
Development of a Nuclear Magnetic Resonance Method and a Near Infrared Calibration Model for the Rapid Determination of Lipid Content in the Field Pea (Pisum sativum)

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
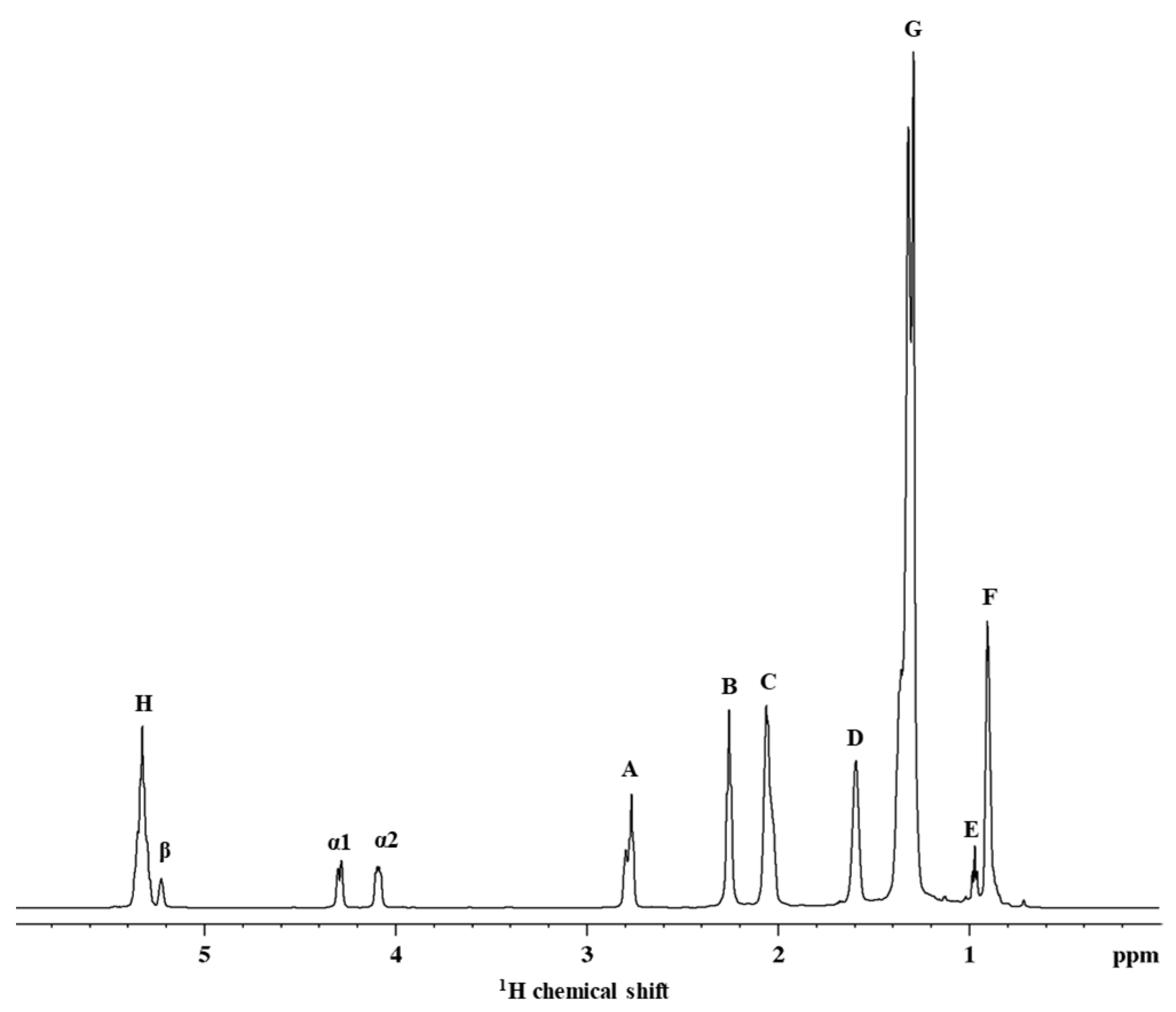
2.1. HR-MAS NMR Spectroscopy
2.1.1. Fatty Acid Composition of Pea Seeds Using 1H HR-MAS NMR
2.1.2. Quantification of Total Lipid Content Using 1H HR-MAS NMR
2.1.3. Seed Viability
2.2. NIR Spectroscopy
3. Materials and Methods
3.1. HR-MAS NMR Spectroscopy
3.1.1. Pea Accessions and Sample Preparation
3.1.2. Instrumental Setup
3.1.3. HR-MAS NMR Spectra Processing
3.1.4. Statistical Analyses of HR-MAS NMR Data
3.2. NIR Spectroscopy
3.2.1. Instrumental Setup and Scan Parameters
3.2.2. Data Pre-Processing
3.2.3. Calibration and Validation Model Design
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Iqbal, A.; Khalil, I.A.; Ateeq, N.; Khan, M.S. Nutritional quality of important food legumes. Food Chem. 2006, 97, 331–335. [Google Scholar] [CrossRef]
- Jezierny, D.; Mosenthin, R.; Bauer, E. The use of grain legumes as a protein source in pig nutrition: A review. Anim. Feed Sci. Technol. 2010, 157, 111–128. [Google Scholar] [CrossRef]
- Karn, A.; Heim, C.; Flint-Garcia, S.; Bilyeu, K.; Gillman, J. Development of rigorous fatty acid near-infrared spectroscopy quantitation methods in support of soybean oil improvement. J. Am. Oil Chem. Soc. 2017, 94, 69–76. [Google Scholar] [CrossRef]
- Ryan, E.; Galvin, K.; O’Connor, T.P.; Maguire, A.R.; O’Brien, N.M. Phytosterol, squalene, tocopherol content and fatty acid profile of selected seeds, grains, and legumes. Plant Foods Hum. Nutr. 2007, 62, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Khodapanahi, E.; Lefsrud, M.; Orsat, V.; Singh, J.; Warkentin, T.D. Study of pea accessions for development of an oilseed pea. Energies 2012, 5, 3788–3802. [Google Scholar] [CrossRef]
- Sarwar, M.F.; Sarwar, M.H.; Sarwar, M.; Qadri, N.A.; Moghal, S. The role of oilseeds nutrition in human health: A critical review. J. Cereal Oil 2013, 4, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Kocer, A.; Albayrak, S. Determination of forage yield and quality of pea (Pisum sativum L.) mixtures with oat and barley. Turkish J. Field Crop. 2012, 17, 96–99. [Google Scholar]
