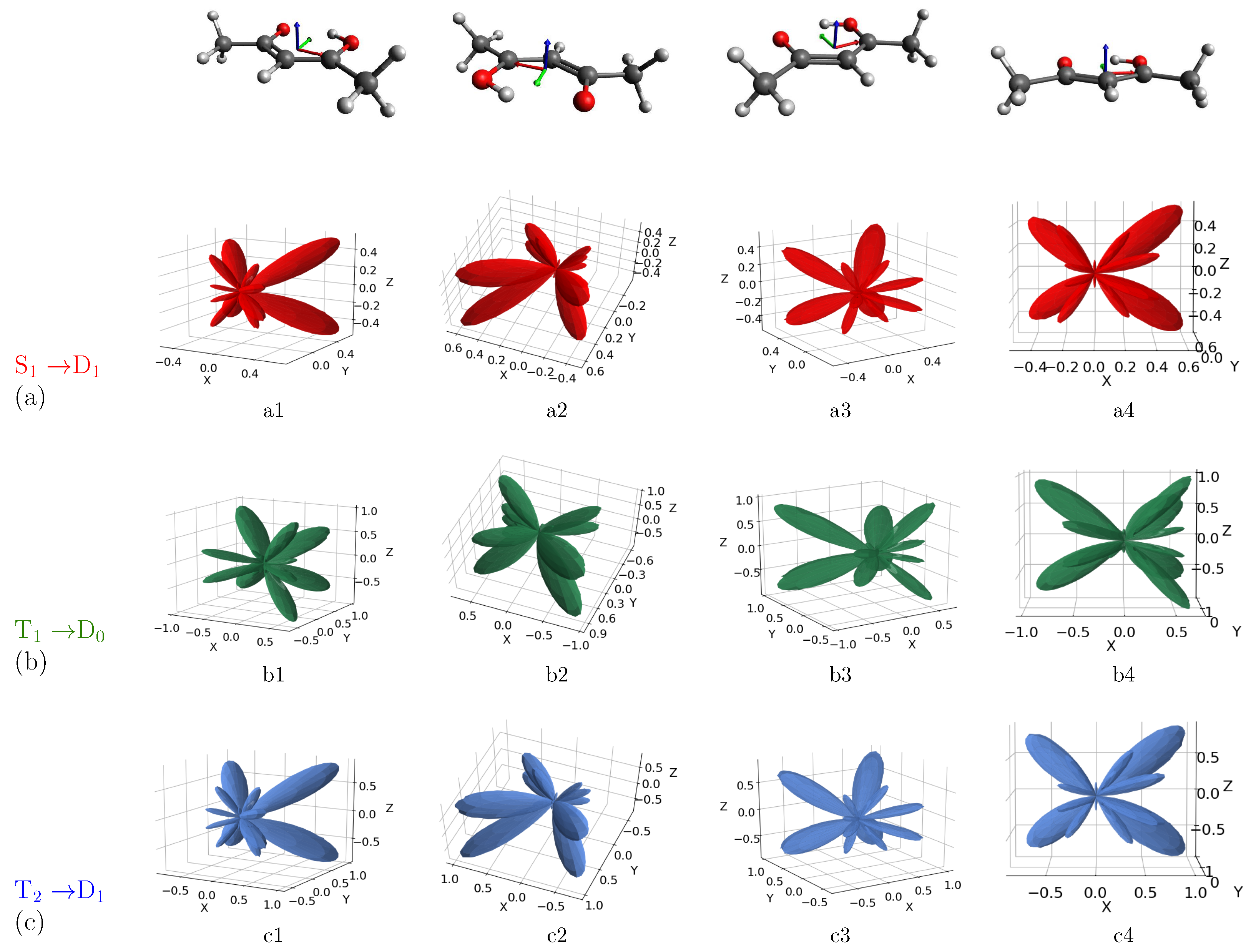
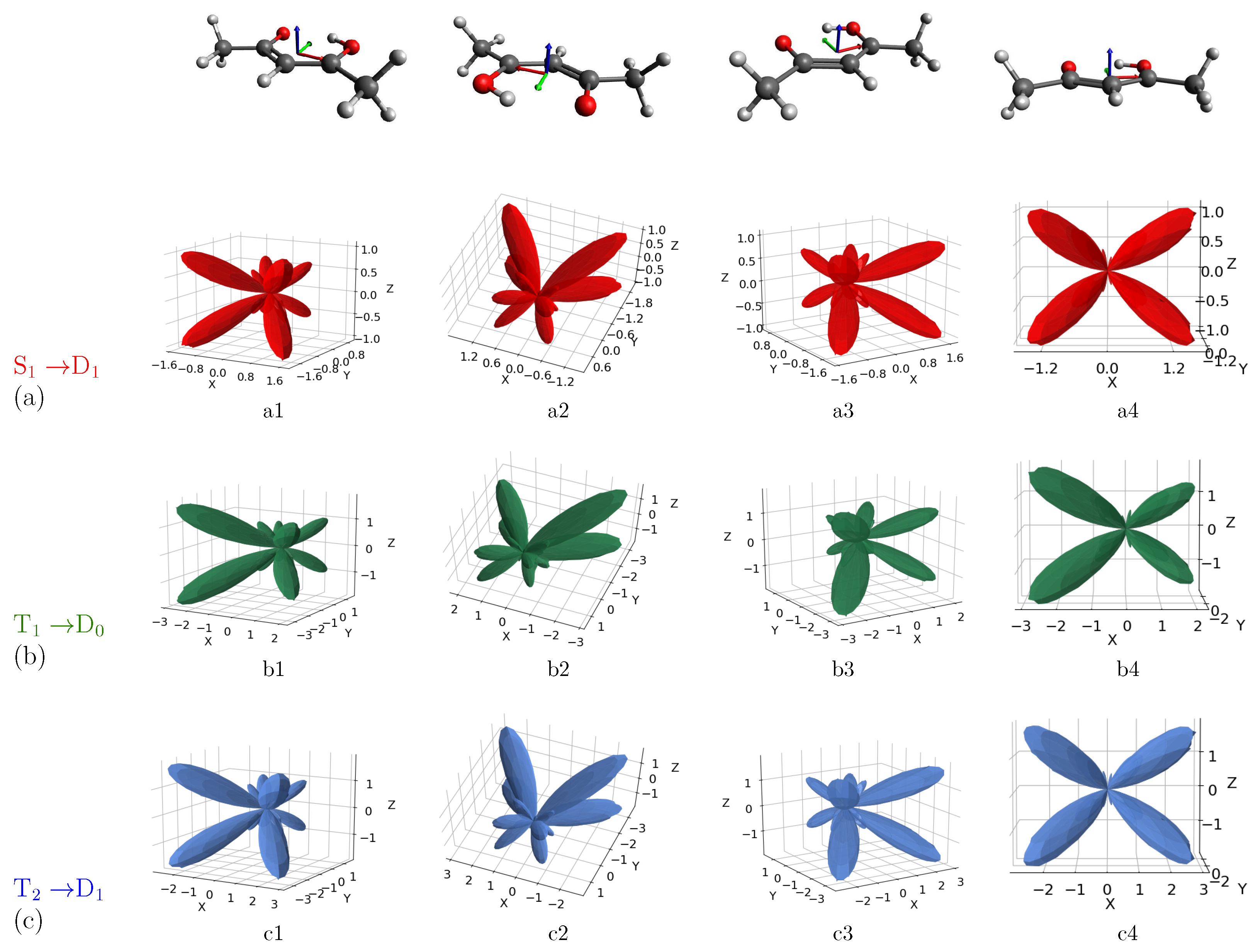
3. Results and Discussion
MFPAD Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cherepkov, N.A.; Kuznetsov, V.V. Photoionization of oriented molecules. Z. Phys. D At. Mol. Clust. 1987, 7, 271–280. [Google Scholar] [CrossRef]
- Chandra, N. Photoelectron spectroscopic studies of polyatomic molecules. I. Theory. J. Phys. B At. Mol. Phys. 1987, 20, 3405–3415. [Google Scholar] [CrossRef]
- Reid, K.L. Photoelectron angular distributions: Developments in applications to isolated molecular systems. Mol. Phys. 2012, 110, 131–147. [Google Scholar] [CrossRef]
- Stapelfeldt, H.; Seideman, T. Colloquium: Aligning molecules with strong laser pulses. Rev. Mod. Phys. 2003, 75, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Dill, D. Fixed-molecule photoelectron angular distributions. J. Chem. Phys. 1976, 65, 1130–1133. [Google Scholar] [CrossRef]
- Dill, D.; Siegel, J.; Dehmer, J.L. Spectral variation of fixed-molecule photoelectron angular distributions. J. Chem. Phys. 1976, 65, 3158–3160. [Google Scholar] [CrossRef]
- Davenport, J.W. Ultraviolet Photoionization Cross Sections for N2 and CO. Phys. Rev. Lett. 1976, 36, 945–949. [Google Scholar] [CrossRef]
- Dörner, R.; Mergel, V.; Jagutzki, O.; Spielberger, L.; Ullrich, J.; Moshammer, R.; Schmidt-Böcking, H. Cold Target Recoil Ion Momentum Spectroscopy: A ‘momentum microscope’ to view atomic collision dynamics. Phys. Rep. 2000, 330, 95–192. [Google Scholar] [CrossRef]
- Borràs, V.J.; González-Vázquez, J.; Argenti, L.; Martín, F. Molecular-Frame Photoelectron Angular Distributions of CO in the Vicinity of Feshbach Resonances: An XCHEM Approach. J. Chem. Theory Comput. 2021, 17, 6330–6339. [Google Scholar]
- Motoki, S.; Adachi, J.; Hikosaka, Y.; Ito, K.; Sano, M.; Soejima, K.; Yagishita, A.; Raseev, G.; Cherepkov, N.A. K-shell photoionization of CO: I. Angular distributions of photoelectrons from fixed-in-space molecules. J. Phys. At. Mol. Opt. Phys. 2000, 33, 4193–4212. [Google Scholar] [CrossRef]
- Geßner, O.; Hikosaka, Y.; Zimmermann, B.; Hempelmann, A.; Lucchese, R.R.; Eland, J.H.D.; Guyon, P.-M.; Becker, U. 4σ−1 Inner Valence Photoionization Dynamics of NO Derived from Photoelectron-Photoion Angular Correlations. Phys. Rev. Lett. 2002, 88, 193002. [Google Scholar] [PubMed]
