
Dissection of the Multichannel Reaction O(3P) + C2H2: Differential Cross-Sections and Product Energy Distributions


Abstract
:1. Introduction
2. Methods
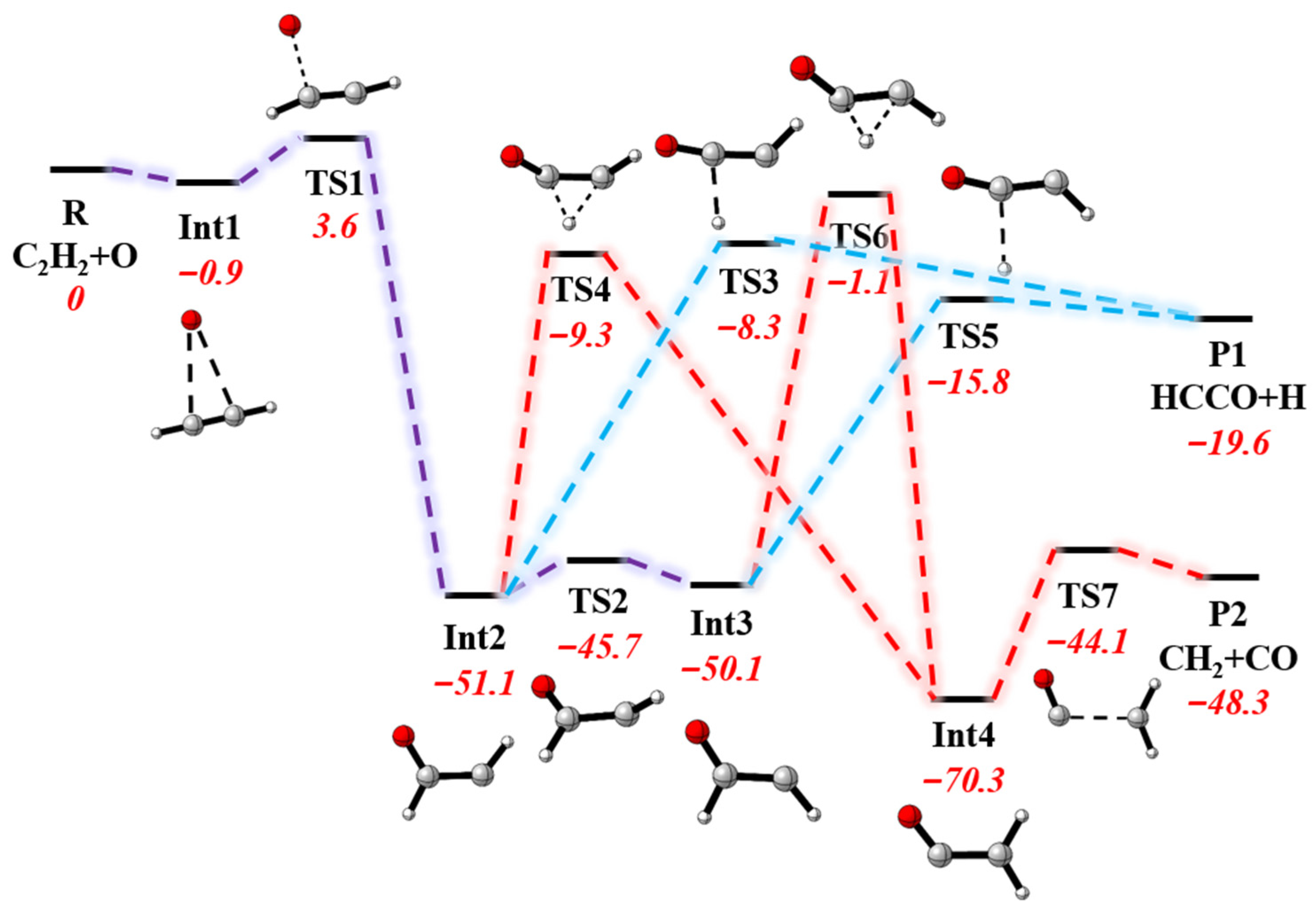
3. Results and Discussion
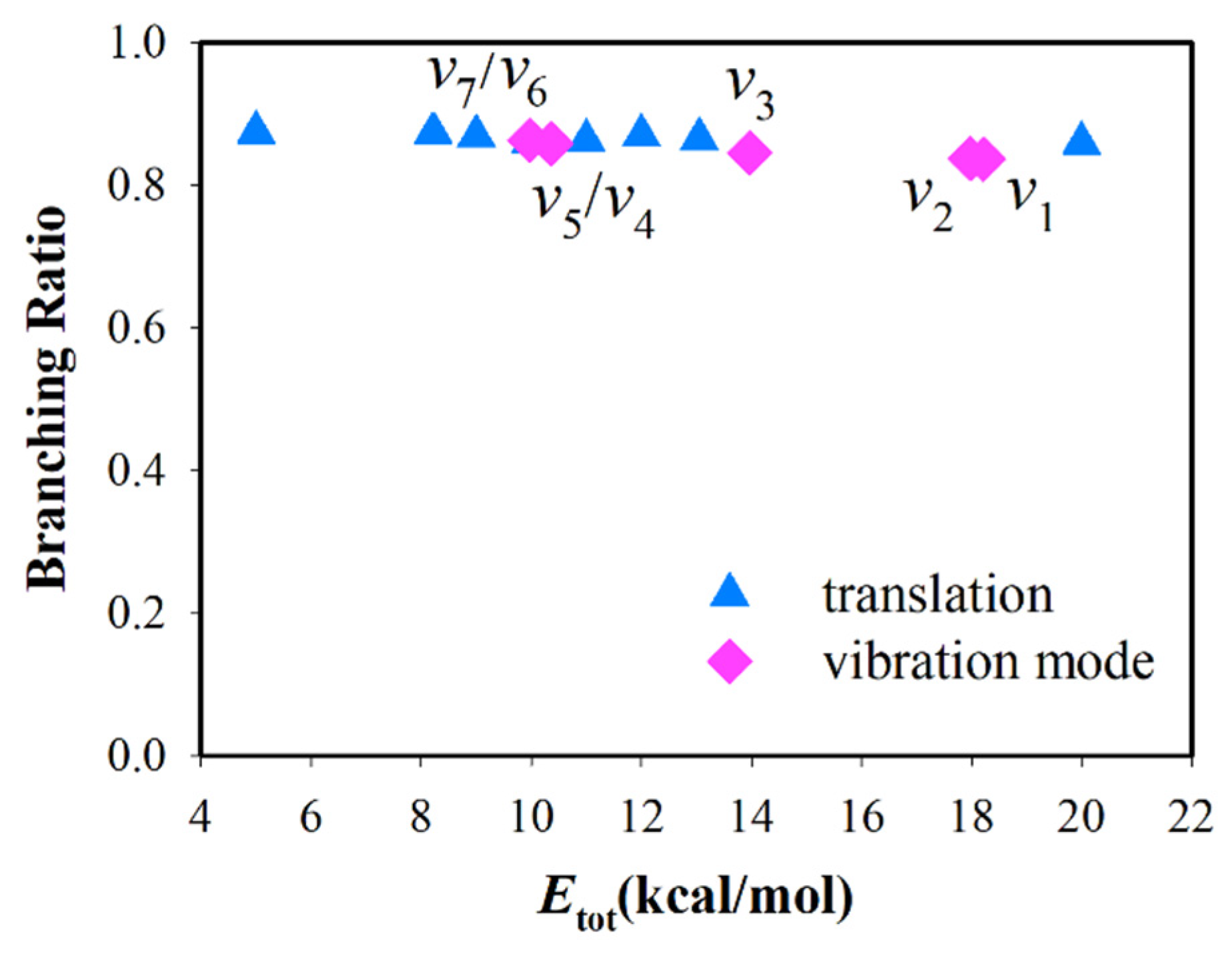
3.1. Mode Specificity on Reactivity
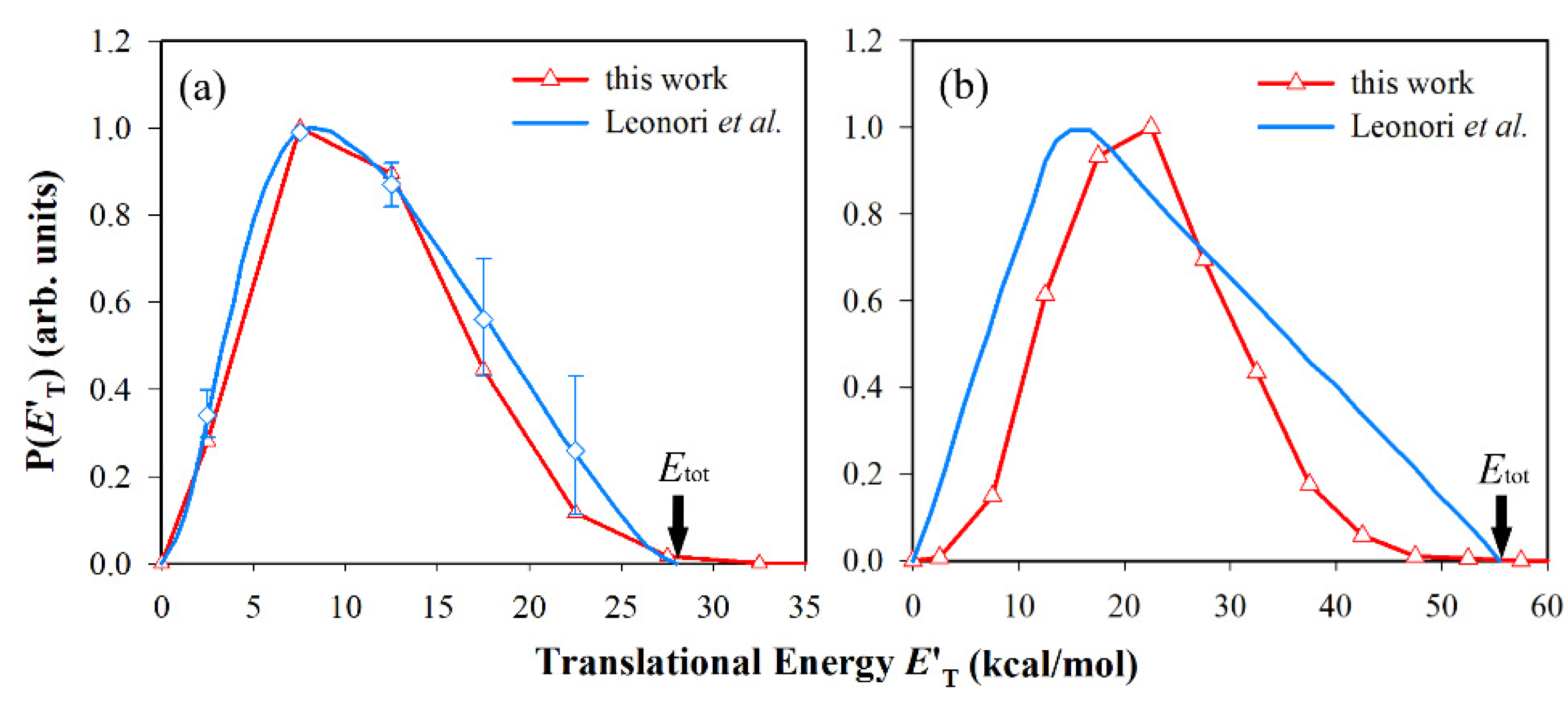
3.2. Differential Cross-Sections and Product Energy Fractions
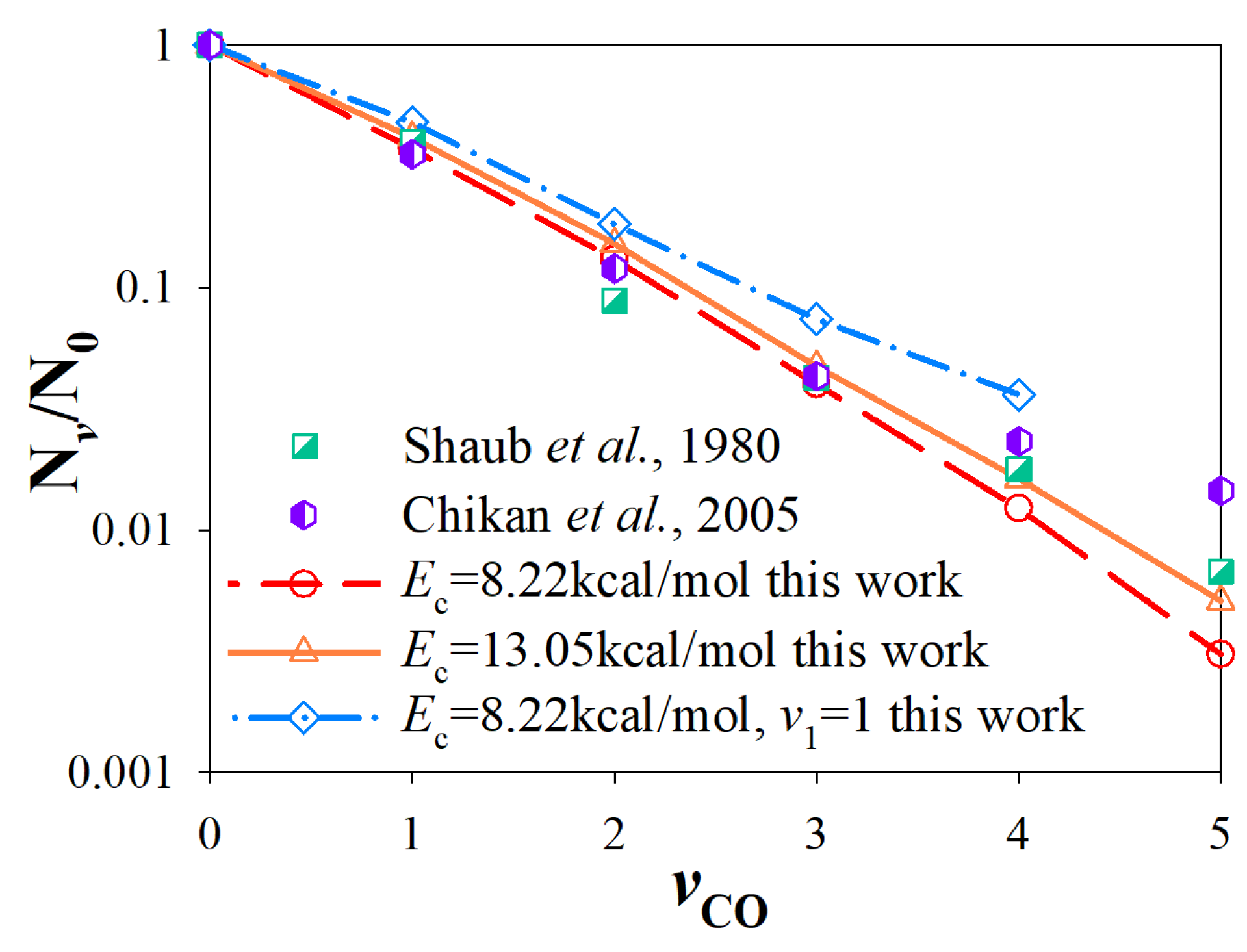
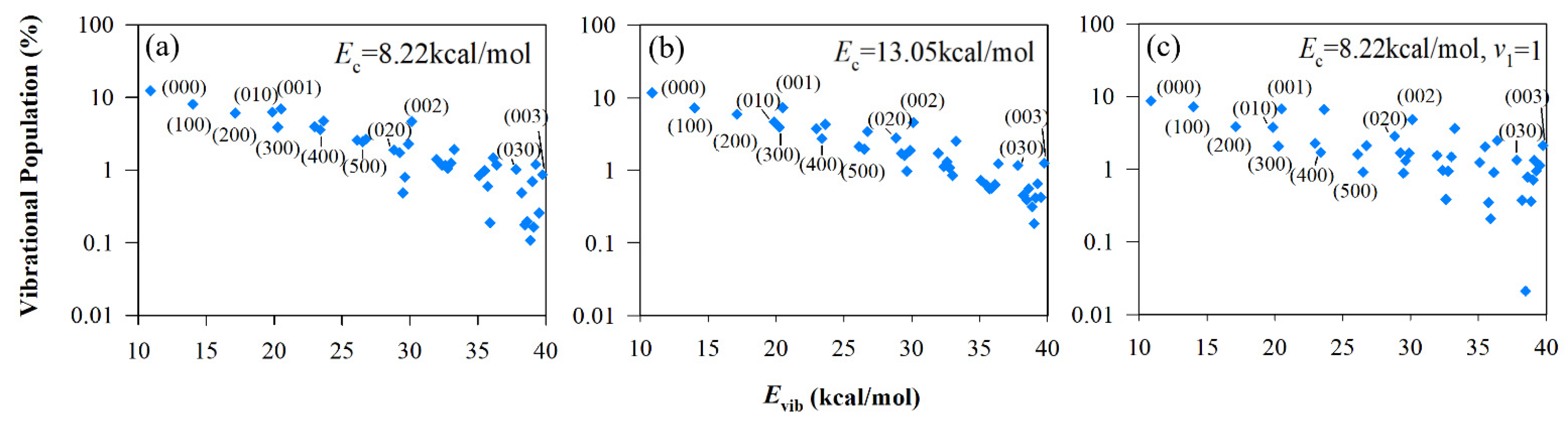
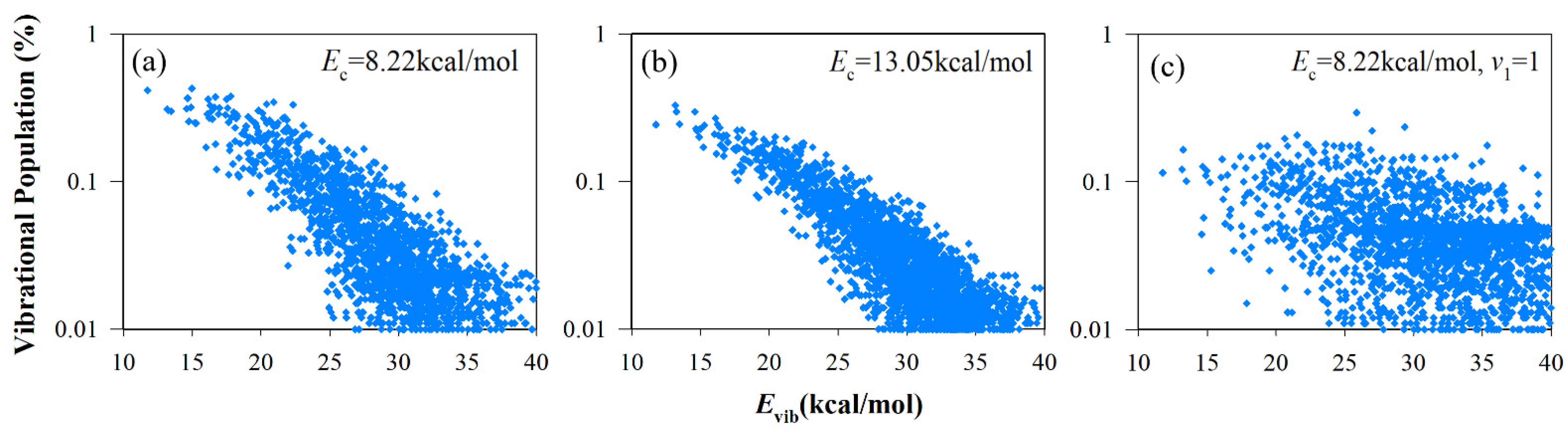
3.3. Vibrational State Distributions of Products
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Williams, A.; Smith, D.B. The Combustion and Oxidation of Acetylene. Chem. Rev. 1970, 70, 267–293. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Dryer, F.L. Chemical Kinetic Modeling of Hydrocarbon Combustion. Prog. Energy Combust. Sci. 1984, 10, 1–57. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed Chemical Kinetic Models for the Combustion of Hydrocarbon Fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Harding, L.B. Theoretical Studies on the Reaction of Atomic Oxygen (3P) with Acetylene. J. Phys. Chem. 1981, 85, 10–11. [Google Scholar] [CrossRef]
- Harding, L.B.; Wagner, A.F. Theoretical Studies on the Reaction of Atomic Oxygen (O(3P)) with Acetylene. 2. J. Phys. Chem. 1986, 90, 2974–2987. [Google Scholar] [CrossRef]