- Solis, M.I.V.; Patel, A.; Orsat, V.; Singh, J.; Lefsrud, M. Fatty acid profiling of the seed oils of some varieties of field peas (Pisum sativum) by RP-LC/ESI-MS/MS: Towards the development of an oilseed pea. Food Chem. 2013, 139, 986–993. [Google Scholar] [CrossRef]
- Yoshida, H.; Tomiyama, Y.; Tanaka, M.; Mizushina, Y. Characteristic profiles of lipid classes, fatty acids and triacylglycerol molecular species of peas (Pisum sativum L.). Eur. J. Lipid Sci. Technol. 2007, 109, 600–607. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 1–32. [Google Scholar] [CrossRef]
- Ahmad, S.; Kaur, S.; Lamb-Palmer, N.D.; Lefsrud, M.; Singh, J. Genetic diversity and population structure of Pisum sativum accessions for marker-trait association of lipid content. Crop J. 2015, 3, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Caprioli, G.; Giusti, F.; Ballini, R.; Sagratini, G.; Vila-Donat, P.; Vittori, S.; Fiorini, D. Lipid nutritional value of legumes: Evaluation of different extraction methods and determination of fatty acid composition. Food Chem. 2016, 192, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.S.; Vicenti, J.R.; Moron-Villarreyes, J.A.; Caldas, S.; Cardoso, L.V.; Freitas, R.F.; D’oca, M.G. Extraction and characterization of lipids from Sarcocornia ambigua meal: A halophyte biomass produced with shrimp farm effluent irrigation. An. Acad. Bras. Ciênc. 2014, 86, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padhi, E.M.; Liu, R.; Hernandez, M.; Tsao, R.; Ramdath, D.D. Total polyphenol content, carotenoid, tocopherol and fatty acid composition of commonly consumed Canadian pulses and their contribution to antioxidant activity. J. Funct. Food. 2017, 38, 602–611. [Google Scholar] [CrossRef]
- Mazurek, B.; Chmiel, M.; Górecka, B. Fatty acids analysis using gas chromatography-mass spectrometer detector (GC/MSD)-method validation based on berry seed extract samples. Food Anal. Method. 2017, 10, 2868–2880. [Google Scholar] [CrossRef] [Green Version]
- Kalia, R. Correlation of Lipid Content and Phenotypic Markers of Canadian Field Peas (Pisum sativum); McGill University Libraries: Montreal, QC, Canada, 2016. [Google Scholar]
- Kleinberg, R.L.; Jackson, J.A. An introduction to the history of NMR well logging. Concepts Magn. Res. 2001, 13, 340–342. [Google Scholar] [CrossRef]