- Cherepkov, N.A.; Semenov, S.K.; Hikosaka, Y.; Ito, K.; Motoki, S.; Yagishita, A. Manifestation of Many-Electron Correlations in Photoionization of the K Shell of N2. Phys. Rev. Lett. 2000, 84, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.; Hockett, P.; Stolow, A.; Makhija, V. Towards molecular frame photoelectron angular distributions in polyatomic molecules from lab frame coherent rotational wavepacket evolution. J. Phys. B At. Mol. Opt. Phys. 2021, 54, 145601. [Google Scholar] [CrossRef]
- Guillemin, R.; Decleva, P.; Stener, M.; Bomme, C.; Marin, T.; Journel, L.; Marchenko, T.; Kushawaha, R.K.; Jänkälä, K.; Trcera, N.; et al. Selecting core-hole localization or delocalization in CS2 by photofragmentation dynamics. Nat. Commun. 2015, 6, 6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuzawa, H.; Lucchese, R.R.; Liu, X.-J.; Sakai, K.; Iwayama, H.; Nagaya, K.; Kreidi, K.; Schöffler, M.S.; Harries, J.R.; Tamenori, Y.; et al. Probing molecular bond-length using molecular-frame photoelectron angular distributions. J. Chem. Phys. 2019, 150, 174306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuzawa, H.; Yamada, S.; Sakakibara, Y.; Tachibana, T.; Ito, Y.; Takanashi, T.; Nishiyama, T.; Sakai, T.; Nagaya, K.; Saito, N.; et al. Probing gaseous molecular structure by molecular-frame photoelectron angular distributions. J. Chem. Phys. 2019, 151, 104302. [Google Scholar] [CrossRef]
- Shigemasa, E.; Adachi, J.; Soejima, K.; Watanabe, N.; Yagishita, A.; Cherepkov, N.A. Direct Determination of Partial Wave Contributions in the σ* Shape Resonance of CO Molecules. Phys. Rev. Lett. 1998, 80, 1622–1625. [Google Scholar] [CrossRef]
- Jahnke, T.; Weber, T.; Landers, A.L.; Knapp, A.; Schössler, S.; Nickles, J.; Kammer, S.; Jagutzki, O.; Schmidt, L.; Czasch, A.; et al. Circular Dichroism in K-Shell Ionization from Fixed-in-Space CO and N2 Molecules. Phys. Rev. Lett. 2002, 88, 73002. [Google Scholar] [CrossRef]
- Motoki, S.; Adachi, J.; Ito, K.; Ishii, K.; Soejima, K.; Yagishita, A.; Semenov, S.K.; Cherepkov, N.A. Complete photoionization experiment in the region of the 2σg→σu shape resonance of the N2 molecule. J. Phys. B At. Mol. Opt. Phys. 2002, 35, 3801–3819. [Google Scholar] [CrossRef]
- Lebech, M.; Houver, J.C.; Dowek, D.; Lucchese, R.R. Molecular Frame Photoelectron Emission in the Presence of Autoionizing Resonances. Phys. Rev. Lett. 2006, 96, 73001. [Google Scholar] [CrossRef]
