
The Chemical Property Position of Bedaquiline Construed by a Chemical Global Positioning System-Natural Product

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
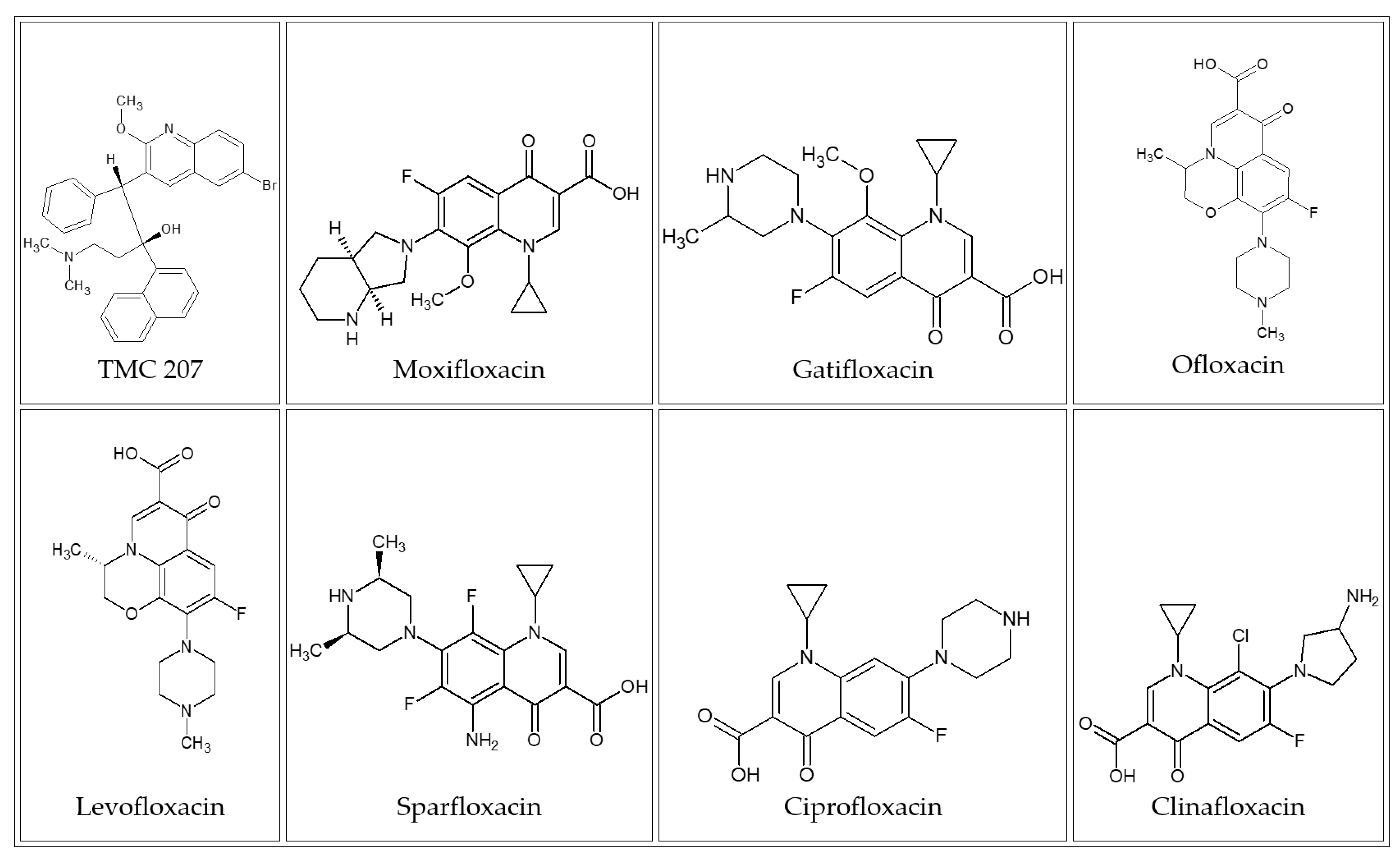
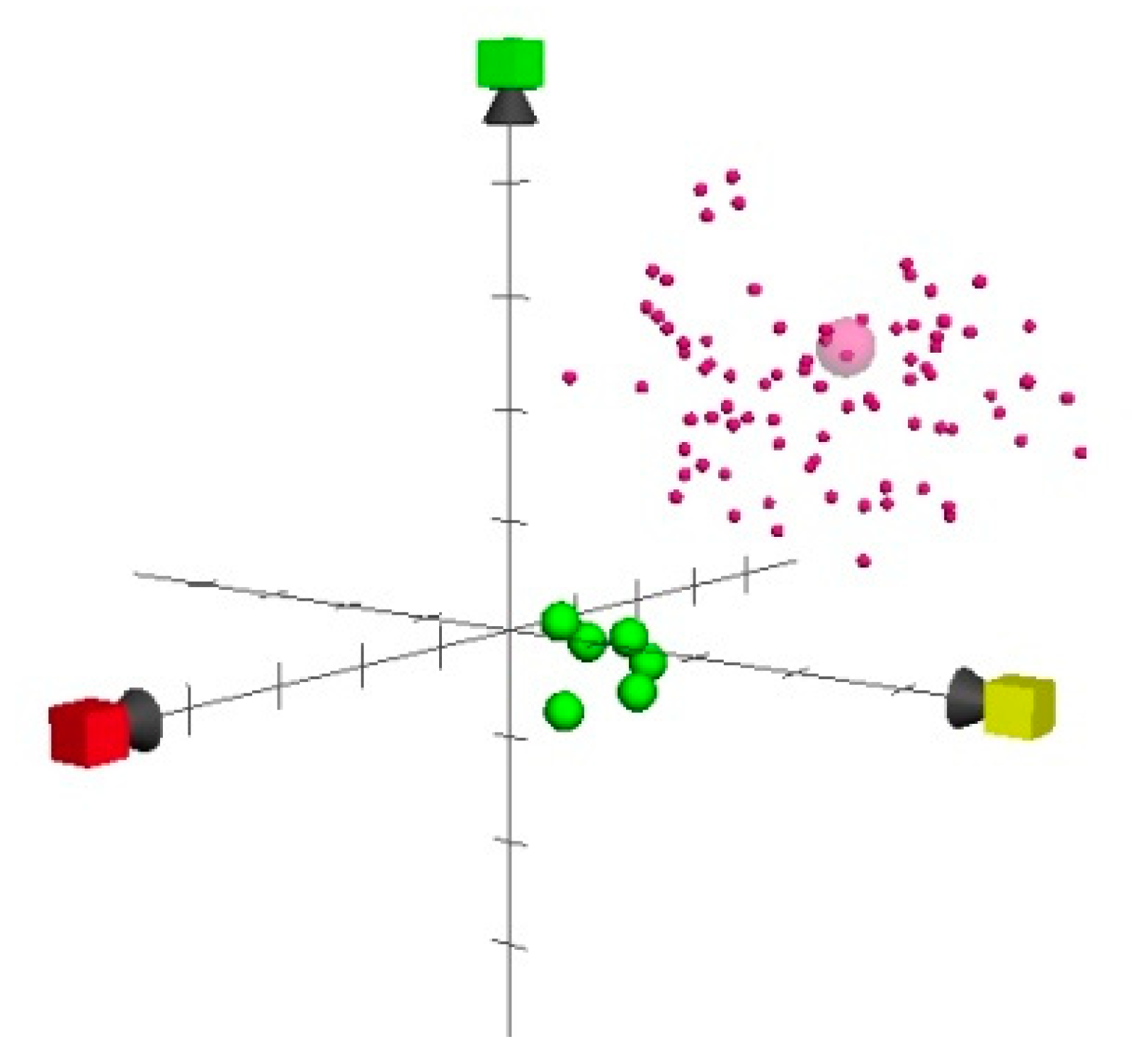
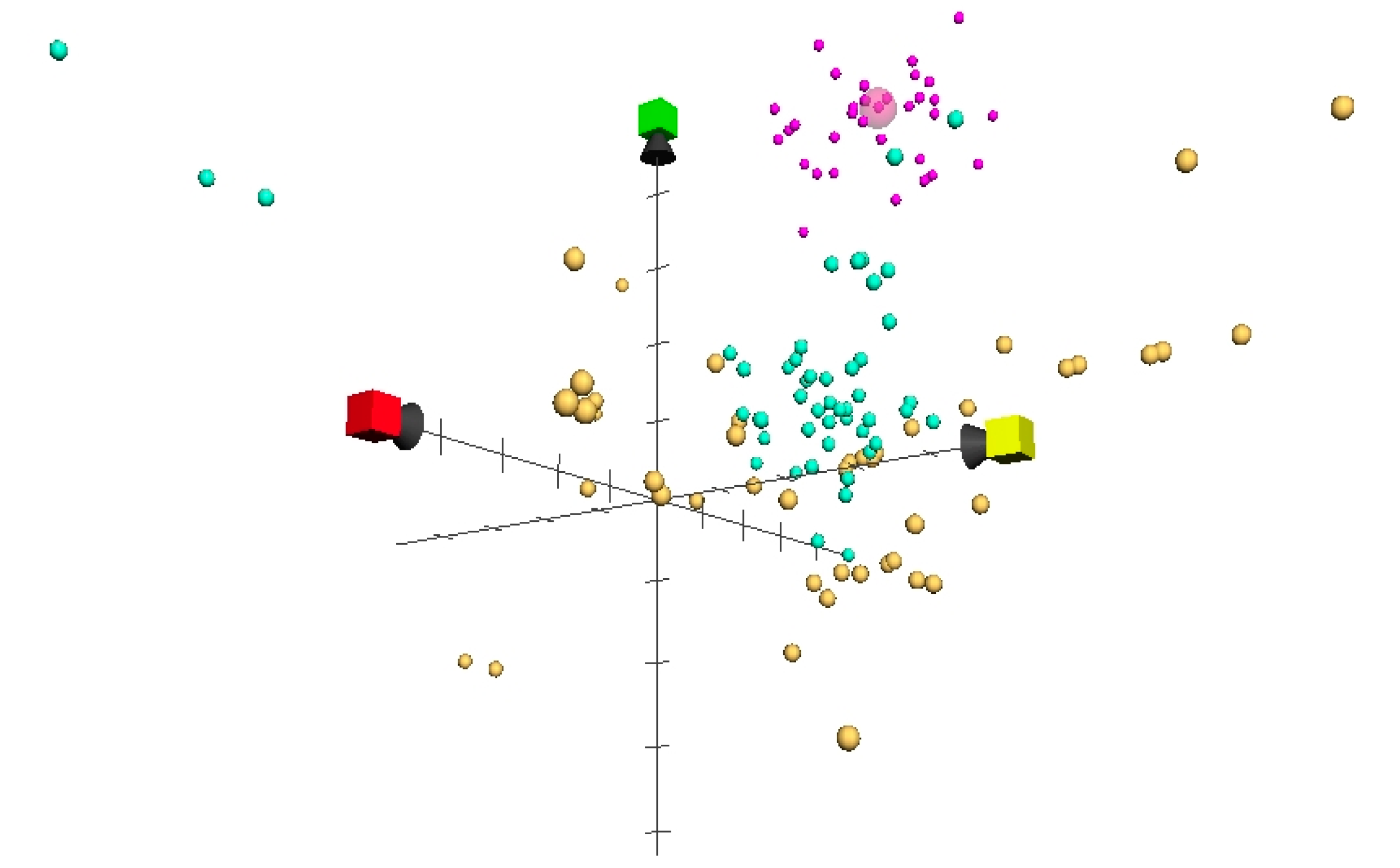
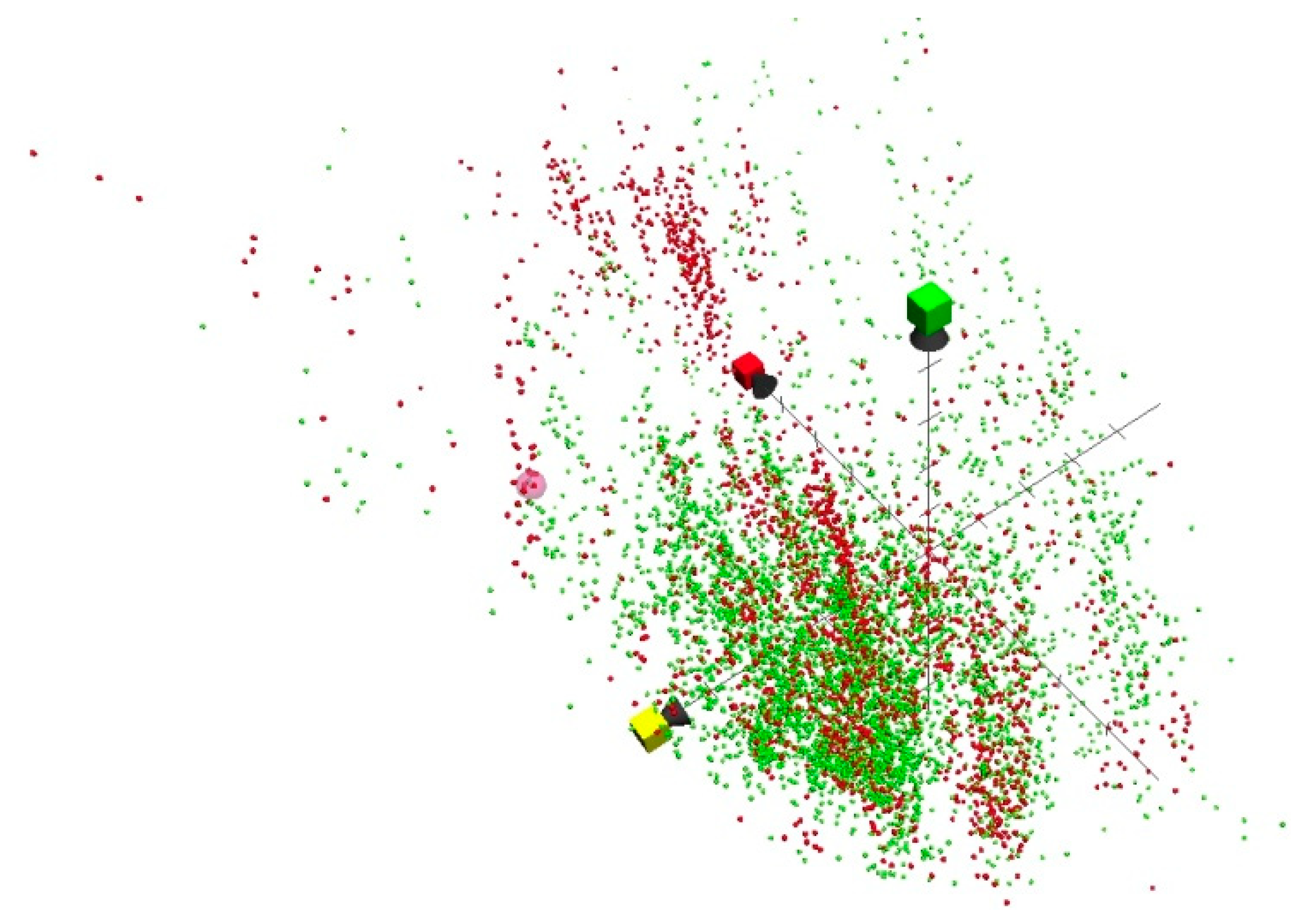
