
Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S)


Abstract
:1. Introduction
2. Results and Discussion
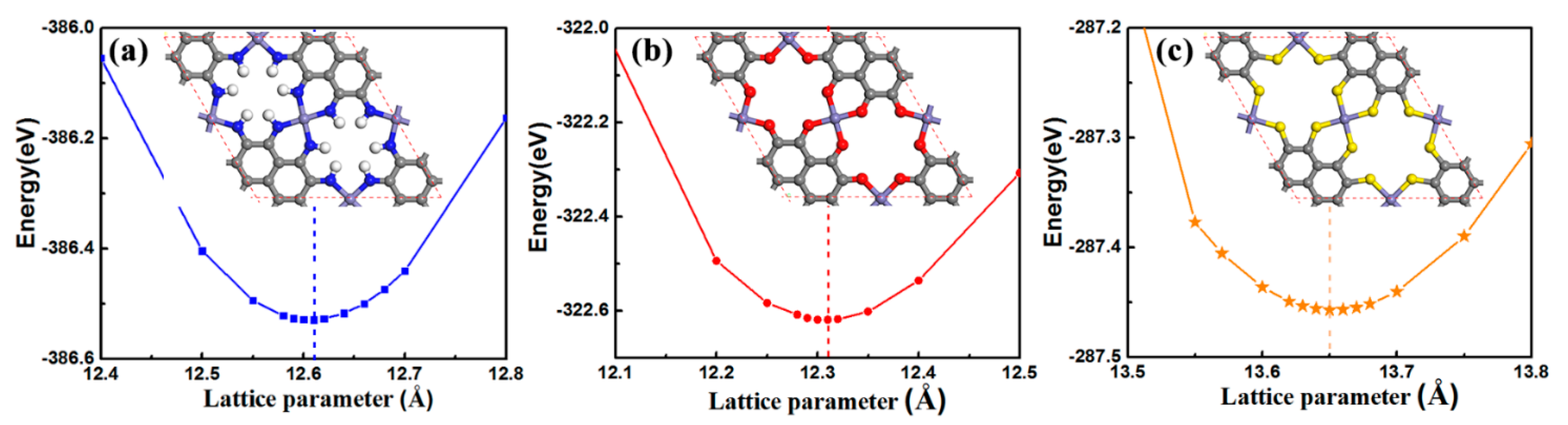
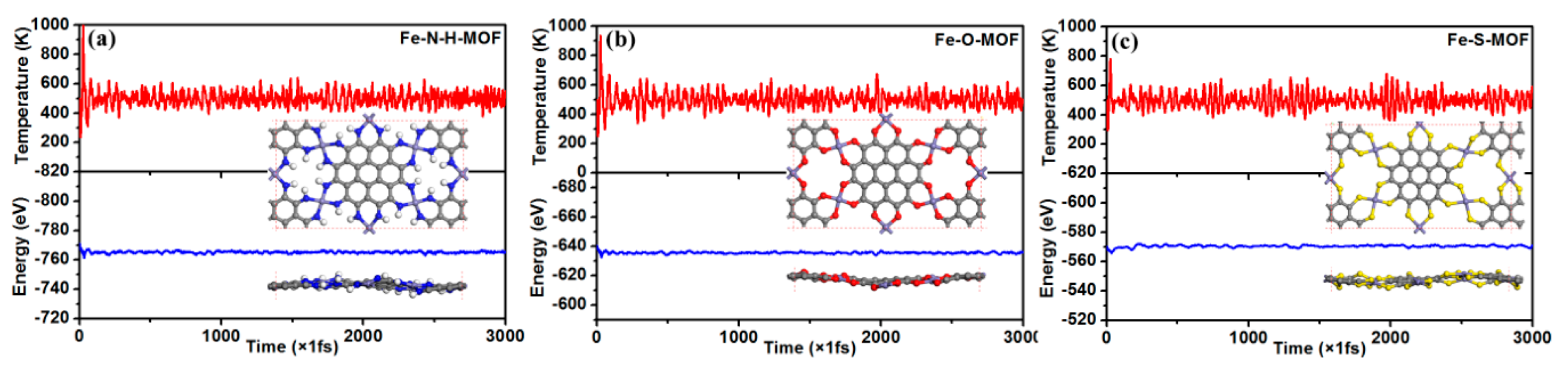
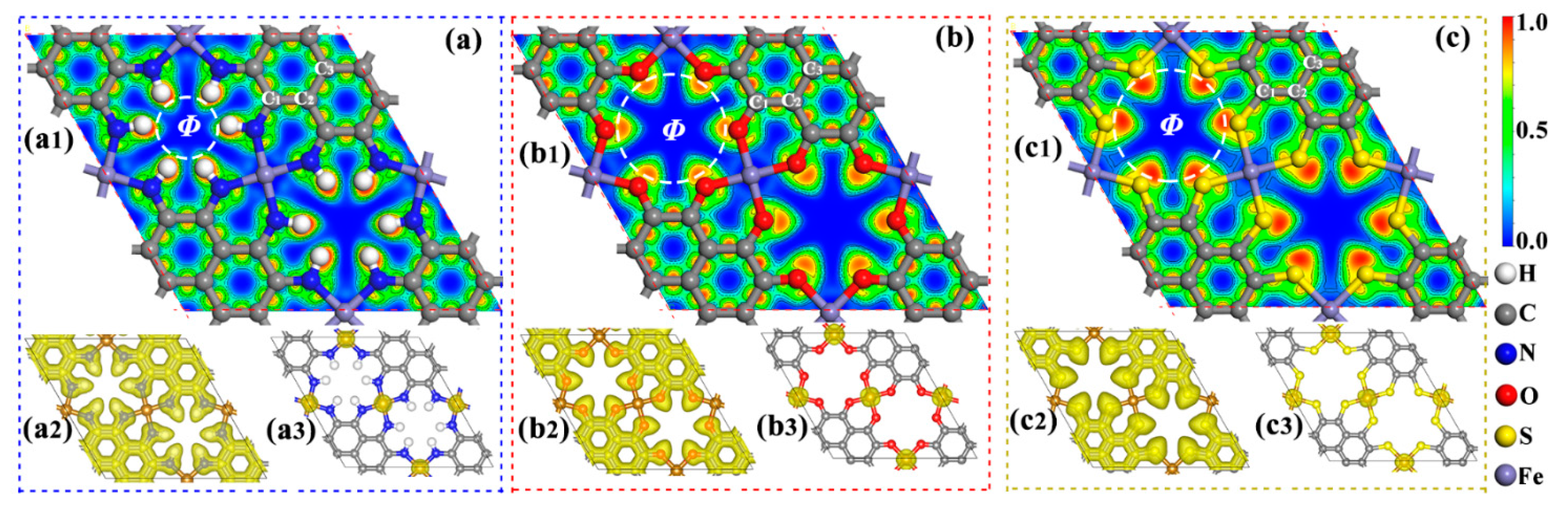
2.1. Geometry and Stability
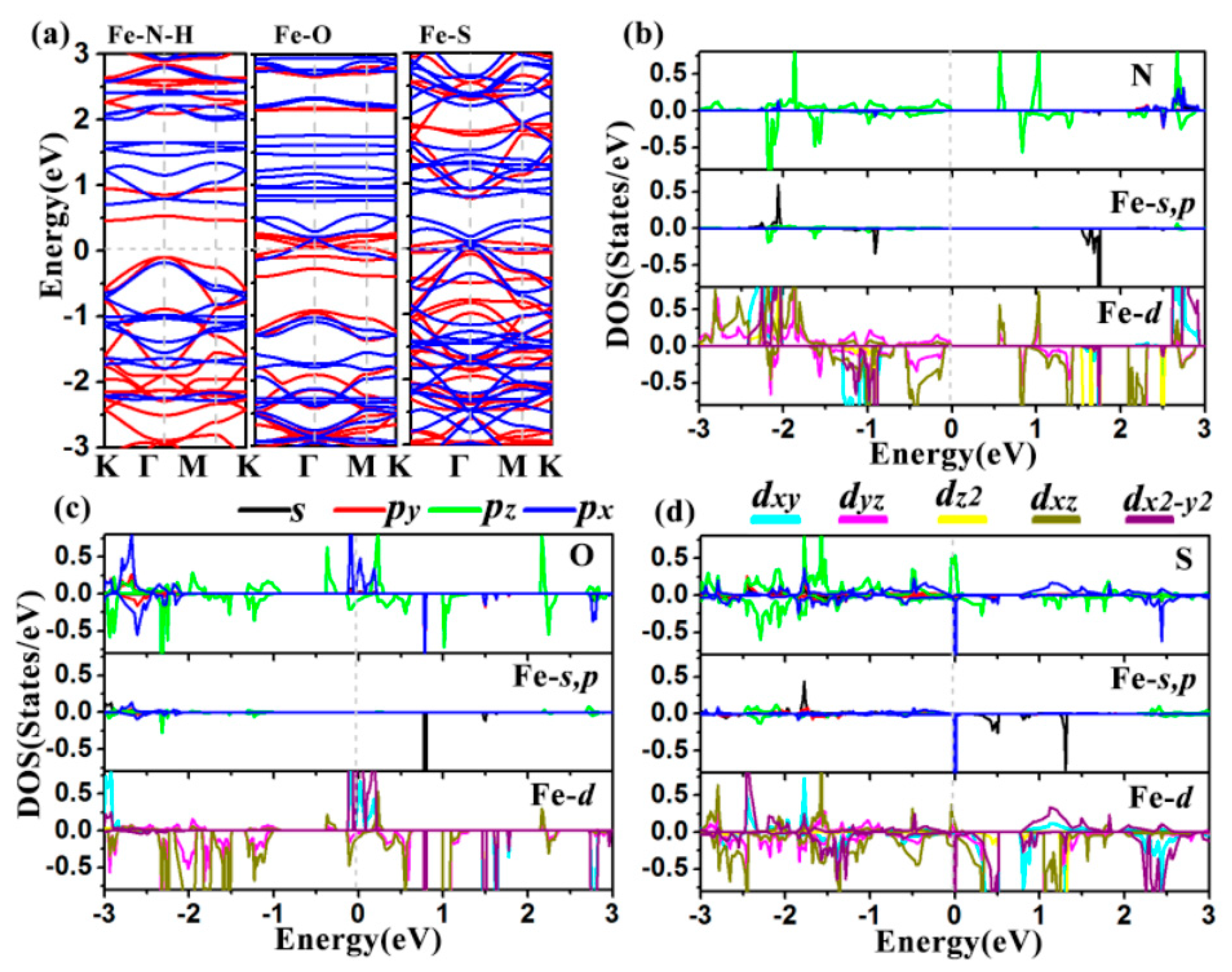
2.2. Electronic Property
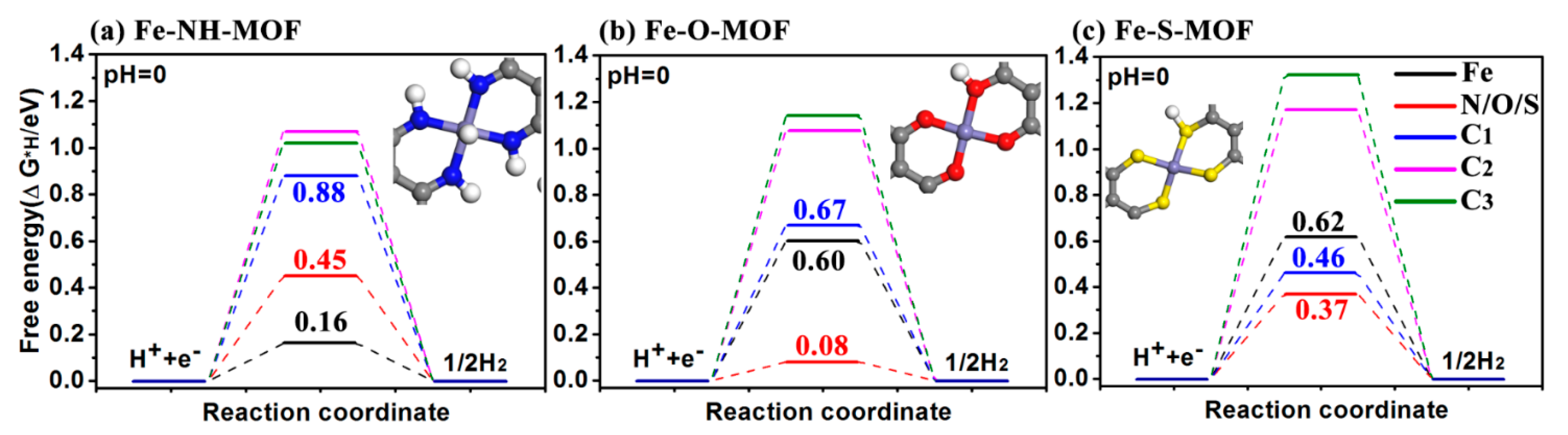
2.3. HER
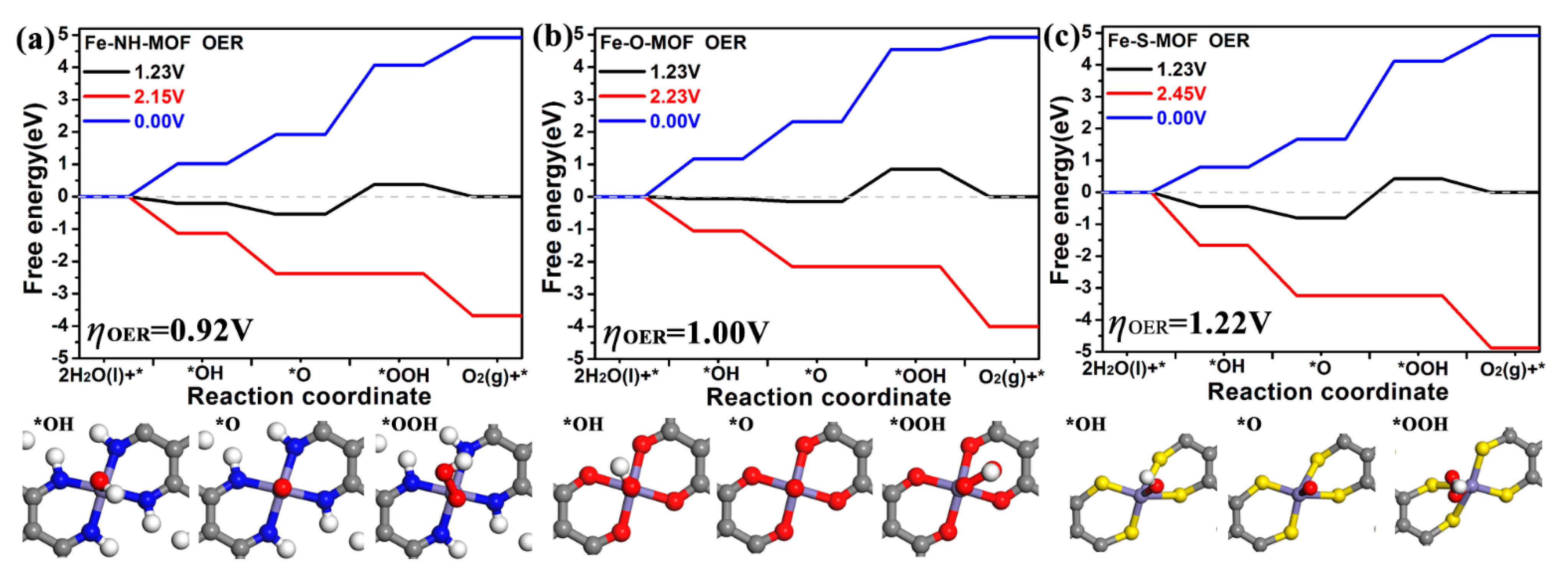
2.4. OER
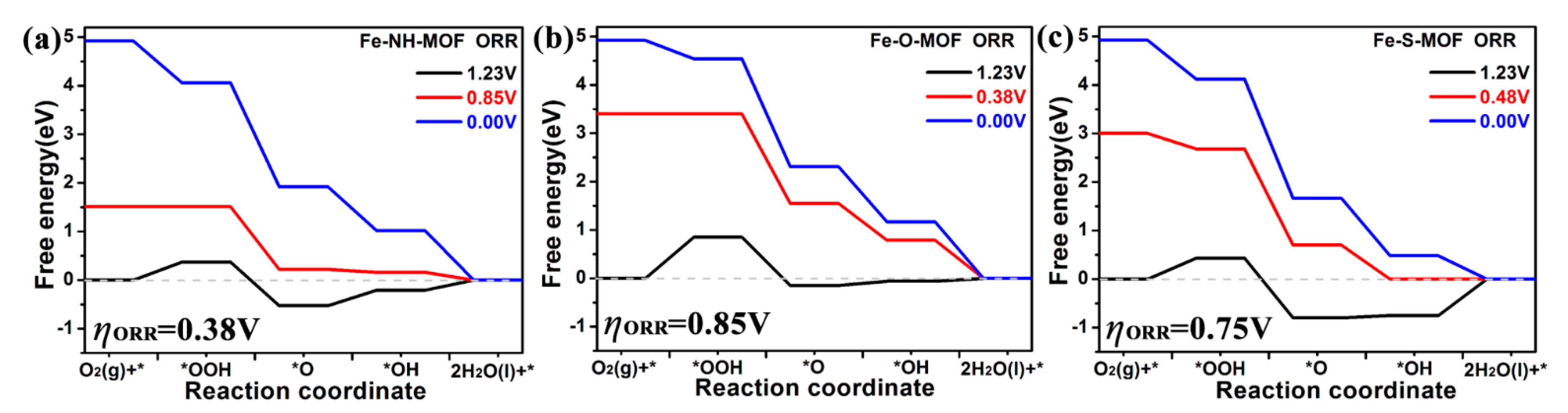
2.5. ORR
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, M.; Zhao, Z.; Zhang, Z.; Ye, S.; Xu, S.; Wang, H.; Li, H. Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells. Nat. Energy 2021, 6, 475–486. [Google Scholar] [CrossRef]
- Turner, J.A. Sustainable hydrogen production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhu, L.; Chen, H.; Qu, J.; Dai, L. Recent advances in carbon-based metal-free electrocatalysts. Adv. Mater. 2019, 31, e1806403. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hu, S.; Yu, W.; Shen, S.; Li, T. Stabilizing mechanism of single-atom catalysts on a defective carbon surface. Npj Comput. Mater. 2020, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, M.; Deng, Y.; Xu, M.; Artiglia, L.; Wen, W.; Gao, R.; Chen, B.; Yao, S.; Zhang, X.; et al. A stable low-temperature H2-production catalyst by crowding Pt on alpha-MoC. Nature 2021, 589, 396–401. [Google Scholar] [CrossRef]