- Hoyermann, K.; Wagner, H.G.; Wolfrum, J. Zur Reaktion O + C2H2 → CO + CH2. Z. Physik. Chem. N. F. 1969, 63, 193–196. [Google Scholar] [CrossRef]
- James, G.S.; Glass, G.P. Some Aspects of Acetylene Oxidation. J. Chem. Phys. 1969, 50, 2268–2269. [Google Scholar] [CrossRef]
- Westenberg, A.A.; De Haas, N. Absolute Measurements of O + C2H2 Rate Coefficient. J. Phys. Chem. 1969, 73, 1181–1186. [Google Scholar] [CrossRef]
- Löhr, R.; Roth, P. Shock Tube Measurements of the Reaction Behavior of Acetylene with O-Atoms. Ber. Bunsenges. Phys. Chem. 1981, 85, 153–158. [Google Scholar] [CrossRef]
- Clemo, A.R.; Duncan, G.L.; Grice, R. Reactive Scattering of a Supersonic Oxygen-Atom Beam: O+C2H4, C2H2. J. Chem. Soc. Faraday Trans. 2 1982, 78, 1231–1238. [Google Scholar] [CrossRef]
- Peeters, J.; Schaekers, M.; Vinckier, C. Ketenyl Radical Yield of the Elementary Reaction of Ethyne with Atomic Oxygen at T = 290–540 K. J. Phys. Chem. 1986, 90, 6552–6557. [Google Scholar] [CrossRef]
- Mahmud, K.; Fontijn, A. A High-Temperature Photochemistry Kinetics Study of the Reaction of O(3P) Atoms with Acetylene from 290 to 1510K. J. Phys. Chem. 1987, 91, 1918–1921. [Google Scholar] [CrossRef]
