
Natural Product-Derived Phosphonic Acids as Corrosion Inhibitors for Iron and Steel


Abstract
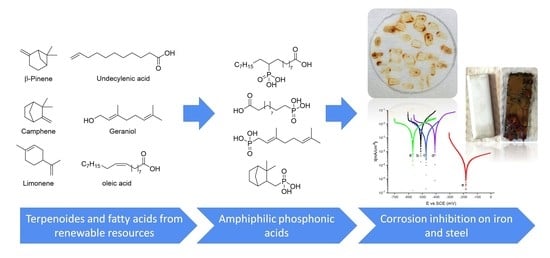
1. Introduction
2. Results
2.1. Synthesis
2.2. Evaluation of Corrosion Inhibition on Steel
2.2.1. Chip Filter Assays
2.2.2. Electrochemical Analysis
2.2.3. Gravimetric Evaluation of Anticorrosive Properties
3. Discussion
4. Materials and Methods
4.1. NMR Spectroscopy
4.2. Mass Spectrometry
4.3. Chip Filter Test
4.4. Gravimetric Corrosion Test
4.5. Electrochemistry
4.6. Palladium-Catalyzed Hydrophosphinylation (General Procedure)
Synthesis of 10-Carboxyundecylphosphonic Acid 3 [56]
4.7. Synthesis of 10-Phosphonooctadecanoic Acid 18 [57]
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Verma, C.; Olasunkanmi, L.O.; Ebenso, E.E.; Quraishi, M.A. Substituents effect on corrosion inhibition performance of organic compounds in aggressive ionic solutions: A review. J. Mol. Liq. 2018, 251, 100–118. [Google Scholar] [CrossRef]
- Groysman, A. Corrosion for Everybody; Springer Science & Business Media: Cham, Germany, 2010; pp. 1–368. [Google Scholar]
- Brinksmeier, E.; Meyer, D.; Huesmann-Cordes, A.G.; Herrmann, C. Metalworking fluids-mechanisms and performance. Cirp Ann.-Manuf. Technol. 2015, 64, 605–628. [Google Scholar] [CrossRef]
- Kuznetsov, Y.I. Organic corrosion inhibitors: Where are we now? A review. Part II. Passivation and the role of chemical structure of carboxylates. Int. J. Corros. Scale Inhib. 2016, 5, 282–318. [Google Scholar] [CrossRef]
- Gunasekaran, G.; Natarajan, R.; Muralidharan, V.S.; Palaniswamy, N.; Appa Rao, B.V. Inhibition by phosphonic acids—An overview. Anti-Corr. Method Mater. 1997, 44, 248–259. [Google Scholar] [CrossRef]
- Finšgar, M.; Jackson, J. Application of corrosion inhibitors for steels in acidic media for the oil and gas industry: A review. Corros. Sci. 2014, 86, 17–41. [Google Scholar] [CrossRef]
- Amar, H.; Benzakour, J.; Derja, A.; Villemin, D.; Moreau, B. A corrosion inhibition study of iron by phosphonic acids in sodium chloride solution. J. Electroanal. Chem. 2003, 558, 131–139. [Google Scholar] [CrossRef]
- Milošev, I.; Zimerl, D.; Carriére, C.; Zanna, S.; Seyeux, A.; Iskra, J.; Stavber, S.; Chiter, F.; Poberžnik, M.; Costa, D.; et al. Editors’ choice—The effect of anchor group and alkyl backbone chain on performance of organic compounds as corrosion inhibitors for aluminum investigated using an integrative experimental-modeling approach. J. Electrochem. Soc. 2020, 167, 061509. [Google Scholar] [CrossRef]
- Kokalj, A. Molecular modeling of organic corrosion inhibitors: Calculations, pitfalls, and conceptualization of molecule–surface bonding. Corros. Sci. 2021, 193, 109650. [Google Scholar] [CrossRef]
- Abohalkuma, T.; Shawish, F.; Telegdi, J. Phosphonic acid derivatives used in self assembled layers against metal corrosion. Int. J. Corros. Scale Inhib. 2014, 3, 151–159. [Google Scholar] [CrossRef]
- Zhu, Y.; Free, M.L.; Woollam, R.; Durnie, W. A review of surfactants as corrosion inhibitors and associated modeling. Prog. Mater. Sci. 2017, 90, 159–223. [Google Scholar] [CrossRef]