- Fang, S.; Zhu, X.; Liu, X.; Gu, J.; Liu, W.; Wang, D.; Zhang, W.; Lin, Y.; Lu, J.; Wei, S.; et al. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 2020, 11, 1029. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Luo, Q.; Chen, J.; Wang, L.; Lin, Y.; Wang, H.; Liu, X.; Shen, X.; Zhang, W.; Liu, W.; et al. Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 2019, 10, 4849. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gao, G.; Kang, J.; Chu, W.; Wang, L.W. Transition metal-embedded two-dimensional C3N as a highly active electrocatalyst for oxygen evolution and reduction reactions. J. Mater. Chem. A 2019, 7, 12050–12059. [Google Scholar] [CrossRef]
- Zhong, W.; Qiu, Y.; Shen, H.; Wang, X.; Yuan, J.; Jia, C.; Bi, S.; Jiang, J. Electronic spin moment as a catalytic descriptor for Fe single-atom catalysts supported on C2N. J. Am. Chem. Soc. 2021, 143, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Liu, W.; He, Z.; Xiao, C.; Yao, T.; Zou, Y.; Wang, C.; Qi, Z.; Tong, W.; Pan, B.; et al. Single atom accelerates ammonia photosynthesis. Sci. China Chem. 2018, 61, 1187–1196. [Google Scholar] [CrossRef]
- Yang, H.; Lin, Q.; Wu, Y.; Li, G.; Hu, Q.; Chai, X.; Ren, X.; Zhang, Q.; Liu, J.; He, C. Highly efficient utilization of single atoms via constructing 3D and free-standing electrodes for CO2 reduction with ultrahigh current density. Nano Energy 2020, 70, 104454. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Li, Q.; Bai, X.; Wang, J. Metal single-atom coordinated graphitic carbon nitride as an efficient catalyst for CO oxidation. Nanoscale 2020, 12, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Dong, R.; Feng, X. Two-dimensional conjugated metal-organic frameworks (2D c-MOFs): Chemistry and function for MOF tronics. Chem. Soc. Rev. 2021, 50, 2764–2793. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Gao, X.; Zhang, Z.; Wang, F. Recent advances in electrocatalysis with phthalocyanines. Chem. Soc. Rev. 2021, 50, 12985–13011. [Google Scholar] [CrossRef]
- Wei, Y.S.; Zhang, M.; Zou, R.; Xu, Q. Metal-organic framework-based catalysts with single metal sites. Chem. Rev. 2020, 120, 12089–12174. [Google Scholar] [CrossRef]
- Wu, Z.; Adekoya, D.; Huang, X.; Kiefel, M.J.; Xie, J.; Xu, W.; Zhang, Q.; Zhu, D.; Zhang, S. Highly conductive two-dimensional metal-organic frameworks for resilient lithium storage with superb rate capability. ACS Nano 2020, 14, 12016–12026. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, P.; Zhang, S.; Zhang, Y.; Chang, H.; Zheng, X.; Jiang, J.; Xu, Q. CO2 coordination-driven top-down synthesis of a 2D non-layered metal–organic framework. Fundam. Res. 2021, in press. [Google Scholar] [CrossRef]