- Peeters, J.; Vanhaelemeersch, S.; Vanhoeymissen, J.; Borms, R.; Vermeylen, D. Spectroscopic Observation of the CH2(ã1A1) Radical in the Reaction of C2H2 with O Atoms. J. Phys. Chem. 1989, 93, 3892–3894. [Google Scholar] [CrossRef]
- Schmoltner, A.M.; Chu, P.M.; Lee, Y.T. Crossed Molecular Beam Study of the Reaction O(3P)+C2H2. J. Chem. Phys. 1989, 91, 5365–5373. [Google Scholar] [CrossRef]
- Bohn, B.; Stuhl, F. Rate Constants of the Reaction O(3P) + C2H2 at Low Temperatures. J. Phys. Chem. 1990, 94, 8010–8011. [Google Scholar] [CrossRef]
- Michael, J.V.; Wagner, A.F. Rate Constants for the Reactions O+C2H2 and O+C2D2 → Products, over the Temperature Range ~850–1950 K, by the Flash Photolysis-Shock Tube Technique. Determination of the Branching Ratio and a Further Theoretical Analysis. J. Phys. Chem. 1990, 94, 2453–2464. [Google Scholar] [CrossRef]
- Boullart, W.; Peeters, J. Product Distributions of the C2H2+O and HCCO+H Reactions. Rate Constant of CH2(3B1)+H. J. Phys. Chem. 1992, 96, 9810–9816. [Google Scholar]
- Huang, X.; Xing, G.; Bersohn, R. Dynamics of the Reaction of O(3P) Atoms with Acetylene. J. Chem. Phys. 1994, 101, 5818–5823. [Google Scholar] [CrossRef]
- Capozza, G.; Segoloni, E.; Leonori, F.; Volpi, G.G.; Casavecchia, P. Soft Electron Impact Ionization in Crossed Molecular Beam Reactive Scattering: The Dynamics of the O(3P)+C2H2 Reaction. J. Chem. Phys. 2004, 120, 4557–4560. [Google Scholar] [CrossRef]