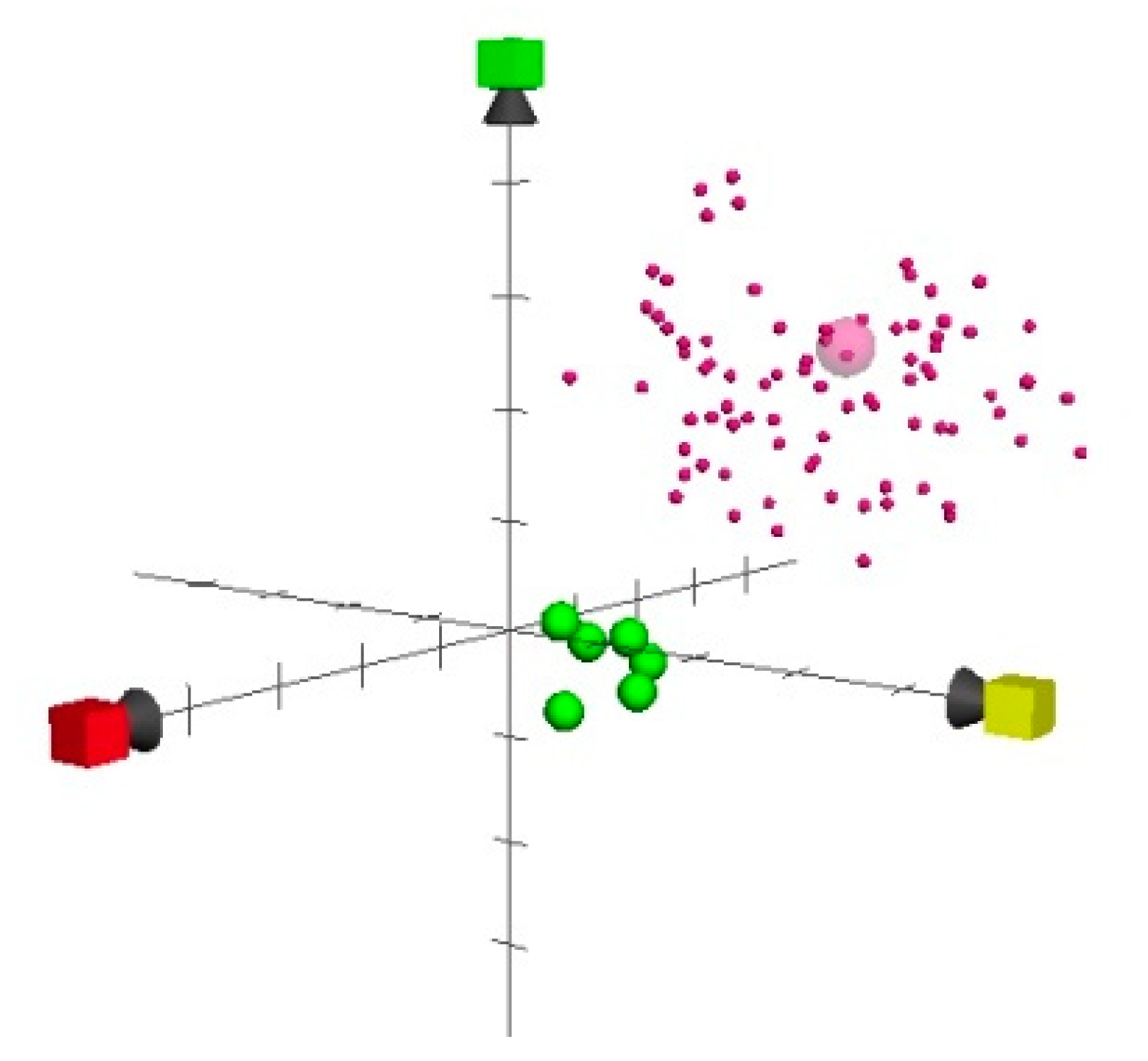
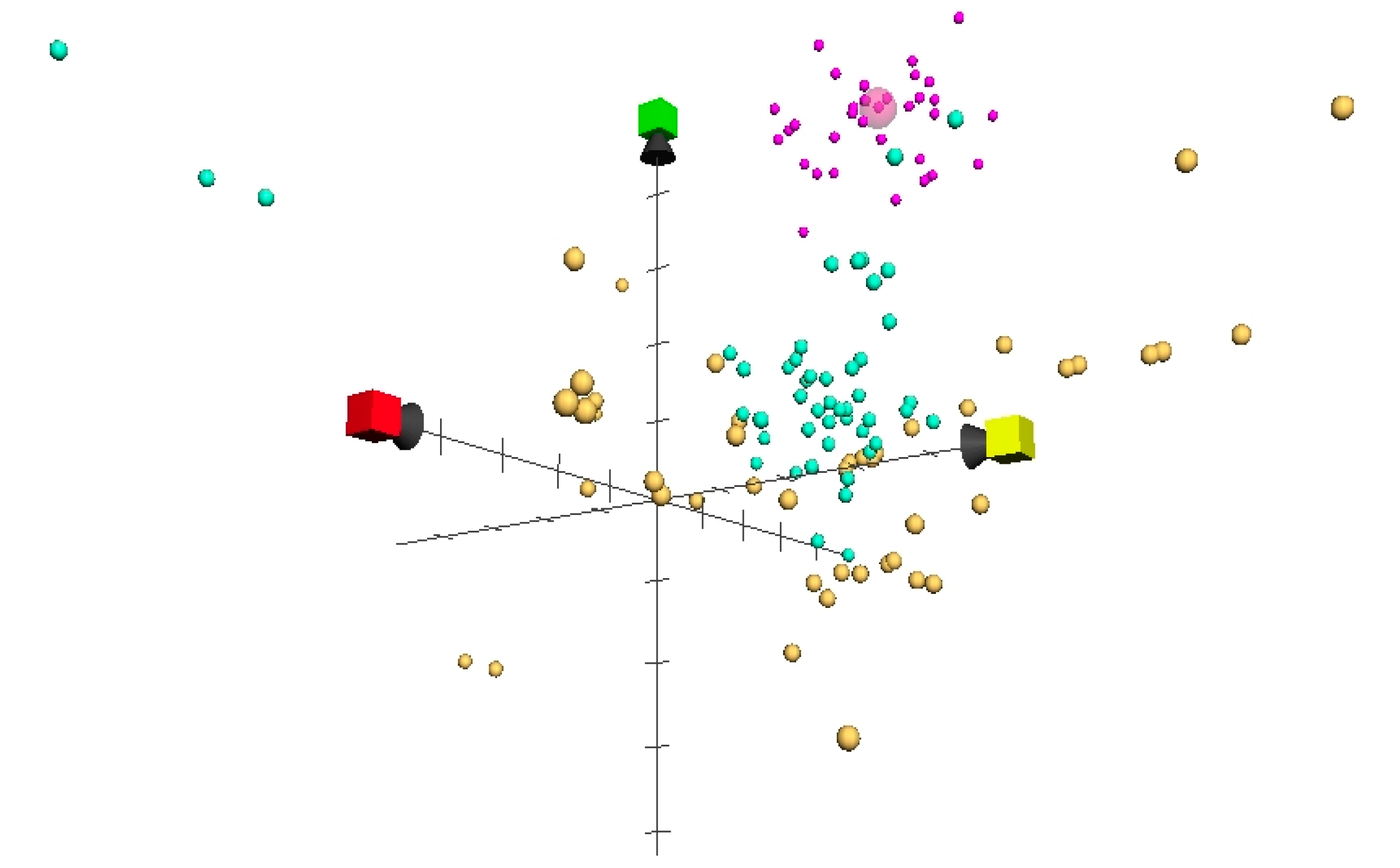
2. Results and Discussion
3. Materials and Methods
3.1. Data Collection and Sources
3.1.1. Basic Antituberculosis Agents
3.1.2. Screening Sets of Antituberculosis Agents
3.1.3. Drug-like Dataset
3.2. Mechanism of Action
3.3. ChemGPS-NP
3.4. Euclidean Distances
3.5. Permeability Prediction
3.6. Toxicity Prediction
3.7. Structural Similarity Search
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2012; World Health Organization: Geneva, Switzerland, 2013; Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 4 June 2014).