- Cui, Q.; Qin, G.; Wang, W.; Geethalakshmi, K.R.; Du, A.; Sun, Q. Mo-based 2D MOF as a highly efficient electrocatalyst for reduction of N2 to NH3: A density functional theory study. J. Mater. Chem. A 2019, 7, 14510–14518. [Google Scholar] [CrossRef]
- Feng, Z.; Yang, Z.; Meng, X.; Li, F.; Guo, Z.; Zheng, S.; Su, G.; Ma, Y.; Tang, Y.; Dai, X. Two-dimensional metal-organic frameworks Mo3(C2O)12 as promising single-atom catalysts for selective nitrogen-to-ammonia. J. Mater. Chem. A 2022. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, W.; Liang, Z.; Zou, R. Metal-organic framework based nanomaterials for electrocatalytic oxygen redox reaction. Sci. China Chem. 2019, 62, 417–429. [Google Scholar] [CrossRef]
- Fan, X.; Tan, S.; Yang, J.; Liu, Y.; Bian, W.; Liao, F.; Lin, H.; Li, Y. From theory to experiment: Cascading of thermocatalysis and electrolysis in oxygen evolution reactions. ACS Energy Lett. 2021, 7, 343–348. [Google Scholar] [CrossRef]
- Yu, M.; Dong, R.; Feng, X. Two-dimensional carbon-rich conjugated frameworks for electrochemical energy applications. J. Am. Chem. Soc. 2020, 142, 12903–12915. [Google Scholar] [CrossRef]
- Feng, Z.; Li, Y.; Ma, Y.; An, Y.; Dai, X. Magnetic and electronic properties of two-dimensional metal-organic frameworks TM3(C2NH)12. Chin. Phys. B 2021, 30, 097102. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, Z.; Tranca, D.C.; Zhou, S.; Wang, M.; Adler, P.; Liao, Z.; Liu, F.; Sun, Y.; Shi, W.; et al. A coronene-based semiconducting two-dimensional metal-organic framework with ferromagnetic behavior. Nat. Commun. 2018, 9, 2637. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Pramanik, A.; Sarkar, P. Graphitic carbon nitride sheet supported single-atom metal-free photocatalyst for oxygen reduction reaction: A first-principles analysis. J. Phys. Chem. Lett. 2021, 12, 2788–2795. [Google Scholar] [CrossRef]
- Chanier, T.; Sargolzaei, M.; Opahle, I.; Hayn, R.; Koepernik, K. LSDA+U versus LSDA: Towards a better description of the magnetic nearest-neighbor exchange coupling in Co- and Mn-doped ZnO. Phys. Rev. B 2006, 73, 134418. [Google Scholar] [CrossRef]
- Kattel, S.; Atanassov, P.; Kiefer, B. Stability, electronic and magnetic properties of in-plane defects in graphene: A first-principles study. J. Phys. Chem. C 2012, 116, 8161–8166. [Google Scholar] [CrossRef]
- Dang, Q.; Lin, H.; Fan, Z.; Ma, L.; Shao, Q.; Ji, Y.; Zheng, F.; Geng, S.; Yang, S.Z.; Kong, N.; et al. Iridium metallene oxide for acidic oxygen evolution catalysis. Nat. Commun. 2021, 12, 6007. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Liu, X.; Chen, F.; Chen, Z.; Fan, X.; Lau, W. Single atoms on a nitrogen-doped boron phosphide monolayer: A new promising bifunctional electrocatalyst for ORR and OER. ACS Appl. Mater. Interfaces 2020, 12, 52549–52559. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wu, Y.; Li, N.; Chen, X.; Zeng, X.; Arramel; Zhao, X.; Jiang, J. Single-metal atoms supported on MBenes for robust electrochemical hydrogen evolution. ACS Appl. Mater. Interfaces 2020, 12, 9261–9267. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Y.; Yuan, J.; Wu, P.; Zhou, W. Scaling law of hydrogen evolution reaction for InSe monolayer with 3d transition metals doping and strain engineering. J. Energy Chem. 2020, 41, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sun, F.; Bai, F.; Xie, Z. DFT study of the two dimensional metal–organic frameworks X3(HITP)2 as the cathode electrocatalysts for fuel cell. Appl. Surf. Sci. 2019, 471, 256–262. [Google Scholar] [CrossRef]
- Qi, S.; Wang, J.; Song, X.; Fan, Y.; Li, W.; Du, A.; Zhao, M. Synergistic trifunctional electrocatalysis of pyridinic nitrogen and single transition-metal atoms anchored on pyrazine-modified graphdiyne. Sci. Bull. 2020, 65, 995–1002. [Google Scholar] [CrossRef]
- Zhao, C.X.; Liu, J.N.; Wang, J.; Ren, D.; Li, B.Q.; Zhang, Q. Recent advances of noble-metal-free bifunctional oxygen reduction and evolution electrocatalysts. Chem. Soc. Rev. 2021, 50, 7745–7778. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2014, 108, 17886–17892. [Google Scholar] [CrossRef]
