
Role of Cholinergic Signaling in Alzheimer’s Disease

Abstract
1. Introduction
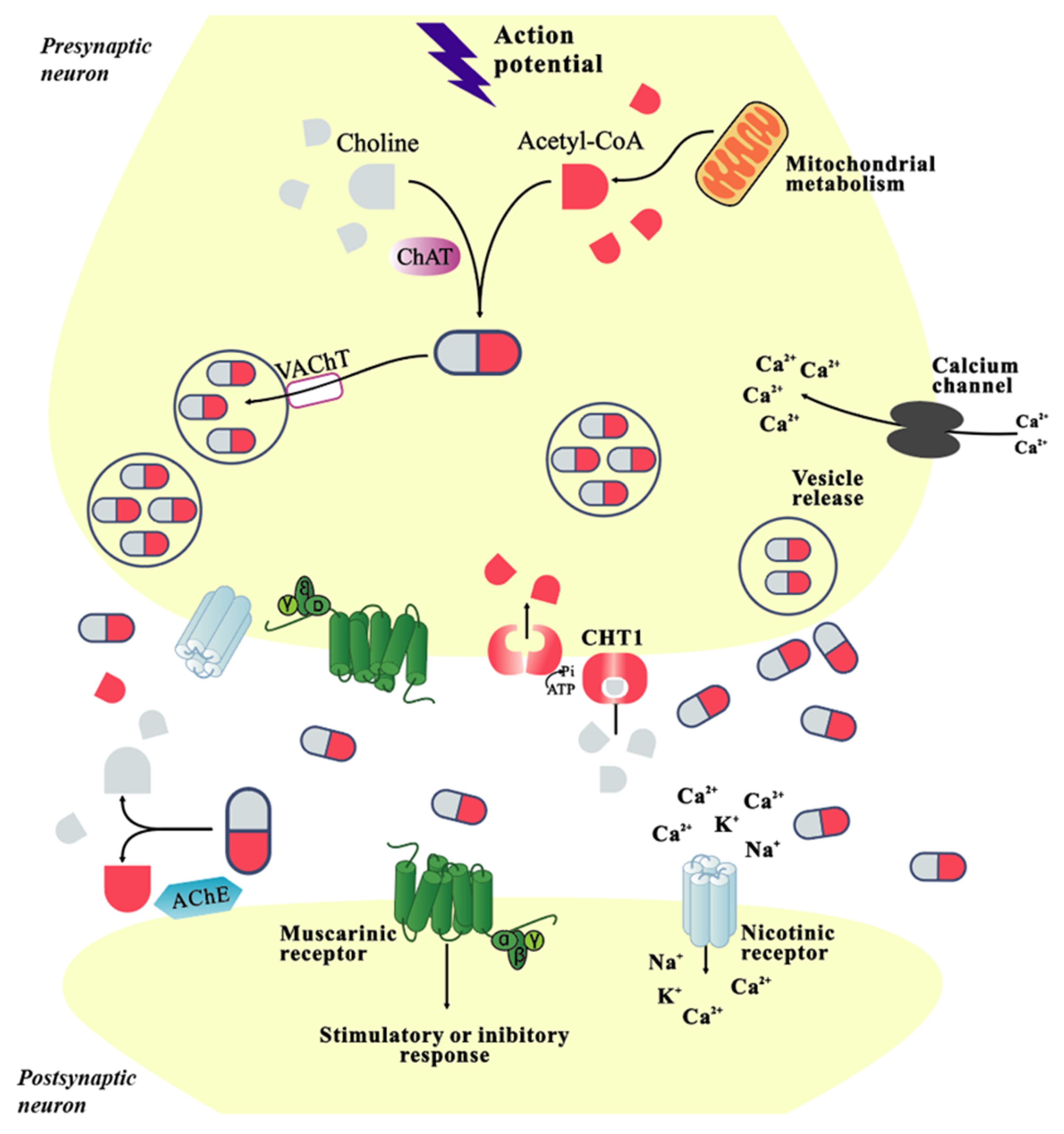
2. Physiological Function of Cholinergic System
3. AD Relation to the Cholinergic System
3.1. Amyloid Protein
3.2. Neuroinflammation and Cholinergic System Abnormalities
3.2.1. Acetylcholine Receptor and Signal Changing
3.2.2. Scopolamine Pathogenesis
3.2.3. Inflammation Pathogenesis
3.2.4. Ion Instability Pathogenesis
3.3. Other Factors
3.3.1. Other Brain Damaging Cofactors
3.3.2. Diabetes Related
3.3.3. Pregnancy Related
4. Relevant Treatment of Alzheimer’s Disease
4.1. Therapy Targeting the Cholinergic System
4.1.1. AChEI
4.1.2. Improvement of Deficiency in ACh
4.1.3. Protection of BFCNs and Regulation of NGFs and BDNF
4.1.4. AChR and Scopolamine
4.2. Relevant Treatment for Neuroinflammation
4.3. Reduction of Deposition of Amyloid Beta (Aβ) and Phosphorylation of Tau
4.4. Other Treatment for AD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Park, H.J.; Kwon, H.; Lee, J.H.; Cho, E.; Lee, Y.C.; Moon, M.; Jun, M.; Kim, D.H.; Jung, W.J. β-Amyrin Ameliorates Alzheimer’s Disease-Like Aberrant Synaptic Plasticity in the Mouse Hippocampus. Biomol. Ther. 2020, 28, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hafez, H.S.; Ghareeb, D.A.; Saleh, S.R.; Abady, M.M.; El Demellawy, M.A.; Hussien, H.; Abdel-Monem, N. Neuro-protective effect of ipriflavone against scopolamine-induced memory impairment in rats. Psychopharmacology 2017, 234, 3037–3053. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Verma, J. Exendin-4 attenuates brain mitochondrial toxicity through PI3K/Akt-dependent pathway in amyloid β (1–42)-induced cognitive deficit rats. Neurochem. Int. 2019, 128, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Semwal, B.C.; Garabadu, D. 5-N-ethyl Carboxamidoadenosine Stimulates Adenosine-2b Receptor-Mediated Mito-gen-Activated Protein Kinase Pathway to Improve Brain Mitochondrial Function in Amyloid Β-Induced Cognitive Deficit Mice. Neuromolecular Med. 2020, 22, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Cerda-Troncoso, C.; Andrade, V.; Maccion, R.B. The Natural Product Curcumin as a Potential Coadjuvant in Alzheimer’s Treatment. J. Alzheimer’s Dis. 2017, 60, 451–460. [Google Scholar] [CrossRef]
- Ishola, I.O.; Jacinta, A.A.; Adeyemi, O.O. Cortico-hippocampal memory enhancing activity of hesperetin on scopola-mine-induced amnesia in mice: Role of antioxidant defense system, cholinergic neurotransmission and expression of BDNF. Metab. Brain Dis. 2019, 34, 979–989. [Google Scholar] [CrossRef]
- Foveau, B.; Correia, A.S.; Hébert, S.S.; Rainone, S.; Potvin, O.; Kergoat, M.-J.; Belleville, S.; Duchesne, S.; LeBlanc, A.C. Stem Cell-Derived Neurons as Cellular Models of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 893–910. [Google Scholar] [CrossRef]
- Thangnipon, W.; Puangmalai, N.; Soi-Ampornkul, R.; Suwanna, N.; Tuchinda, P.; Nobsathian, S. Neuroprotection of N-benzylcinnamide on scopolamine-induced cholinergic dysfunction in human SH-SY5Y neuroblastoma cells. Neural Regen. Res. 2017, 12, 1492–1498. [Google Scholar] [CrossRef]
- Thompson, K.J.; Tobin, A.B. Crosstalk between the M1 muscarinic acetylcholine receptor and the endocannabinoid system: A relevance for Alzheimer’s disease? Cell Signal 2020, 70, 109545. [Google Scholar] [CrossRef]
- Bekdash, R. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Qu, Z.Y.; Cao, S.Y.; Li, Q.; Ma, L.; Krencik, R.; Xu, M.; Liu, Y. Directed differentiation of basal forebrain cholinergic neurons from human pluripotent stem cells. J. Neurosci. Methods 2016, 266, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Latina, V.; Caioli, S.; Zona, C.; Ciotti, M.T.; Borreca, A.; Calissano, P.; Amadoro, G. NGF-Dependent Changes in Ubiquitin Homeostasis Trigger Early Cholinergic Degeneration in Cellular and Animal AD-Model. Front. Cell Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- Cafe-Mendes, C.C.; Garay-Malpartida, H.M.; Malta, M.B.; de Sa Lima, L.; Scavone, C.; Ferreira, Z.S.; Markus, R.P.; Marcourakis, T. Chronic nicotine treatment decreases LPS signaling through NF-kappaB and TLR-4 modulation in the hippocampus. Neurosci. Lett. 2017, 636, 218–224. [Google Scholar] [CrossRef]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Campanari, M.L.; Garcia-Ayllon, M.S.; Belbin, J.O.; Galceran, A.L.; Saez-Valero, J. Acetylcholinesterase modulates presenilin-1 levels and gamma-secretase activity. J. Alzheimer’s Dis. 2014, 41, 911–924. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef]