- Moser, E.; Laistler, E.; Schmitt, F.; Kontaxis, G. Ultra-High Field NMR and MRI-The Role of Magnet Technology to Increase Sensitivity and Specificity. Front. Phys. 2017, 5, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Baias, M.; Dumez, J.-N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De novo determination of the crystal structure of a large drug molecule by crystal structure prediction-based powder NMR crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, A.P.; Testone, G.; Santoro, F.; Nicolodi, C.; Iannelli, M.A.; Amato, M.E.; Ianniello, A.; Brosio, E.; Giannino, D.; Mannina, L. Quality traits of conventional and transgenic lettuce (Lactuca sativa L.) at harvesting by NMR metabolic profiling. J. Agric. Food Chem. 2010, 58, 6928–6936. [Google Scholar] [CrossRef]
- Baianu, I.; You, T.; Costescu, D.; Lozano, P.; Prisecaru, V.; Nelson, R.L. Determination of soybean oil, protein and amino acid residues in soybean seeds by high resolution nuclear magnetic resonance (NMRS) and near infrared (NIRS). Nature 2012, 1, 1–62. [Google Scholar] [CrossRef]
- Cen, H.; He, Y. Theory and application of near infrared reflectance spectroscopy in determination of food quality. Trends Food Sci. Technol. 2007, 18, 72–83. [Google Scholar] [CrossRef]
- Shi, H.; Lei, Y.; Prates, L.L.; Yu, P. Evaluation of near-infrared (NIR) and Fourier transform mid-infrared (ATR-FT/MIR) spectroscopy techniques combined with chemometrics for the determination of crude protein and intestinal protein digestibility of wheat. Food Chem. 2019, 272, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, S.; Wu, X.; Xing, C.; Yuan, J. Determination of soybean routine quality parameters using near-infrared spectroscopy. Food Sci. Nutr. 2018, 6, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Porep, J.U.; Kammerer, D.R.; Carle, R. On-line application of near infrared (NIR) spectroscopy in food production. Trends Food Sci. Technol. 2015, 46, 211–230. [Google Scholar] [CrossRef]
- Barison, A.; Pereira da Silva, C.W.; Campos, F.R.; Simonelli, F.; Lenz, C.A.; Ferreira, A.G. A simple methodology for the determination of fatty acid composition in edible oils through 1H NMR spectroscopy. Magn. Reson. Chem. 2010, 48, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Salinero, C.; Feás, X.; Mansilla, J.P.; Seijas, J.A.; Vázquez-Tato, M.P.; Vela, P.; Sainz, M.J. 1H-nuclear magnetic resonance analysis of the triacylglyceride composition of cold-pressed oil from Camellia japonica. Molecules 2012, 17, 6716–6727. [Google Scholar] [CrossRef] [Green Version]