- Feng, Z.; Li, R.; Ma, Y.; Li, Y.; Wei, D.; Tang, Y.; Dai, X. Molecule-level graphdiyne coordinated transition metals as a new class of bifunctional electrocatalysts for oxygen reduction and oxygen evolution reactions. Phys. Chem. Chem. Phys. 2019, 21, 19651–19659. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; Wiley: New York, NY, USA, 2007. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Pe, B. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Stoeffler, D.; Etz, C. Ab initioelectronic structure and magnetism in Sr2XMoO6(X = Fe or Co) double perovskite systems: A GGA and GGA+ U comparative study. J. Phys. Condens. Mat. 2006, 18, 11291–11300. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical gga-type density functional constructed with a longrange dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. The NIST chemistry WebBook: A chemical data resource on the internet. J. Chem. Eng. Data 2001, 46, 1059–1063. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | la (Å) | DFe-N (O,S) (Å) | DC-N (O,S) (Å) | Φ (Å) | Mtot (μB) | QFe (e) | QN,O,S (e) |
|---|---|---|---|---|---|---|---|
| Fe-NH-MOF | 12.61 | 1.85 | 1.35 | 3.44 | 6.00 | −1.26 | +0.83 |
| Fe-O-MOF | 12.31 | 1.83 | 1.30 | 5.17 | 10.45 | −1.51 | +1.07 |
| Fe-S-MOF | 13.65 | 2.15 | 1.74 | 5.74 | 9.25 | −0.58 | +0.13 |
| Materials | ΔGH (eV) | ηOER (V) | ηORR (V) | Materials | ΔGH (eV) | ηOER (V) | ηORR (V) |