- Alonso Frank, M.; Meltzer, C.; Braunschweig, B.; Peukert, W.; Boccaccini, A.R.; Virtanen, S. Functionalization of steel surfaces with organic acids: Influence on wetting and corrosion behavior. Appl. Surf. Sci. 2017, 404, 326–333. [Google Scholar] [CrossRef]
- Kuznetsov, Y.I. Organic corrosion inhibitors: Where are we now? A review. Part IV. Passivation and the role of mono- and diphosphonates. Int. J. Corros. Scale Inhib. 2017, 6, 384–427. [Google Scholar]
- Kosian, M.; Smulders, M.M.; Zuilhof, H. Structure and long-term stability of alkylphosphonic acid monolayers on SS316L stainless steel. Langmuir 2016, 32, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, G.; Alauzun, J.G.; Granier, M.; Laurencin, D.; Mutin, P.H. Phosphonate coupling molecules for the control of surface/interface properties and the synthesis of nanomaterials. Dalton Trans. 2013, 42, 12569–12585. [Google Scholar] [CrossRef] [PubMed]
- Queffelec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface Modification Using Phosphonic Acids and Esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Raman, A.; Dubey, M.; Gouzman, I.; Gawalt, E.S. Formation of self-assembled monolayers of alkylphosphonic acid on the native oxide surface of SS316L. Langmuir 2006, 22, 6469–6472. [Google Scholar] [CrossRef]
- Boissezon, R.; Muller, J.; Beaugeard, V.; Monge, S.; Robin, J.-J. Organophosphonates as anchoring agents onto metal oxide-based materials: Synthesis and applications. RSC Adv. 2014, 4, 35690–35707. [Google Scholar] [CrossRef]
- Watanabe, S. Preparations and properties of anti-corrosion additives of water-soluble metal working fluids for aluminum alloy materials. J. Oleo Sci. 2008, 57, 1–10. [Google Scholar] [CrossRef][Green Version]
- Kuznetsov, Y.I. Organic corrosion inhibitors: Where are we now? A review. Part III. Passivation and the role of the chemical structure of organophosphates. Int. J. Corros. Scale Inhib. 2017, 6, 209–239. [Google Scholar]
- Demadis, K.D.; Katarachia, S.D. Metal-phosphonate chemistry: Synthesis, crystal structure of calcium-aminotris-(methylene phosphonate) and inhibition of CaCO3 crystal growth. Phosphorus Sulfur 2004, 179, 627–648. [Google Scholar] [CrossRef]
- Umoren, S.A.; Solomon, M.M.; Obot, I.B.; Suleiman, R.K. A critical review on the recent studies on plant biomaterials as corrosion inhibitors for industrial metals. J. Ind. Eng. Chem. 2019, 76, 91–115. [Google Scholar] [CrossRef]
- Choi, D.-J.; You, S.-J.; Kim, J.-G. Development of an environmentally safe corrosion, scale, and microorganism inhibitor for open recirculating cooling systems. Mater. Sci. Eng. A 2002, 335, 228–235. [Google Scholar] [CrossRef]
- Hamadi, L.; Mansouri, S.; Oulmi, K.; Kareche, A. The use of amino acids as corrosion inhibitors for metals: A review. Egyptian J. Petrol. 2018, 27, 1157–1165. [Google Scholar] [CrossRef]
- Verma, C.; Verma, D.K.; Ebenso, E.E.; Quraishi, M.A. Sulfur and phosphorus heteroatom-containing compounds as corrosion inhibitors: An overview. Heteroat. Chem. 2018, 29, e21437. [Google Scholar] [CrossRef]
- Anastas, P.T. Benign by design chemistry. In Benign by Design; American Chemical Society: Washington, DC, USA, 1994; Volume 577, pp. 2–22. [Google Scholar]