- Li, Y.; Fan, H.; Sun, J.; Ni, M.; Zhang, L.; Chen, C.; Hong, X.; Fang, F.; Zhang, W.; Ma, P. Circular RNA expression profile of Alzheimer’s disease and its clinical significance as biomarkers for the disease risk and progression. Int. J. Biochem. Cell Biol. 2020, 123, 105747. [Google Scholar] [CrossRef]
- Lecrux, C.; Sandoe, C.; Neupane, S.; Kropf, P.; Toussay, X.; Tong, X.-K.; Lacalle-Aurioles, M.; Shmuel, A.; Hamel, E. Impact of Altered Cholinergic Tones on the Neurovascular Coupling Response to Whisker Stimulation. J. Neurosci. 2017, 37, 1518–1531. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Simchovitz, A.; Heneka, M.T.; Soreq, H. Personalized genetics of the cholinergic blockade of neuroinflammation. J. Neurochem. 2017, 142, 178–187. [Google Scholar] [CrossRef]
- Singh, S.P.; Gupta, D. Discovery of potential inhibitor against human acetylcholinesterase: A molecular docking and molecular dynamics investigation. Comput. Biol. Chem. 2017, 68, 224–230. [Google Scholar] [CrossRef]
- Fukunaga, K.; Yabuki, Y. SAK3-Induced Neuroprotection Is Mediated by Nicotinic Acetylcholine Receptors. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Akaike, A., Shimohama, S., Misu, Y., Eds.; Springer: Singapore, 2018; pp. 159–171. [Google Scholar]
- Muramatsu, I.; Yoshiki, H.; Uwada, J.; Masuoka, T.; Sada, K.; Taniguchi, T.; Nishio, M. Pharmacological evidence of specific acetylcholine transport in rat cerebral cortex and other brain regions. J. Neurochem. 2016, 139, 566–575. [Google Scholar] [CrossRef]
- Frinchi, M.; Scaduto, P.; Cappello, F.; Belluardo, N.; Mudo, G. Heat shock protein (Hsp) regulation by muscarinic acetyl-choline receptor (mAChR) activation in the rat hippocampus. J. Cell Physiol. 2018, 233, 6107–6116. [Google Scholar] [CrossRef]
- Anni, D.; Weiss, E.M.; Guhathakurta, D.; Akdas, Y.E.; Klueva, J.; Zeitler, S.; Andres-Alonso, M.; Huth, T.; Fejtova, A. Aβ1-16 controls synaptic vesicle pools at excitatory synapses via cholinergic modulation of synapsin phosphorylation. Cell Mol. Life Sci. 2021, 78, 4973–4992. [Google Scholar] [CrossRef]
- Shekari, A.; Fahnestock, M. Cholinergic neurodegeneration in Alzheimer disease mouse models. Handb. Clin. Neurol. 2021, 182, 191–209. [Google Scholar] [CrossRef]
- Gamage, R.; Wagnon, I.; Rossetti, I.; Childs, R.; Niedermayer, G.; Chesworth, R.; Gyengesi, E. Cholinergic Modulation of Glial Function During Aging and Chronic Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 577912. [Google Scholar] [CrossRef]
- Eivani, M.; Alijanpour, S.; Arefian, E.; Rezayof, A. Corticolimbic analysis of microRNAs and protein expressions in scopolamine-induced memory loss under stress. Neurobiol. Learn. Mem. 2019, 164, 107065. [Google Scholar] [CrossRef]
- Yegla, B.; Parikh, V. Developmental suppression of forebrain trkA receptors and attentional capacities in aging rats: A longitudinal study. Behav. Brain. Res. 2017, 335, 111–121. [Google Scholar] [CrossRef]
- Martínez-Rubio, C.; Paulk, A.C.; McDonald, E.J.; Widge, A.S.; Eskandar, E.N. Multimodal Encoding of Novelty, Reward, and Learning in the Primate Nucleus Basalis of Meynert. J. Neurosci. 2018, 38, 1942–1958. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gardeazabal, J.; Gonzalez de San Roman, E.; Moreno-Rodriguez, M.; Llorente-Ovejero, A.; Manuel, I.; Rodriguez-Puertas, R. Lipid mapping of the rat brain for models of disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1548–1557. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Β-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Patil, P.; Thakur, A.; Sharma, A.; Flor, S.J.S. Natural products and their derivatives as multifunctional ligands against Alzheimer’s disease. Drug Dev. Res. 2020, 81, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative Therapy for Alzheimer’s Disease-with Focus on Biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Majdi, A.; Sadigh-Eteghad, S.; Rahigh Aghsan, S.; Farajdokht, F.; Vatandoust, S.M.; Namvaran, A.; Mahmoudi, J. Amyloid-β, tau, and the cholinergic system in Alzheimer’s disease: Seeking direction in a tangle of clues. Rev. Neurosci. 2020, 31, 391–413. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Β-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Bungau, S. Multifaceted Alzheimer’s Disease: Building a Roadmap for Advancement of Novel Therapies. Neurochem. Res. 2021, 46, 2832–2851. [Google Scholar] [CrossRef]