- Martín, F.; Fernández, J.; Havermeier, T.; Foucar, L.; Weber, T.; Kreidi, K.; Schöffler, M.; Schmidt, L.; Jahnke, T.; Jagutzki, O.; et al. Single photon-induced symmetry breaking of H2 dissociation. Science 2007, 315, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Golovin, A.V.; Heiser, F.; Quayle, C.J.K.; Morin, P.; Simon, M.; Gessner, O.; Guyon, P.-M.; Becker, U. Observation of Site-Specific Electron Emission in the Decay of Superexcited O2. Phys. Rev. Lett. 1997, 79, 4554–4557. [Google Scholar] [CrossRef]
- Holzmeier, F.; Joseph, J.; Houver, J.C.; Lebech, M.; Dowek, D.; Lucchese, R.R. Influence of shape resonances on the angular dependence of molecular photoionization delays. Nat. Commun. 2021, 12, 7343. [Google Scholar] [CrossRef]
- Rist, J.; Klyssek, K.; Novikovskiy, N.M.; Kircher, M.; Vela-Pérez, I.; Trabert, D.; Grundmann, S.; Tsitsonis, D.; Siebert, J.; Geyer, A.; et al. Measuring the photoelectron emission delay in the molecular frame. Nat. Commun. 2021, 12, 6657. [Google Scholar] [CrossRef]
- Koch, C.P.; Lemeshko, M.; Sugny, D. Quantum control of molecular rotation. Rev. Mod. Phys. 2019, 91, 35005. [Google Scholar] [CrossRef] [Green Version]
- Boll, R.; Rouzée, A.; Adolph, M.; Anielski, D.; Aquila, A.; Bari, S.; Bomme, C.; Bostedt, C.; Bozek, J.D.; Chapman, H.N.; et al. Imaging molecular structure through femtosecond photoelectron diffraction on aligned and oriented gas-phase molecules. Faraday Discuss. 2014, 171, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, J.; Moshammer, R.; Dorn, A.; Dörner, R.; Schmidt, L.P.H.; Schmidt-Böcking, H. Recoil-ion and electron momentum spectroscopy: Reaction-microscopes. Rep. Prog. Phys. 2003, 66, 1463–1545. [Google Scholar] [CrossRef]
- Kastirke, G.; Schöffler, M.S.; Weller, M.; Rist, J.; Boll, R.; Anders, N.; Baumann, T.M.; Eckart, S.; Erk, B.; De Fanis, A.; et al. Photoelectron Diffraction Imaging of a Molecular Breakup Using an X-ray Free-Electron Laser. Phys. Rev. X 2020, 10, 21052. [Google Scholar] [CrossRef]
- Pier, A.; Fehre, K.; Grundmann, S.; Vela-Perez, I.; Strenger, N.; Kircher, M.; Tsitsonis, D.; Williams, J.B.; Senftleben, A.; Baumert, T.; et al. Chiral photoelectron angular distributions from ionization of achiral atomic and molecular species. Phys. Rev. Res. 2020, 2, 33209. [Google Scholar] [CrossRef]