- Chikan, V.; Leone, S.R. Vibrational Distributions of the CO(ν) Products of the C2H2+O(3P) and HCCO+O(3P) Reactions Studied by FTIR Emission. J. Phys. Chem. A 2005, 109, 2525–2533. [Google Scholar] [CrossRef]
- Lahankar, S.A.; Zhang, J.; Garashchuk, S.; Schatz, G.C.; Minton, T.K. Electronic Population Inversion in HCCO/DCCO Products from Hyperthermal Collisions of O(3P) with HCCH/DCCD. J. Phys. Chem. Lett. 2013, 4, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Leonori, F.; Balucani, N.; Capozza, G.; Segoloni, E.; Volpi, G.G.; Casavecchia, P. Dynamics of the O(3P) + C2H2 Reaction from Crossed Molecular Beam Experiments with Soft Electron Ionization Detection. Phys. Chem. Chem. Phys. 2014, 16, 10008–10022. [Google Scholar] [CrossRef] [PubMed]
- Yarkony, D.R. On the Mechanism of the Spin-Nonconserving Chemical Reaction O(3P)+HCCH → CH2(ã1A1)+CO(X1Σ+). I. Feasibility. J. Phys. Chem. A 1998, 102, 5305–5311. [Google Scholar] [CrossRef]
- Girard, Y.; Chaquin, P. Addition Reactions of 1D and 3P Atomic Oxygen with Acetylene. Potential Energy Surfaces and Stability of the Primary Products. Is Oxirene Only a Triplet Molecule? A Theoretical Study. J. Phys. Chem. A 2003, 107, 10462–10470. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Vereecken, L.; Peeters, J. Quantum Chemical and Theoretical Kinetics Study of the O(3P)+C2H2 Reaction: A Multistate Process. J. Phys. Chem. A 2006, 110, 6696–6706. [Google Scholar] [CrossRef]