- Guillén, M.D.; Ruiz, A. Monitoring of heat-induced degradation of edible oils by proton NMR. Eur. J. Lipid Sci. Technol. 2008, 110, 52–60. [Google Scholar] [CrossRef]
- Lanzmann-Petithory, D. Alpha-linolenic acid and cardiovascular diseases. J. Nutr. Health Aging 2001, 5, 179–183. [Google Scholar]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Riccioli, C.; Sun, D.W. An overview on nondestructive spectroscopic techniques for lipid and lipid oxidation analysis in fish and fish products. Compr. Rev. Food Sci. Food Saf. 2015, 14, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Moreau, R.A.; Powell, M.J.; Singh, V. Pressurized liquid extraction of polar and nonpolar lipids in corn and oats with hexane, methylene chloride, isopropanol, and ethanol. J. Oil Am. Chem. Soc. 2003, 80, 1063–1067. [Google Scholar] [CrossRef]
- Ossowski, P. An Overview of Results Associated with the Experiments Performed for the Development of an Oilseed Pea; McGill University Libraries: Montreal, QC, Canada, 2017. [Google Scholar]
- Baianu, I.; Guo, J. NIR Calibrations for Soybean Seeds and Soy Food Composition Analysis: Total Carbohydrates, Oil, Proteins and Water Contents. Nat. Preced. 2011. [Google Scholar] [CrossRef]
- Armstrong, P. Rapid single-kernel NIR measurement of grain and oil-seed attributes. Appl. Eng. Agric. 2006, 22, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, H.N.; Dalrymple, D.M.; Serajuddin, A.T. A comparative evaluation of mono-, di-and triglyceride of medium chain fatty acids by lipid/surfactant/water phase diagram, solubility determination and dispersion testing for application in pharmaceutical dosage form development. Pharm. Res. 2012, 29, 285–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillén, M.D.; Ruiz, A. 1H nuclear magnetic resonance as a fast tool for determining the composition of acyl chains in acylglycerol mixtures. Eur. J. Lipid Sci. Technol. 2003, 105, 502–507. [Google Scholar] [CrossRef]