- Chan, B.; Khadem, T.M.; Brown, J. A review of tuberculosis: Focus on bedaquiline. Am. J. Heal. Pharm. 2013, 70, 1984–1994. [Google Scholar] [CrossRef]
- Lounis, N.; Guillemont, J.; Veziris, N.; Koul, A.; Jarlier, V.; Andries, K. R207910 (TMC207): A new antibiotic for the treatment of tuberculosis. Med. Mal. Infect. 2010, 40, 383–390. [Google Scholar] [CrossRef]
- Tiberi, S.; De Lorenzo, S.; Centis, R.; Viggiani, P.; D’ambrosio, L.; Migliori, G.B. Bedaquiline in MDR/XDR-TB cases: First experience on compassionate use. Eur. Respir. J. 2013, 43, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Alajlani, M.M.; Backlund, A. Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning Sys-tem-Natural Product Analysis. Molecules 2020, 25, 945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K. Where Tuberculosis Meets Computation: 10 Points of Intersection. Biomed. Comput. Rev. Summer 2012, 20–28. [Google Scholar]
- Sarker, M.; Talcott, C.; Madrid, P.; Chopra, S.; Bunin, B.A.; Lamichhane, G.; Freundlich, J.S.; Ekins, S. Combining Cheminformatics Methods and Pathway Analysis to Identify Molecules with Whole-Cell Activity Against Mycobacterium Tuberculosis. Pharm. Res. 2012, 29, 2115–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballester, P.J.; Mangold, M.; Howard, N.I.; Robinson, R.M.; Abell, C.; Blumberger, J.; Mitchell, J. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. Interface 2012, 9, 3196–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekins, S.; Reynolds, R.C.; Kim, H.; Koo, M.-S.; Ekonomidis, M.; Talaue, M.; Paget, S.D.; Woolhiser, L.K.; Lenaerts, A.J.; Bunin, B.A.; et al. Bayesian Models Leveraging Bioactivity and Cytotoxicity Information for Drug Discovery. Chem. Biol. 2013, 20, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prathipati, P.; Ma, N.L.; Keller, T.H. Global Bayesian models for the prioritization of antitubercular agents. J. Chem. Inf. Model. 2008, 48, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drlica, K.; Malik, M. Fluoroquinolones: Action and resistance. Curr. Top. Med. Chem. 2003, 3, 249–282. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Trygg, J.; Wikström, C.; Wold, S. Multiand Megavariate Data Analysis Part I Basic Principles and Applications, 2nd ed.; Umetrics AB: Umea, Sweden, 2006. [Google Scholar]
- Merget, B.; Zilian, D.; Müller, T.; Sotriffer, C.A. MycPermCheck: The Mycobacterium tuberculosis permeability prediction tool for small molecules. Bioinformatics 2012, 29, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kazius, J.; McGuire, R.; Bursi, R. Derivation and Validataion of Toxicophores for Mutagenicity Prediction. J. Med. Chem. 2005, 48, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alajlani, M.M. The Chemical Property Position of Bedaquiline Construed by a Chemical Global Positioning System-Natural Product. Molecules 2022, 27, 753. https://doi.org/10.3390/molecules27030753
Alajlani MM. The Chemical Property Position of Bedaquiline Construed by a Chemical Global Positioning System-Natural Product. Molecules. 2022; 27(3):753. https://doi.org/10.3390/molecules27030753
Chicago/Turabian StyleAlajlani, Muaaz Mutaz. 2022. "The Chemical Property Position of Bedaquiline Construed by a Chemical Global Positioning System-Natural Product" Molecules 27, no. 3: 753. https://doi.org/10.3390/molecules27030753
APA StyleAlajlani, M. M. (2022). The Chemical Property Position of Bedaquiline Construed by a Chemical Global Positioning System-Natural Product. Molecules, 27(3), 753. https://doi.org/10.3390/molecules27030753