- Gao, H.L.; Li, C.; Nabeka, H.; Shimokawa, T.; Wang, Z.Y.; Cao, Y.M.; Matsuda, S. An 18-mer Peptide Derived from Prosaposin Ameliorates the Effects of Aβ1-42 Neurotoxicity on Hippocampal Neurogenesis and Memory Deficit in Mice. J. Alzheimer’s Dis. 2016, 53, 1173–1192. [Google Scholar] [CrossRef]
- Mufson, E.J.; He, B.; Ginsberg, S.D.; Carper, B.A.; Bieler, G.S.; Crawford, F.; Alvarez, V.E.; Huber, B.R.; Stein, T.D.; McKee, A.C. Gene Profiling of Nucleus Basalis Tau Containing Neurons in Chronic Traumatic Encephalopathy: A Chronic Effects of Neurotrauma Consortium Study. J. Neurotrauma 2018, 35, 1260–1271. [Google Scholar] [CrossRef]
- George, A.A.; Vieira, J.M.; Xavier-Jackson, C.; Gee, M.T.; Cirrito, J.R.; Bimonte-Nelson, H.A.; Picciotto, M.R.; Lukas, R.J.; Whiteaker, P. Implications of Oligomeric Amyloid-Β (oAβ42) Signaling through α7β2-Nicotinic Acetylcholine Receptors (nAChRs) on Basal Forebrain Cholinergic Neuronal Intrinsic Excitability and Cognitive Decline. J. Neurosci. 2021, 41, 555–575. [Google Scholar] [CrossRef]
- Ren, Z.; Dong, Z.; Xie, P.; Lv, J.; Hu, Y.; Guan, Z.; Zhang, C.; Yu, W. PNU282987 inhibits amyloidβ aggregation by up-regulating astrocytic endogenous αBcrystallin and HSP70 via regulation of the α7AChR, PI3K/Akt/HSF1 signaling axis. Mol. Med. Rep. 2020, 22, 201–208. [Google Scholar] [CrossRef]
- Akhtar, A.; Bishnoi, M.; Sah, S.P. Sodium orthovanadate improves learning and memory in intracerebroventricular-streptozotocin rat model of Alzheimer’s disease through modulation of brain insulin resistance induced tau pathology. Brain Res. Bull. 2020, 164, 83–97. [Google Scholar] [CrossRef]
- Varshney, V.; Garabadu, D. Ang (1-7)/Mas receptor-axis activation promotes amyloid β-induced altered mitochondrial bioenergetics in discrete brain regions of Alzheimer’s disease-like rats. Neuropeptides 2021, 86, 102122. [Google Scholar] [CrossRef]
- Huang, X.; Wang, C.; Chen, L.; Zhang, T.; Leung, K.L.; Wong, G. Human amyloid β and α-synuclein co-expression in neurons impair behavior and recapitulate features for Lewy body dementia in Caenorhabditis elegans. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166203. [Google Scholar] [CrossRef]
- Farhat, S.M.; Ahmed, T. Neuroprotective and Neurotoxic Implications of α7 Nicotinic Acetylcholine Receptor and Aβ Interaction: Therapeutic Options in Alzheimer’s Disease. Curr. Drug Targets 2017, 18, 1537–1544. [Google Scholar] [CrossRef]
- Xu, W.; Weissmiller, A.M.; White, J.A., 2nd; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef]
- Semwal, B.C.; Garabadu, D. Amyloid β (1-42) downregulates adenosine-2b receptors in addition to mitochondrial impairment and cholinergic dysfunction in memory-sensitive mouse brain regions. J. Recept. Signal Transduct. Res. 2020, 40, 531–540. [Google Scholar] [CrossRef]
- Kumar, R.; Nordberg, A.; Darreh-Shori, T. Amyloid-β peptides act as allosteric modulators of cholinergic signalling through formation of soluble BAβACs. Brain 2016, 139, 174–192. [Google Scholar] [CrossRef]
- Calvo-Flores Guzman, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef]
- Sadananda, G.; Subramaniam, J.R. Absence of metabotropic glutamate receptor homolog(s) accelerates acetylcholine neuro-transmission in Caenorhabditis elegans. Neurosci. Lett. 2021, 746, 135666. [Google Scholar] [CrossRef]
- Schwarthoff, S.; Tischer, N.; Sager, H.; Schatz, B.; Rohrbach, M.M.; Raztsou, I.; Robaa, D.; Gaube, F.; Arndt, H.D.; Winckler, T.; et al. Evaluation of gamma-carboline-phenothiazine conjugates as simultaneous NMDA receptor blockers and cholinesterase inhibitors. Bioorg. Med. Chem. 2021, 46, 116355. [Google Scholar] [CrossRef]
- De Medeiros, L.M.; De Bastiani, M.A.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbé-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; et al. Cholinergic Differentiation of Human Neuroblastoma SH-SY5Y Cell Line and Its Potential Use as an In vitro Model for Alzheimer’s Disease Studies. Mol. Neurobiol. 2019, 56, 7355–7367. [Google Scholar] [CrossRef]
- Lian, W.; Fang, J.; Xu, L.; Zhou, W.; Kang, W.; Xiong, H.J.; Liu, A.L.; Du, G.H. DL0410 Ameliorates Memory and Cognitive Impairments Induced by Scopolamine via Increasing Cholinergic Neurotransmission in Mice. Molecules 2017, 22, 410. [Google Scholar] [CrossRef]