- Albers, M.J.; Butler, T.N.; Rahwa, I.; Bao, N.; Keshari, K.R.; Swanson, M.G.; Kurhanewicz, J. Evaluation of the ERETIC method as an improved quantitative reference for 1H HR-MAS spectroscopy of prostate tissue. Magn. Reson. Med. 2009, 61, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. TrAC-Trend. Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sample | Form | Linolenic Fatty Acid (%) | Linoleic Fatty Acid (%) | Oleic Fatty Acid (%) | Saturated Fatty Acid (%) |
|---|---|---|---|---|---|
| F1 | Seed portion | 26.62 ± 1.65 a | 23.57 ± 4.60 c | 44.49 ± 3.86 a | 5.32 ± 4.26 b |
| Ground | 24.56 ± 0.56 a | 20.28 ± 5.07 c | 45.64 ± 5.52 a | 9.51 ± 0.90 b | |
| 112351 | Seed portion | 23.84 ± 0.56 ab | 31.42 ± 1.42 b | 32.33 ± 0.65 bc | 12.42 ± 1.42 b |
| Ground | 22.35 ± 2.69 ab | 30.81 ± 0.77 b | 34.89 ± 0.54 bc | 11.96 ± 2.63 b | |
| F2 | Seed portion | 22.29 ± 1.15 b | 34.16 ± 2.40 ab | 31.98 ± 1.10 bc | 11.56 ± 3.05 b |
| Ground | 22.95 ± 2.15 b | 36.50 ± 3.60 ab | 29.28 ± 2.31 bc | 11.28 ± 0.20 b | |
| 42819 | Seed portion | 23.42 ± 0.52 ab | 43.98 ± 7.73 a | 21.71 ± 7.72 c | 10.89 ± 0.21 b |
| Ground | 23.86 ± 0.26 ab | 35.79 ± 3.45 a | 30.59 ± 3.77 c | 9.76 ± 0.79 b | |
| 43016 | Seed portion | 20.88 ± 0.86 b | 35.30 ± 5.24 ab | 34.01 ± 4.90 bc | 9.81 ± 0.69 b |
| Ground | 21.47 ± 0.92 b | 35.78 ± 5.32 ab | 32.29 ± 6.18 bc | 10.46 ± 0.45 b | |
| 29600 | Seed portion | 23.27 ± 1.35 ab | 31.13 ± 3.05 b | 40.99 ± 3.42 ab | 4.61 ± 2.14 b |
| Ground | 22.85 ± 1.61 ab | 28.97 ± 7.28 b | 41.03 ± 7.25 ab | 7.15 ± 1.25 b | |
| 45760 | Seed portion | 21.24 ± 1.64 b | 36.63 ± 1.33 ab | 31.37 ± 1.30 bc | 10.76 ± 1.63 b |
| Ground | 20.81 ± 2.72 b | 34.81 ± 3.13 ab | 31.71 ± 3.81 bc | 12.66 ± 0.94 b | |
| 29579 | Seed portion | 24.08 ± 0.58 ab | 20.19 ± 0.52 c | 44.63 ± 3.75 a | 11.09 ± 3.24 b |
| Ground | 22.61 ± 2.85 ab | 13.89 ± 3.45 c | 55.42 ± 1.17 a | 8.08 ± 4.74 b | |
| 29526 | Seed portion | 25.52 ± 1.04 a | 21.00 ± 7.40 c | 25.24 ± 3.57 bc | 28.24 ± 4.50 a |
| Ground | 25.45 ± 7.46 a | 18.42 ± 7.85 c | 34.37 ± 3.52 bc | 21.76 ± 2.83 a |
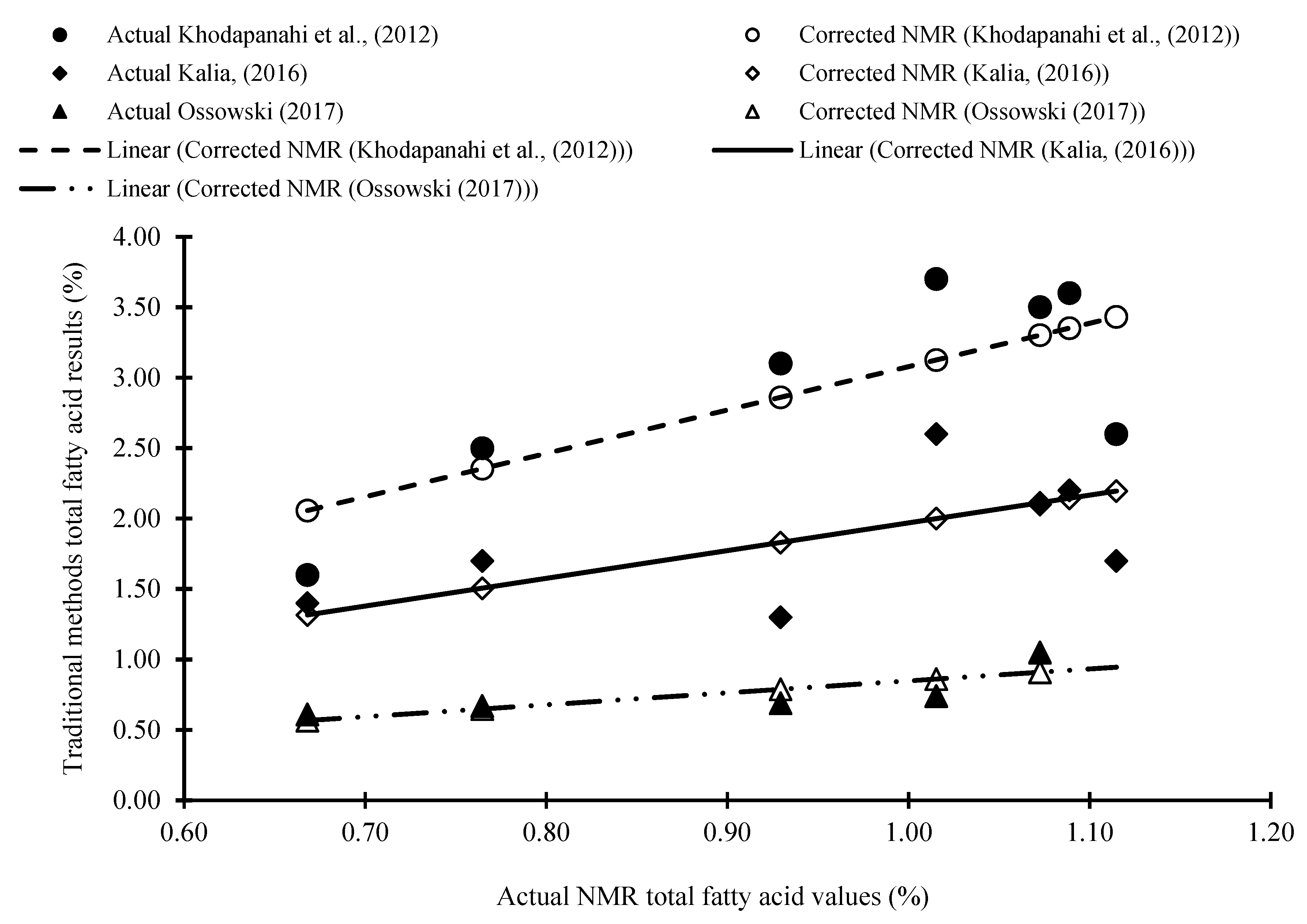
| Samples | Actual NMR Data | Khodapanahi et al. (2012) | Kalia (2016) | Ossowski (2017) | |||
|---|---|---|---|---|---|---|---|
| Actual Soxhlet (Butanol) | Corrected NMR | Actual Soxhlet (Hexane-Isopropanol) | Corrected NMR | Actual Soxhlet (Petroleum Ether) | Corrected NMR | ||
| Soybean | 4.60 ± 0.23 b | 13.90 | 14.17 | NR | NR | NR | NR |
| F1 | 0.80 ± 0.06 a | NR | NR | NR | NR | NR | NR |