- Rajak, K.; Maiti, B. Communications: Direct Dynamics Study of the O(3P) + C2H2 Reaction: Contribution from Spin Nonconserving Route. J. Chem. Phys. 2010, 133, 011101. [Google Scholar] [CrossRef]
- Garashchuk, S.; Rassolov, V.A.; Braams, B.J. Analytical Potential Energy Surface for O + C2H2 System. Chem. Phys. Lett. 2013, 588, 22–26. [Google Scholar] [CrossRef]
- Rajak, K.; Maiti, B. Trajectory Surface Hopping Study of the O(3P) + C2H2 Reaction Dynamics: Effect of Collision Energy on the Extent of Intersystem Crossing. J. Chem. Phys. 2014, 140, 044314. [Google Scholar] [CrossRef]
- Zuo, J.; Chen, Q.; Hu, X.; Guo, H.; Xie, D. Dissection of the Multichannel Reaction of Acetylene with Atomic Oxygen: From the Global Potential Energy Surface to Rate Coefficients and Branching Dynamics. Phys. Chem. Chem. Phys. 2019, 21, 1408–1416. [Google Scholar] [CrossRef]
- Creek, D.M.; Melliar-Smith, C.M.; Jonathan, N. Infrared Emission from Reaction of Atomic Oxygen with Acetylene. J. Chem. Soc. A 1970, 646–651. [Google Scholar] [CrossRef]
- Shaub, W.M.; Burks, T.L.; Lin, M.C. Dynamics of Reactions of O(3P) Atoms with 1-Alkynes as Studied by a CO Laser Resonance Absorption Technique. Chem. Phys. 1980, 45, 455–460. [Google Scholar] [CrossRef]