- Pérez-Torres, J.F.; Sanz-Vicario, J.L.; Veyrinas, K.; Billaud, P.; Picard, Y.J.; Elkharrat, C.; Poullain, S.M.; Saquet, N.; Lebech, M.; Houver, J.C.; et al. Circular dichroism in molecular-frame photoelectron angular distributions in the dissociative photoionization of H2 and D2 molecules. Phys. Rev. A 2014, 90, 43417. [Google Scholar] [CrossRef] [Green Version]
- Golovin, A.V.; Anielski, D.; Gordeev, S.V.; Lagodinski, V.M.; Chizhov, Y.V.; Küpper, J.; Rolles, D. The effect of elliptical polarization in MSXα calculations of the molecular-frame photoelectron angular distributions of CO C(1s) ionization. Eur. Phys. J. D 2019, 73, 131. [Google Scholar] [CrossRef]
- Hockett, P.; Frumker, E.; Villeneuve, D.M.; Corkum, P.B. Time delay in molecular photoionization. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 95602. [Google Scholar] [CrossRef]
- Dowek, D.; Lucchese, R.R. Photoionization dynamics: Photoemission in the molecular frame of small molecules ionized by linearly and elliptically polarized light. In Dynamical Processes in Atomic and Molecular Physics; Ogurtsov, G., Dowek, D., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2012; pp. 57–95. [Google Scholar]
- Squibb, R.J.; Sapunar, M.; Ponzi, A.; Richter, R.; Kivimäki, A.; Plekan, O.; Finetti, P.; Sisourat, N.; Zhaunerchyk, V.; Marchenko, T.; et al. Acetylacetone photodynamics at a seeded free-electron laser. Nat. Commun. 2018, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Ponzi, A.; Angeli, C.; Cimiraglia, R.; Coriani, S.; Decleva, P. Dynamical photoionization observables of the CS molecule: The role of electron correlation. J. Chem. Phys. 2014, 140, 204304. [Google Scholar] [CrossRef]
- Ponzi, A.; Sapunar, M.; Angeli, C.; Cimiraglia, R.; Došlić, N.; Decleva, P. Photoionization of furan from the ground and excited electronic states. J. Chem. Phys. 2016, 144, 84307. [Google Scholar] [CrossRef]
- Toffoli, D.; Stener, M.; Fronzoni, G.; Decleva, P. Convergence of the multicenter B-spline DFT approach for the continuum. Chem. Phys. 2002, 276, 25–43. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Phys. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2016. SCM, Theoretical Chemistry. Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com/doc.2016/ADF/index.html (accessed on 20 February 2022).