| 112351 | 0.67 ± 0.05 a | 1.60 | 2.06 | 1.40 | 1.32 | 0.61 | 0.57 |
| F2 | 1.09 ± 0.05 a | NR | NR | NR | NR | NR | NR |
| 42819 | 0.77 ± 0.10 a | 2.50 | 2.37 | 1.70 | 1.52 | 0.67 | 0.65 |
| 43016 | 1.09± 0.09 a | 3.60 | 3.36 | 2.20 | 2.15 | NR | NR |
| 29600 | 1.12 ± 0.08 a | 2.60 | 3.45 | 1.70 | 2.21 | NR | NR |
| 45760 | 1.07 ± 0.12 a | 3.50 | 3.30 | 2.10 | 2.11 | 1.05 | 0.91 |
| 29579 | 1.02 ± 0.06 a | 3.70 | 3.14 | 2.60 | 2.01 | 0.74 | 0.87 |
| 29526 | 0.93 ± 0.10 a | 3.10 | 2.86 | 1.30 | 1.83 | 0.69 | 0.79 |
| Pre-Processing Step(s) | Factors | R2 | SECV | Reference Std. Dev. | RPD |
|---|---|---|---|---|---|
| [a] | 4 | 0.96 | 0.13 | 0.50 | 3.97 |
| [b] | 3 | 0.98 | 0.11 | 0.50 | 4.62 |
| [b] [c] | 4 | 0.99 | 0.11 | 0.50 | 4.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addo, P.W.; Ossowski, P.; MacPherson, S.; Gravel, A.E.; Kaur, R.; Hoyos-Villegas, V.; Singh, J.; Orsat, V.; Dumont, M.-J.; Lefsrud, M. Development of a Nuclear Magnetic Resonance Method and a Near Infrared Calibration Model for the Rapid Determination of Lipid Content in the Field Pea (Pisum sativum). Molecules 2022, 27, 1642. https://doi.org/10.3390/molecules27051642
Addo PW, Ossowski P, MacPherson S, Gravel AE, Kaur R, Hoyos-Villegas V, Singh J, Orsat V, Dumont M-J, Lefsrud M. Development of a Nuclear Magnetic Resonance Method and a Near Infrared Calibration Model for the Rapid Determination of Lipid Content in the Field Pea (Pisum sativum). Molecules. 2022; 27(5):1642. https://doi.org/10.3390/molecules27051642
Chicago/Turabian StyleAddo, Philip Wiredu, Philip Ossowski, Sarah MacPherson, Andrée E. Gravel, Rajvinder Kaur, Valerio Hoyos-Villegas, Jaswinder Singh, Valérie Orsat, Marie-Josée Dumont, and Mark Lefsrud. 2022. "Development of a Nuclear Magnetic Resonance Method and a Near Infrared Calibration Model for the Rapid Determination of Lipid Content in the Field Pea (Pisum sativum)" Molecules 27, no. 5: 1642. https://doi.org/10.3390/molecules27051642
APA StyleAddo, P. W., Ossowski, P., MacPherson, S., Gravel, A. E., Kaur, R., Hoyos-Villegas, V., Singh, J., Orsat, V., Dumont, M.-J., & Lefsrud, M. (2022). Development of a Nuclear Magnetic Resonance Method and a Near Infrared Calibration Model for the Rapid Determination of Lipid Content in the Field Pea (Pisum sativum). Molecules, 27(5), 1642. https://doi.org/10.3390/molecules27051642