- Safron, S.A.; Weinstein, N.D.; Tully, J.C.; Herschbach, D.R. Transition State Theory for Collision Complexes: Product Translational Energy Distributions. Chem. Phys. Lett. 1972, 12, 564–568. [Google Scholar] [CrossRef]
- Hu, X.; Hase, W.L.; Pirraglia, T. Vectorization of the General Monte Carlo Classical Trajectory Program Venus. J. Comput. Chem. 1991, 12, 1014–1024. [Google Scholar] [CrossRef]
- Hase, W.L.; Duchovic, R.J.; Hu, X.; Komornicki, A.; Lim, K.F.; Lu, D.H.; Peslherbe, G.H.; Swamy, K.N.; Linde, S.R.R.V.; Varandas, A. Venus96: A General Chemical Dynamics Computer Program. J. Quantum Chem. Program Exch. Bull. 1996, 16, 671. [Google Scholar]
- Guo, Y.; Thompson, D.L.; Sewell, T.D. Analysis of the Zero-Point Energy Problem in Classical Trajectory Simulations. J. Chem. Phys. 1996, 104, 576–582. [Google Scholar] [CrossRef]
- Gutzwiller, M.C. Chaos in Classical and Quantum Mechanics; Springer Science + Business Media: New York, NY, USA, 1990. [Google Scholar]
- Czakó, G.; Bowman, J.M. Quasiclassical Trajectory Calculations of Correlated Product Distributions for the F+CHD3(ν1=0,1) Reactions Using an Ab Initio Potential Energy Surface. J. Chem. Phys. 2009, 131, 244302. [Google Scholar] [CrossRef] [Green Version]