- Ali, B.; Jamal, Q.M.; Shams, S.; Al-Wabel, N.A.; Siddiqui, M.U.; Alzohairy, M.A.; Al Karaawi, M.A.; Kesari, K.K.; Mushtaq, G.; Kamal, M.A. In Silico Analysis of Green Tea Polyphenols as Inhibitors of AChE and BChE Enzymes in Alzheimer’s Disease Treatment. CNS Neurol. Disord. Drug Targets 2016, 15, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Fu, W.-M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- Klaassens, B.L.; van Gerven, J.M.A.; Klaassen, E.S.; van der Grond, J.; Rombouts, S. Cholinergic and serotonergic modulation of resting state functional brain connectivity in Alzheimer’s disease. Neuroimage 2019, 199, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.L.; Barrantes, F.J. Autoimmune Attack of the Neuromuscular Junction in Myasthenia Gravis: Nicotinic Acetylcholine Receptors and Other Targets. ACS Chem. Neurosci. 2019, 10, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.; Harvey, A.N.; Concheiro-Guisan, M.; Huestis, M.A.; Ross, T.J.; Stein, E.A. Nicotinic receptor modulation of the default mode network. Psychopharmacology 2020, 238, 589–597. [Google Scholar] [CrossRef]
- Ren, Z.; Yang, M.; Guan, Z.; Yu, W. Astrocytic α7 Nicotinic Receptor Activation Inhibits Amyloid-β Aggregation by Upregulating Endogenous αB-crystallin through the PI3K/Akt Signaling Pathway. Curr. Alzheimer Res. 2019, 16, 39–48. [Google Scholar] [CrossRef]
- Randáková, A.; Jakubík, J. Functionally selective and biased agonists of muscarinic receptors. Pharmacol. Res. 2021, 169, 105641. [Google Scholar] [CrossRef]
- Montani, C.; Canella, C.; Schwarz, A.J.; Li, J.; Gilmour, G.; Galbusera, A.; Wafford, K.; Gutierrez-Barragan, D.; McCarthy, A.; Shaw, D.; et al. The M1/M4 preferring muscarinic agonist xanomeline modulates functional connectivity and NMDAR antago-nist-induced changes in the mouse brain. Neuropsychopharmacology 2021, 46, 1194–1206. [Google Scholar] [CrossRef]
- Ghoshal, A.; Moran, S.P.; Dickerson, J.W.; Joffe, M.E.; Grueter, B.A.; Xiang, Z.; Lindsley, C.W.; Rook, J.M.; Conn, P.J. Role of mGlu5 Receptors and Inhibitory Neurotransmission in M1 Dependent Muscarinic LTD in the Prefrontal Cortex: Implications in Schizophrenia. ACS Chem. Neurosci. 2017, 8, 2254–2265. [Google Scholar] [CrossRef]
- Chang, W.; Pedroni, A.; Bertuzzi, M.; Kizil, C.; Simon, A.; Ampatzis, K. Locomotion dependent neuron-glia interactions control neurogenesis and regeneration in the adult zebrafish spinal cord. Nat. Commun. 2021, 12, 4857. [Google Scholar] [CrossRef]
- Llorente-Ovejero, A.; Manuel, I.; Lombardero, L.; Giralt, M.T.; Ledent, C.; Gimenez-Llort, L.; Rodriguez-Puertas, R. Endocannabinoid and Muscarinic Signaling Crosstalk in the 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 117–136. [Google Scholar] [CrossRef]
- Szutowicz, A.; Bielarczyk, H.; Zysk, M.; Dys, A.; Ronowska, A.; Gul-Hinc, S.; Klimaszewska-Lata, J. Early and Late Pathomechanisms in Alzheimer’s Disease: From Zinc to Amyloid-β Neurotoxicity. Neurochem. Res. 2017, 42, 891–904. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, W.S.; Suo, D.Q.; Li, Y.; Peng, L.; Xu, L.X.; Zeng, K.Y.; Ren, T.; Wang, Y.; Zhou, Y.; et al. Moringa oleifera Seed Ex-tract Alleviates Scopolamine-Induced Learning and Memory Impairment in Mice. Front. Pharm. 2018, 9, 389. [Google Scholar] [CrossRef]
- Bujan, A.; Lister, J.J.; O’Brien, J.L.; Edwards, J.D. Cortical auditory evoked potentials in mild cognitive impairment: Evidence from a temporal-spatial principal component analysis. Psychophysiology 2019, 56, e13466. [Google Scholar] [CrossRef]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharm. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Venkatesan, R.; Subedi, L.; Yeo, E.J.; Kim, S.Y. Lactucopicrin ameliorates oxidative stress mediated by scopolamine-induced neurotoxicity through activation of the NRF2 pathway. Neurochem. Int. 2016, 99, 133–146. [Google Scholar] [CrossRef]
- Tabrizian, K.; Amelinia, F.; Belaran, M.; Pourheidar, S.; Mirzaei, H.; Fanoudi, S. Tadalafil Reversed H-89- and Scopolamine-Induced Spatial Learning Impairments in Male Rats. Drug. Res. 2021, 71, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Canu, N.; Amadoro, G.; Triaca, V.; Latina, V.; Sposato, V.; Corsetti, V.; Severini, C.; Ciotti, M.T.; Calissano, P. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int. J. Mol. Sci. 2017, 18, 1319. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Milne, M.R.; Waldvogel, H.J.; Faull, R.L. Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 2122. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Heinsen, H.; Amaro, E.; Grinberg, L.T.; Teipel, S.J. Cognitive Correlates of Basal Forebrain Atrophy and Associated Cortical Hypometabolism in Mild Cognitive Impairment. Cereb. Cortex 2015, 26, 2411–2426. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Ovejero, A.; Martínez-Gardeazabal, J.; Moreno-Rodríguez, M.; Lombardero, L.; Román, E.G.D.S.; Manuel, I.; Giralt, M.T.; Rodríguez-Puertas, R. Specific Phospholipid Modulation by Muscarinic Signaling in a Rat Lesion Model of Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2167–2181. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.M.; Ginsberg, S.D.; Alldred, M.J.; Strupp, B.J.; Mufson, E.J. Maternal Choline Supplementation Alters Basal Forebrain Cholinergic Neuron Gene Expression in the Ts65Dn Mouse Model of Down Syndrome. Dev. Neurobiol. 2019, 79, 664–683. [Google Scholar] [CrossRef] [PubMed]
- Goshadrou, F.; Sadeghi, B. Nucleus basalis of Meynert modulates signal processing in rat layer 5 somatosensory cortex but leads to memory impairment and tactile discrimination deficits following lesion. Behav. Brain Res. 2020, 386, 112608. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jia, X.; Qi, Z.; Fan, X.; Ma, T.; Ni, H.; Li, C.-S.R.; Li, K. Altered Functional Connectivity of the Basal Nucleus of Meynert in Mild Cognitive Impairment: A Resting-State fMRI Study. Front. Aging Neurosci. 2017, 9, 127. [Google Scholar] [CrossRef]
- Tiernan, C.T.; Ginsberg, S.D.; He, B.; Ward, M.S.; Guillozet-Bongaarts, A.L.; Kanaan, N.M.; Mufson, E.J.; Counts, S.E. Pre-tangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer’s disease. Neurobiol. Dis. 2018, 117, 125–136. [Google Scholar] [CrossRef]
- Luo, H.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Li, G.; Han, L.; Qin, X. Apelin-13 Suppresses Neuroinflammation Against Cognitive Deficit in a Streptozotocin-Induced Rat Model of Alzheimer’s Disease Through Activation of BDNF-TrkB Signaling Pathway. Front. Pharm. 2019, 10, 395. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Zheng, X.; Fang, T.; Yang, X.; Luo, X.; Guo, A.; Newell, K.A.; Huang, X.-F.; Yu, Y. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflamm. 2018, 15, 112. [Google Scholar] [CrossRef]
- Giil, L.M.; Aarsland, D.; Hellton, K.; Lund, A.; Heidecke, H.; Schulze-Forster, K.; Riemekasten, G.; Vik-Mo, A.O.; Kristoffersen, E.O.; Vedeler, C.A.; et al. Antibodies to Multiple Receptors are Associated with Neuropsychiatric Symptoms and Mortality in Alzheimer’s Disease: A Longitudinal Study. J. Alzheimer’s Dis. 2018, 64, 761–774. [Google Scholar] [CrossRef]
- Wu, A.J.; Tong, B.C.; Huang, A.S.; Li, M.; Cheung, K.H. Mitochondrial Calcium Signaling as a Therapeutic Target for Alzheimer’s Disease. Curr. Alzheimer. Res. 2020, 17, 329–343. [Google Scholar] [CrossRef]
- Triaca, V.; Calissano, P. Impairment of the nerve growth factor pathway driving amyloid accumulation in cholinergic neurons: The incipit of the Alzheimer’s disease story? Neural. Regen. Res. 2016, 11, 1553–1556. [Google Scholar] [CrossRef]
- Li, L.; Xu, S.; Liu, L.; Feng, R.; Gong, Y.; Zhao, X.; Li, J.; Cai, J.; Feng, N.; Wang, L.; et al. Multifunctional Compound AD-35 Im-proves Cognitive Impairment and Attenuates the Production of TNF-α and IL-1β in an Aβ25-35-induced Rat Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 1403–1417. [Google Scholar] [CrossRef]
- Olasehinde, T.A.; Olaniran, A.O.; Okoh, A.I. Neuroprotective effects of some seaweeds against Zn-Induced neuronal damage in HT-22 cells via modulation of redox imbalance, inhibition of apoptosis and acetylcholinesterase activity. Metab. Brain Dis. 2019, 34, 1615–1627. [Google Scholar] [CrossRef]
- Tao, L.X.; Huang, X.T.; Chen, Y.T.; Tang, X.C.; Zhang, H.Y. Acetylcholinesterase-independent protective effects of huperzine A against iron overload-induced oxidative damage and aberrant iron metabolism signaling in rat cortical neurons. Acta Pharm. Sin. 2016, 37, 1391–1400. [Google Scholar] [CrossRef]
- Ijomone, O.M.; Aluko, O.M.; Okoh, C.O.A.; Martins, A.C., Jr.; Aschner, M. Role for calcium signaling in manganese neurotoxicity. J. Trace Elem. Med. Biol. 2019, 56, 146–155. [Google Scholar] [CrossRef]
- Gómez-Gonzalo, M.; Martin-Fernandez, M.; Martínez-Murillo, R.; Mederos, S.; Hernández-Vivanco, A.; Jamison, S.; Fernandez, A.P.; Serrano, J.; Calero, P.; Futch, H.S.; et al. Neuron-astrocyte signaling is preserved in the aging brain. Glia 2017, 65, 569–580. [Google Scholar] [CrossRef]
- Talwar, P.; Kushwaha, S.; Gupta, R.; Agarwal, R. Systemic Immune Dyshomeostasis Model and Pathways in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 290. [Google Scholar] [CrossRef]