- Kümmerer, K. Sustainable from the very beginning: Rational design of molecules by life cycle engineering as an important approach for green pharmacy and green chemistry. Green Chem. 2007, 9, 899–907. [Google Scholar] [CrossRef]
- Maison, W.; Kipphardt, H.; Ruf, E. Naturstoffbasierte Phosphonsäuren als Saure Korrosionsinhibitoren. WIPO Patent Application No. WO/2021/165313, 17 February 2021. [Google Scholar]
- Sevrain, C.M.; Berchel, M.; Couthon, H.; Jaffres, P.A. Phosphonic acid: Preparation and applications. Beilstein J. Org. Chem. 2017, 13, 2186–2213. [Google Scholar] [CrossRef]
- Bouilhac, C.; Travelet, C.; Graillot, A.; Monge, S.; Borsali, R.; Robin, J.J. Synthesis of fatty phosphonic acid based polymethacrylamide by RAFT polymerization and self-assembly in solution. Polym. Chem. 2014, 5, 2756–2767. [Google Scholar] [CrossRef]
- Bravo-Altamirano, K.; Montchamp, J.-L. A novel approach to phosphonic acids from hypophosphorous acid. Tetrahedron Lett. 2007, 48, 5755–5759. [Google Scholar] [CrossRef]
- Coudray, L.; Bravo-Altamirano, K.; Montchamp, J.L. Allylic phosphinates via palladium-catalyzed allylation of H-phosphinic acids with allylic alcohols. Org. Lett. 2008, 10, 1123–1126. [Google Scholar] [CrossRef]
- Bravo-Altamirano, K.; Abrunhosa-Thomas, I.; Montchamp, J.-L. Palladium-catalyzed reactions of hypophosphorous compounds with allenes, dienes, and allylic electrophiles: Methodology for the synthesis of allylic H-phosphinates. J. Org. Chem. 2008, 73, 2292–2301. [Google Scholar] [CrossRef]
- Berger, O.; Winters, K.R.; Sabourin, A.; Dzyuba, S.V.; Montchamp, J.-L. On the cost of academic methodologies. Org. Chem. Front. 2019, 6, 2095–2108. [Google Scholar] [CrossRef]
- Kelley, T.W.; Boardman, L.D.; Dunbar, T.D.; Muyres, D.V.; Pellerite, M.J.; Smith, T.P. High-performance OTFTs using surface-modified alumina dielectrics. J. Phys. Chem. B 2003, 107, 5877–5881. [Google Scholar] [CrossRef]
- Belbachir, M.; Boutevin, B.; Pietrasanta, Y.; Rigal, G. Synthesis and chemical-transformations of telomers from methyl 10-unidecenoate. 1. Makromol. Chem.-Macromol. Chem. Phys. 1982, 183, 2347–2358. [Google Scholar] [CrossRef]
- Griffin, C.E.; Wells, H.J. Phosphonic acids and esters. 1. Radical initiated addition of phosphorous acid to olefins. J. Org. Chem. 1959, 24, 2049–2051. [Google Scholar] [CrossRef]
- Deprele, S.; Montchamp, J.L. Triethylborane-initiated room temperature radical addition of hypophosphites to olefins: Synthesis of monosubstituted phosphinic acids and esters. J. Org. Chem. 2001, 66, 6745–6755. [Google Scholar] [CrossRef]
- Francois, H.; Lalande, R. Radical-addition of phosphorus derivatives to pinenes. C. R. Acad. Sci. C Chim. 1974, 279, 117–119. [Google Scholar]
- Eddingsaas, N.C.; Loza, C.L.; Yee, L.D.; Seinfeld, J.H.; Wennberg, P.O. α-pinene photooxidation under controlled chemical conditions—Part 1: Gas-phase composition in low- and high-NOx environments. Atmos. Chem. Phys. 2012, 12, 6489–6504. [Google Scholar] [CrossRef]
- Biresaw, G.; Bantchev, G.B. Tribological properties of limonene bisphosphonates. Tribol. Lett. 2015, 60, 11. [Google Scholar] [CrossRef]