- Corchado, J.C.; Espinosa-Garcia, J. Product Vibrational Distributions in Polyatomic Species Based on Quasiclassical Trajectory Calculations. Phys. Chem. Chem. Phys. 2009, 11, 10157–10164. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J. Quasi-Classical Trajectory Study of the Hydrogen Abstraction F+CHD3 Reaction: A State-to-State Dynamics Analysis. Chem. Phys. Lett. 2008, 454, 158–162. [Google Scholar] [CrossRef]
- Li, J.; Corchado, J.C.; Espinosa-Garcia, J.; Guo, H. Final State-Resolved Mode Specificity in HX + OH → X + H2O (X = F and Cl) Reactions: A Quasi-Classical Trajectory Study. J. Chem. Phys. 2015, 142, 084314. [Google Scholar] [CrossRef]
- Ping, L.; Tian, L.; Song, H.; Yang, M. New Method to Extract Final-State Information of Polyatomic Reactions Based on Normal Mode Analysis. J. Phys. Chem. A 2018, 122, 6997–7005. [Google Scholar] [CrossRef]
- Bonnet, L.; Rayez, J.C. Quasiclassical Trajectory Method for Molecular Scattering Processes: Necessity of a Weighted Binning Approach. Chem. Phys. Lett. 1997, 277, 183–190. [Google Scholar] [CrossRef]
- Bonnet, L.; Rayez, J.C. Gaussian Weighting in the Quasiclassical Trajectory Method. Chem. Phys. Lett. 2004, 397, 106–109. [Google Scholar] [CrossRef]
- Bonnet, L. Classical Dynamics of Chemical Reactions in a Quantum Spirit. Int. Rev. Phys. Chem. 2013, 32, 171–228. [Google Scholar] [CrossRef]