- Garcez, M.; Jacobs, K.; Guillemin, G.J. Microbiota Alterations in Alzheimer’s Disease: Involvement of the Kynurenine Pathway and Inflammation. Neurotox. Res. 2019, 36, 424–436. [Google Scholar] [CrossRef]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef]
- Olive, I.; Makris, N.; Densmore, M.; McKinnon, M.C.; Lanius, R.A. Altered basal forebrain BOLD signal variability at rest in posttraumatic stress disorder: A potential candidate vulnerability mechanism for neurodegeneration in PTSD. Hum. Brain Mapp. 2021, 42, 3561–3575. [Google Scholar] [CrossRef]
- Mohamed, R.A.; Abdallah, D.M.; El-Brairy, A.I.; Ahmed, K.A.; El-Abhar, H.S. Palonosetron/Methyllycaconitine Deactivate Hippocampal Microglia 1, Inflammasome Assembly and Pyroptosis to Enhance Cognition in a Novel Model of Neuroinflammation. Molecules 2021, 26, 5068. [Google Scholar] [CrossRef]
- Letra, L.; Rodrigues, T.; Matafome, P.; Santana, I.; Seica, R. Adiponectin and sporadic Alzheimer’s disease: Clinical and molecular links. Front. Neuroendocr. 2019, 52, 1–11. [Google Scholar] [CrossRef]
- Sorial, M.E.; El Dine El Sayed, N.S. Protective effect of valproic acid in streptozotocin-induced sporadic Alzheimer’s disease mouse model: Possible involvement of the cholinergic system. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2017, 390, 581–593. [Google Scholar] [CrossRef]
- Chakrabarti, M.; McDonald, A.J.; Will Reed, J.; Moss, M.A.; Das, B.C.; Ray, S.K. Molecular Signaling Mechanisms of Natural and Synthetic Retinoids for Inhibition of Pathogenesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 50, 335–352. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Zhang, D.; Dragomir, A.; Akay, Y.M. Gene expression profiling of midbrain dopamine neurons upon gestational nicotine exposure. Med. Biol. Eng. Comput. 2017, 55, 467–482. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Nebieridze, N.; Velisek, L.; Veliskova, J. Estrogen Protects Neurotransmission Transcriptome During Status Epilepticus. Front. Neurosci. 2018, 12, 332. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.T., 2nd; Dell’Acqua, S.; Thach, D.Q.; Sessler, J.L. Classics in Chemical Neuroscience: Donepezil. ACS Chem. Neurosci. 2019, 10, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Messiha, B.A.S.; Ali, M.R.A.; Khattab, M.M.; Abo-Youssef, A.M. Perindopril ameliorates experimental Alzheimer’s disease progression: Role of amyloid β degradation, central estrogen receptor and hyperlipidemic-lipid raft signaling. Inflammopharmacology 2020, 28, 1343–1364. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. AChE and the amyloid precursor protein (APP)—Cross-talk in Alzheimer’s disease. Chem. Biol. Interact. 2016, 259, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Maloney, B.; Sambamurti, K.; Karnati, H.K.; Nelson, P.T.; Greig, H.N.; Lahiri, D.K. Rivastigmine modifies the α-secretase pathway and potentially early Alzheimer’s disease. Transl. Psychiatry 2020, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Perumal, Y.; Bansal, S.; Arora, S.; Chopra, K. Phycocyanin alleviates ICV-STZ induced cognitive and molecular deficits via PI3-Kinase dependent pathway. Food Chem. Toxicol. 2020, 145, 111684. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Abe, K. Nicotinic acetylcholine receptor in Alzheimer’s disease. Jpn. J. Clin. Med. 2013, 71, 743–750. [Google Scholar]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Damar, U.; Gersner, R.; Johnstone, J.T.; Schachter, S.; Rotenberg, A. Huperzine A as a neuroprotective and antiepileptic drug: A review of preclinical research. Expert Rev. Neurother. 2016, 16, 671–680. [Google Scholar] [CrossRef]
- Machhi, J.; Sinha, A.; Patel, P.; Kanhed, A.M.; Upadhyay, P.; Tripathi, A.; Parikh, Z.S.; Chruvattil, R.; Pillai, P.P.; Gupta, S.; et al. Neuroprotective Potential of Novel Multi-Targeted Isoalloxazine Derivatives in Rodent Models of Alzheimer’s Disease Through Activation of Canonical Wnt/β-Catenin Signalling Pathway. Neurotox. Res. 2016, 29, 495–513. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 10, 852–862. [Google Scholar] [CrossRef]
- Soto-Mercado, V.; Mendivil-Perez, M.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Multi-Target Effects of the Cannabinoid CP55940 on Familial Alzheimer’s Disease PSEN1 E280A Cholinergic-Like Neurons: Role of CB1 Receptor. J. Alzheimer’s Dis. 2021, 82, S359–S378. [Google Scholar] [CrossRef]
- Gregory, J.; Vengalasetti, Y.V.; Bredesen, D.E.; Rao, R.V. Neuroprotective Herbs for the Management of Alzheimer’s Disease. Biomolecules 2021, 11, 543. [Google Scholar] [CrossRef]