- Brosolo, M.; Decleva, P.; Lisini, A. Continuum wavefunctions calculations with least-squares schemes in a B-splines basis. Comput. Phys. Commun. 1992, 71, 207–214. [Google Scholar] [CrossRef]
- Van Leeuwen, R.; Baerends, E.J. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Stener, M.; Furlan, S.; Decleva, P. Density functional calculations of photoionization with an exchange-correlation potential with the correct asymptotic behaviour. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 1081–1102. [Google Scholar] [CrossRef]
- Ponzi, A.; Quadri, N.; Angeli, C.; Decleva, P. Electron correlation effects in the photoionization of CO and isoelectronic diatomic molecules. Phys. Chem. Chem. Phys. 2019, 21, 1937–1951. [Google Scholar] [CrossRef] [PubMed]
- Piteša, T.; Sapunar, M.; Ponzi, A.; Gelin, M.F.; Došlić, N.; Domcke, W.; Decleva, P. Combined Surface-Hopping, Dyson orbital, and B-Spline approach for the computation of time-resolved photoelectron spectroscopy signals: The internal conversion in pyrazine. J. Chem. Theory Comput. 2021, 17, 5098–5109. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schutz, M. MOLPRO, Version 2010.1, a Package of Ab Initio Programs. Available online: http://wild.life.nctu.edu.tw/~jsyu/compchem/molpro-2010.1-manual.pdf (accessed on 20 February 2022).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponzi, A.; Sapunar, M.; Došlić, N.; Decleva, P. Discrimination of Excited States of Acetylacetone through Theoretical Molecular-Frame Photoelectron Angular Distributions. Molecules 2022, 27, 1811. https://doi.org/10.3390/molecules27061811
Ponzi A, Sapunar M, Došlić N, Decleva P. Discrimination of Excited States of Acetylacetone through Theoretical Molecular-Frame Photoelectron Angular Distributions. Molecules. 2022; 27(6):1811. https://doi.org/10.3390/molecules27061811
Chicago/Turabian StylePonzi, Aurora, Marin Sapunar, Nadja Došlić, and Piero Decleva. 2022. "Discrimination of Excited States of Acetylacetone through Theoretical Molecular-Frame Photoelectron Angular Distributions" Molecules 27, no. 6: 1811. https://doi.org/10.3390/molecules27061811
APA StylePonzi, A., Sapunar, M., Došlić, N., & Decleva, P. (2022). Discrimination of Excited States of Acetylacetone through Theoretical Molecular-Frame Photoelectron Angular Distributions. Molecules, 27(6), 1811. https://doi.org/10.3390/molecules27061811