- Blau, N.F.; Wang, T.T.S.; Buess, C.M. Potential Inhibitors of cholesterol biosynthesis phosphonates derived from geraniol and congeners. J. Chem. Eng. Data 1970, 15, 206–208. [Google Scholar] [CrossRef]
- Popjak, G.; Hadley, C. Inhibition of liver prenyltransferase by citronellyl and geranyl phosphonate and phosphonylphosphate. J. Lipid Res. 1985, 26, 1151. [Google Scholar] [CrossRef]
- Yoo, S.-H.; Kim, Y.-W.; Shin, J.; Kim, N.-K.; Kim, J.-S. Effects of the chain length of tris(carboxyalkylamino)triazine on corrosion inhibition properties. Bull. Korean Chem. Soc. 2015, 36, 346–355. [Google Scholar] [CrossRef]
- Furó, I. NMR spectroscopy of micelles and related systems. J. Mol. Liq. 2005, 117, 117–137. [Google Scholar] [CrossRef]
- Felhősi, I.; Telegdi, J.; Pálinkás, G.; Kálmán, E. Kinetics of self-assembled layer formation on iron. Electrochim. Acta 2002, 47, 2335–2340. [Google Scholar] [CrossRef]
- Földes, E.; Tóth, A.; Kálmán, E.; Fekete, E.; Tomasovszky–Bobák, á. Surface changes of corona-discharge-treated polyethylene films. J. Appl. Polym. Sci. 2000, 76, 1529–1541. [Google Scholar] [CrossRef]
- Felhösi, I.; Kálmán, E.; Póczik, P. Corrosion protection by self-assembly. Russ. J. Electrochem. 2002, 38, 230–237. [Google Scholar] [CrossRef]
- Paszternák, A.; Stichleutner, S.; Felhősi, I.; Keresztes, Z.; Nagy, F.; Kuzmann, E.; Vértes, A.; Homonnay, Z.; Pető, G.; Kálmán, E. Surface modification of passive iron by alkyl-phosphonic acid layers. Electrochim. Acta 2007, 53, 337–345. [Google Scholar] [CrossRef]
- Shi, X.; Wang, Y.; Li, H.; Zhang, S.; Zhao, R.; Li, G.; Zhang, R.; Sheng, Y.; Cao, S.; Zhao, Y.; et al. Corrosion resistance and biocompatibility of calcium-containing coatings developed in near-neutral solutions containing phytic acid and phosphoric acid on AZ31B alloy. J. Alloys Compd. 2020, 823, 153721. [Google Scholar] [CrossRef]
- Soleymanibrojeni, M.; Shi, H.; Udoh, I.I.; Liu, F.; Han, E.-H. Microcontainers with 3-amino-1,2,4-triazole-5-thiol for Enhancing anticorrosion waterborne coatings for AA2024-T3. Prog. Org. Coat. 2019, 137, 105336. [Google Scholar] [CrossRef]
- Hefter, G.T.; North, N.A.; Tan, S.H. Organic corrosion inhibitors in neutral solutions; Part 1—Inhibition of steel, copper, and aluminum by straight chain carboxylates. Corrosion 1997, 53, 657–667. [Google Scholar] [CrossRef]
- Pawsey, S.; McCormick, M.; De Paul, S.; Graf, R.; Lee, Y.S.; Reven, L.; Spiess, H.W. 1H fast MAS NMR studies of hydrogen-bonding interactions in self-assembled monolayers. J. Am. Chem. Soc. 2003, 125, 4174–4184. [Google Scholar] [CrossRef]
- Amar, H.; Benzakour, J.; Derja, A.; Villemin, D.; Moreau, B.; Braisaz, T. Piperidin-1-yl-phosphonic acid and (4-phosphono-piperazin-1-yl) phosphonic acid: A new class of iron corrosion inhibitors in sodium chloride 3% media. Appl. Surf. Sci. 2006, 252, 6162–6172. [Google Scholar] [CrossRef]
- Ormellese, M.; Lazzari, L.; Goidanich, S.; Fumagalli, G.; Brenna, A. A study of organic substances as inhibitors for chloride-induced corrosion in concrete. Corros. Sci. 2009, 51, 2959–2968. [Google Scholar] [CrossRef]