- Braidy, N.; Behzad, S.; Habtemariam, S.; Ahmed, T.; Daglia, M.; Nabavi, S.M.; Sobarzo-Sanchez, E.; Nabavi, S.F. Neuropro-tective Effects of Citrus Fruit-Derived Flavonoids, Nobiletin and Tangeretin in Alzheimer’s and Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 387–397. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, A.; Li, X.; Qin, D.; Jin, B.; Liu, J.; Tang, Y.; Wu, J.; Yu, C. The seed of Litchi chinensis fraction ameliorates hippo-campal neuronal injury in an Aβ25-35-induced Alzheimer’s disease rat model via the AKT/GSK-3β pathway. Pharm. Biol. 2020, 58, 35–43. [Google Scholar] [CrossRef]
- Fukunaga, K.; Izumi, H.; Yabuki, Y.; Shinoda, Y.; Shioda, N.; Han, F. Alzheimer’s disease therapeutic candidate SAK3 is an enhancer of T-type calcium channels. J. Pharm. Sci. 2019, 139, 51–58. [Google Scholar] [CrossRef]
- Alldred, M.J.; Chao, H.M.; Lee, S.H.; Beilin, J.; Powers, B.E.; Petkova, E.; Strupp, B.J.; Ginsberg, S.D. CA1 pyramidal neuron gene expression mosaics in the Ts65Dn murine model of Down syndrome and Alzheimer’s disease following maternal choline supplementation. Hippocampus 2018, 28, 251–268. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, Y.; Gao, K.; Rudd, J.A.; Fang, M. Ovarian hormones ameliorate memory impairment, cholinergic deficit, neuronal apoptosis and astrogliosis in a rat model of Alzheimer’s disease. Exp. Ther. Med. 2016, 11, 89–97. [Google Scholar] [CrossRef][Green Version]
- McKeever, P.M.; Kim, T.; Hesketh, A.; MacNair, L.; Miletic, D.; Favrin, G.; Oliver, S.G.; Zhang, Z.; George-Hyslop, P.S.; Robertson, J. Cholinergic neuron gene expression differences captured by translational profiling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 57, 104–119. [Google Scholar] [CrossRef]
- Fahimi, A.; Noroozi, M.; Salehi, A. Enlargement of early endosomes and traffic jam in basal forebrain cholinergic neurons in Alzheimer’s disease. Handb. Clin. Neurol. 2021, 179, 207–218. [Google Scholar] [CrossRef]
- Xhima, K.; Markham-Coultes, K.; Nedev, H.; Heinen, S.; Saragovi, H.U.; Hynynen, K.; Aubert, I. Focused ultrasound delivery of a selective TrkA agonist rescues cholinergic function in a mouse model of Alzheimer’s disease. Sci. Adv. 2020, 6, eaax6646. [Google Scholar] [CrossRef] [PubMed]
- Faiq, M.A.; Wollstein, G.; Schuman, J.S.; Chan, K.C. Cholinergic nervous system and glaucoma: From basic science to clinical applications. Prog. Retin. Eye Res. 2019, 72, 100767. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Niu, S.; Zhao, H.; Li, S.; Jiao, J. Donepezil improves the cognitive impairment in a tree shrew model of Alzheimer’s disease induced by amyloid-β1-40 via activating the BDNF/TrkB signal pathway. Metab. Brain Dis. 2018, 33, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-H.; Li, S.-H.; Gao, Z.; Zou, S.-F.; Li, H.-Y.; Tao, Z.-Y.; Song, J.; Yang, J.-X. Neurotrophin-3 promotes proliferation and cholinergic neuronal differentiation of bone marrow-derived neural stem cells via notch signaling pathway. Life Sci. 2016, 166, 131–138. [Google Scholar] [CrossRef]
- Labban, S.; Alghamdi, B.S.; Alshehri, F.S.; Kurdi, M. Effects of melatonin and resveratrol on recognition memory and passive avoidance performance in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2021, 402, 113100. [Google Scholar] [CrossRef]
- Kim, S.K.; Ko, Y.H.; Lee, S.Y.; Jang, G.C. Memory-enhancing effects of 7,3′,4′-trihydroxyisoflavone by regulation of cholinergic function and BDNF signaling pathway in mice. Food Chem. Toxicol. 2020, 137, 111160. [Google Scholar] [CrossRef]
- Moran, S.P.; Maksymetz, J.; Conn, P.J. Targeting Muscarinic Acetylcholine Receptors for the Treatment of Psychiatric and Neurological Disorders. Trends Pharmacol. Sci. 2019, 40, 1006–1020. [Google Scholar] [CrossRef]
- Yin, W.; Mamashli, F.; Buhl, D.L.; Khudyakov, P.; Volfson, D.; Martenyi, F.; Gevorkyan, H.; Rosen, L.; Simen, A.A. Safety, pharmacokinetics and quantitative EEG modulation of TAK-071, a novel muscarinic M1 receptor positive allosteric modulator, in healthy subjects. Br. J. Clin. Pharmacol. 2021, 88, 600–612. [Google Scholar] [CrossRef]
- Bradley, S.J.; Molloy, C.; Valuskova, P.; Dwomoh, L.; Scarpa, M.; Rossi, M.; Finlayson, L.; Svensson, K.A.; Chernet, E.; Barth, V.N.; et al. Biased M1-muscarinic-receptor-mutant mice inform the design of next-generation drugs. Nat. Chem. Biol. 2020, 16, 240–249. [Google Scholar] [CrossRef]