|---|---|---|---|---|---|---|---|
| Fe-NH-MOF | 0.16 | 0.92 | 0.38 | IrO2 [31] | - | 0.45–0.59 | - |
| Fe-O-MOF | 0.08 | 1.00 | 0.85 | Co-BP [32] | - | 0.42 | 0.36 |
| Fe-S-MOF | 0.37 | 1.22 | 0.75 | Ni-BP [32] | - | 0.44 | 0.29 |
| V-W2B2O2 [33] | 0.01–0.15 | - | - | Pt-BP [32] | - | 0.25 | 0.32 |
| ZnW2B2O2 [33] | 0.14–0.26 | - | - | Fe-BHT [24] | - | 0.88 | - |
| Zn@InSe [34] | 0.02 | - | - | Ir3(HITP)2 [35] | - | - | 0.31 |
| Ni@PR-GDY [36] | −0.05 | 0.29 | 0.38 | Rh3(HITP)2 [35] | - | - | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Feng, Z.; Guo, Z. Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S). Molecules 2022, 27, 1528. https://doi.org/10.3390/molecules27051528
Yang X, Feng Z, Guo Z. Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S). Molecules. 2022; 27(5):1528. https://doi.org/10.3390/molecules27051528
Chicago/Turabian StyleYang, Xiaohang, Zhen Feng, and Zhanyong Guo. 2022. "Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S)" Molecules 27, no. 5: 1528. https://doi.org/10.3390/molecules27051528
APA StyleYang, X., Feng, Z., & Guo, Z. (2022). Theoretical Investigation on the Hydrogen Evolution, Oxygen Evolution, and Oxygen Reduction Reactions Performances of Two-Dimensional Metal-Organic Frameworks Fe3(C2X)12 (X = NH, O, S). Molecules, 27(5), 1528. https://doi.org/10.3390/molecules27051528