- Chavane, V. Chimie Organique-Synthese de Quelques Acides Phosphoniques Amines de Formule Generale H3n+ (-)(Ch2) N-Po3h. Compt. Rend. Hebd. Seances L Acad. Sci. 1947, 224, 406–408. [Google Scholar]
- Millet, F.; Auvergne, R.; Caillol, S.; David, G.; Manseri, A.; Pébère, N. Improvement of corrosion protection of steel by incorporation of a new phosphonated fatty acid in a phosphorus-containing polymer coating obtained by UV curing. Prog. Org. Coat. 2014, 77, 285–291. [Google Scholar] [CrossRef]
- Siu, J.C.; Parry, J.B.; Lin, S. Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc. 2019, 141, 2825–2831. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acid, c (mmol/L) | jcorr (nA/cm2) | IEIcorr (%) | Ecorr (mV) | βa (mV/dec) | βc (mV/dec) | CR (mm/y) |
|---|---|---|---|---|---|---|---|
| 1 | −[a] | 37.5 × 103 | − | −584 | 544 | 1540 | 436 × 10−3 |
| 2 | TC [b], 64.4 | 11.4 | 99.97 | −153 | 174 | 65.3 | 132 × 10−6 |
| 3 | TC [b], 2.2 | 56.2 | 99.85 | −141 | 184 | 76.9 | 653 × 10−6 |
| 4 | 3 [b], 112 | 5.79 | 99.98 | −149 | 211 | 62.7 | 67.3 × 10−6 |
| 5 | 3 [b], 3.8 | 23.6 | 99.94 | −189 | 80.0 | 49.2 | 274 × 10−6 |
| 6 | 3 [b], 18.8 | 5.46 | 99.99 | −130 | 202 | 64.6 | 63.5 × 10−6 |
| 7 | 18 [b], 18.8 | 1.79 × 103 | 95.23 | −368 | 402 | 244 | 20.8 × 10−3 |
| 8 | OPA [c], 18.8 | 1.40 × 103 | 96.27 | −305 | 92.0 | 110 | 16.2 × 10−3 |
| 9 | OPA [d], 18.8 | 10.7 | 99.97 | −107 | 297 | 62.2 | 125 × 10−6 |
| Entry | Acid, c (mmol/L) | jcorr (nA/cm2) | IEIcorr (%) | Ecorr (mV) | βa (mV/dec) | βc (mV/dec) | CR (mm/y) |
|---|---|---|---|---|---|---|---|
| 3.0% NaCl | |||||||
| 1 | −[a] | 26.9 × 103 | - | −511 | 136 | 1.44 × 106 | 313 × 10−3 |
| 2 | TC [b], 64 | 18.2 × 103 | 32.34 | −472 | 98.2 | 986 × 103 | 209 × 10−3 |
| 3 | OPA [c], 154 | 63.5 × 103 | −[d] | −569 | 915 | 2.57 × 103 | 738 × 10−3 |
| 4 | 3 [b], 64 | 68.8 | 99.74 | −190 | 356 | 87.2 | 800 × 10−6 |
| 5 | 18 [b], 112 | 2.31 × 103 | 14.13 | −408 | 88.4 | 123 | 26.9 × 10−3 |
| 0.5% NaCl | |||||||
| 6 | 18 [b], 40 | 37.5 | 99.86 | −240 | 319 | 70.9 | 436 × 10−6 |
| 7 | 3 [b], 20 | 11.4 | 99.96 | −190 | 302 | 36.7 | 134 × 10−6 |
| 8 | TC [b], 64 | 2.26 × 103 | 91.60 | −258 | 449 | 94.5 | 26.2 × 10−3 |
| 0.1% NaCl | |||||||
| 9 | TC [b], 64 | 129 | 99.52 | −170 | 382 | 78.4 | 152 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruf, E.; Naundorf, T.; Seddig, T.; Kipphardt, H.; Maison, W. Natural Product-Derived Phosphonic Acids as Corrosion Inhibitors for Iron and Steel. Molecules 2022, 27, 1778. https://doi.org/10.3390/molecules27061778
Ruf E, Naundorf T, Seddig T, Kipphardt H, Maison W. Natural Product-Derived Phosphonic Acids as Corrosion Inhibitors for Iron and Steel. Molecules. 2022; 27(6):1778. https://doi.org/10.3390/molecules27061778
Chicago/Turabian StyleRuf, Erik, Tim Naundorf, Tom Seddig, Helmut Kipphardt, and Wolfgang Maison. 2022. "Natural Product-Derived Phosphonic Acids as Corrosion Inhibitors for Iron and Steel" Molecules 27, no. 6: 1778. https://doi.org/10.3390/molecules27061778
APA StyleRuf, E., Naundorf, T., Seddig, T., Kipphardt, H., & Maison, W. (2022). Natural Product-Derived Phosphonic Acids as Corrosion Inhibitors for Iron and Steel. Molecules, 27(6), 1778. https://doi.org/10.3390/molecules27061778