- Sato, T.; Ohi, Y.; Kato, D.; Mizuno, M.; Takase, H.; Kanamori, T.; Borlongan, C.V.; Haji, A.; Matsukawa, N. Hippocampal Cholinergic Neurostimulating Peptide as a Possible Modulating Factor against Glutamatergic Neuronal Disability by Amyloid Oligomers. Cell Transplant. 2017, 26, 1542–1550. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Andreasen, J.T.; Arvaniti, M.; Kohlmeier, K.A. Nicotinic Acetylcholine Receptors in the Pathophysiology of Al zheimer’s Disease: The Role of Protein-Protein Interactions in Current and Future Treatment. Curr. Pharm. Des. 2016, 22, 2015–2034. [Google Scholar] [CrossRef]
- Ishola, I.O.; Olubodun-Obadun, T.G.; Ojulari, M.A.; Adeyemi, O.O. Rutin ameliorates scopolamine-induced learning and memory impairments through enhancement of antioxidant defense system and cholinergic signaling. Drug Metab. Pers. Ther. 2020, 36, 53–61. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Li, Z.; Liu, L.; Tang, W.-X.; Wang, Y.; Dong, M.-R.; Xiao, C. Curcumin Attenuates Β-Amyloid-Induced Neuroinflammation via Activation of Peroxisome Proliferator-Activated Receptor-Gamma Function in a Rat Model of Alzheimer’s Disease. Front. Pharmacol. 2016, 7, 261. [Google Scholar] [CrossRef]
- Jha, A.B.; Panchal, S.S.; Shah, A. Ellagic acid: Insights into its neuroprotective and cognitive enhancement effects in sporadic Alzheimer’s disease. Pharmacol. Biochem. Behav. 2018, 175, 33–46. [Google Scholar] [CrossRef]
- Yu, H.; Yuan, B.; Chu, Q.; Wang, C.; Bi, H. Protective roles of isoastilbin against Alzheimer’s disease via Nrf2mediated antioxidation and antiapoptosis. Int. J. Mol. Med. 2019, 43, 1406–1416. [Google Scholar]
- He, J.; Liao, T.; Zhong, G.X.; Zhang, J.D.; Chen, Y.P.; Wang, Q.; Zeng, Q.P. Alzheimer’s Disease-like Early-phase Brain Pathogenesis: Self-curing Amelioration of Neurodegeneration from Pro-inflammatory ‘Wounding’ to Anti-inflammatory ‘Healing’. Curr. Alzheimer Res. 2017, 14, 1123–1135. [Google Scholar] [CrossRef]
- Roman, G.C.; Jackson, R.E.; Gadhia, R.; Roman, A.N.; Reis, J. Mediterranean diet: The role of long-chain omega-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef]
- Xia, E.; Xu, F.; Hu, C.; Kumal, J.P.P.; Tang, X.; Mao, D.; Li, Y.; Wu, D.; Zhang, R.; Wu, S.; et al. Young Blood Rescues the Cog-nition of Alzheimer’s Model Mice by Restoring the Hippocampal Cholinergic Circuit. Neuroscience 2019, 417, 57–69. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Classification | Name | Property | Reported Correlation with AD | Adverse Reaction | References |
|---|---|---|---|---|---|
| First generation | Tacrine | Reversibility, Lipid solubility | Inhibits AChE in plasma and tissues. Promotes ACh release through M1 receptor. | Hepatotoxicity | [106] |
| Second generation | Donepezil | Reversibility, High selectivity | The only inhibitor that can act on both the peripheral and central catalytic sites of AChE. Increases the concentration of AChE by reversibly inhibiting the ACh hydrolysis induced by AChE. Increases the protein levels of PINK 1, NFASC, MYLK2 and NRAS in the hippocampus. | Nausea, Vomiting Diarrhea Fatigue | [102] |
| Second generation | Rivastigmine | Reversibility | Guides APP treatment away from BACE1 and toward A secretase. Increases the concentration of AChE by reversibly inhibiting the ACh hydrolysis induced by AChE. | Dizziness Vertigo Upper respiratory tract infection | [21,105] |
| Second generation | Galantamine | Reversibility | Allosteric activation effect on nicotinic ACh receptors. Activates MARK, PI3K and other cell signal transduction pathways to play an anti-inflammatory effect. Promotes the release of neurotransmitters associated with glutamate, norepinephrine and memory and mood. Protects nerves against oxidative damage caused by hydrogen peroxide. | Salivation Bradycardia Dizziness Abdominal pain | [107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. https://doi.org/10.3390/molecules27061816
Chen Z-R, Huang J-B, Yang S-L, Hong F-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules. 2022; 27(6):1816. https://doi.org/10.3390/molecules27061816
Chicago/Turabian StyleChen, Zhi-Ru, Jia-Bao Huang, Shu-Long Yang, and Fen-Fang Hong. 2022. "Role of Cholinergic Signaling in Alzheimer’s Disease" Molecules 27, no. 6: 1816. https://doi.org/10.3390/molecules27061816
APA StyleChen, Z.-R., Huang, J.-B., Yang, S.-L., & Hong, F.-F. (2022). Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules, 27(6), 1816. https://doi.org/10.3390/molecules27061816