- Levine, R.D. Molecular Reaction Dynamics; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Jiang, B.; Guo, H. Control of Mode/Bond Selectivity and Product Energy Disposal by the Transition State: X + H2O (X = H, F, O(3P), and Cl) Reactions. J. Am. Chem. Soc. 2013, 135, 15251–15256. [Google Scholar] [CrossRef]
- Jiang, B.; Guo, H. Relative Efficacy of Vibrational vs. Translational Excitation in Promoting Atom-Diatom Reactivity: Rigorous Examination of Polanyi’s Rules and Proposition of Sudden Vector Projection (SVP) Model. J. Chem. Phys. 2013, 138, 234104. [Google Scholar] [CrossRef]
- Robinson, G.N.; Continetti, R.E.; Lee, Y.T. The Translational Energy Dependence of the F+C2H4→H+C2H3F Reaction Cross Section near Threshold. J. Chem. Phys. 1990, 92, 275–284. [Google Scholar] [CrossRef]
- Fu, B.; Han, Y.-C.; Bowman, J.M.; Angelucci, L.; Balucani, N.; Leonori, F.; Casavecchia, P. Intersystem Crossing and Dynamics in O(3P)+C2H4 Multichannel Reaction: Experiment Validates Theory. Proc. Natl. Acad. Sci. USA 2012, 109, 9733–9738. [Google Scholar] [CrossRef] [Green Version]
- Brouard, M.; Vallance, C. Tutorials in Molecular Reaction Dynamics; The Royal Society of Chemistry: Oxford, UK, 2012. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | Frequencies (cm−1) | SVP Values |
|---|---|---|
| Translation | - | 0.723 |
| ν1 (C-H symmetrical stretching) | 3499.1 | 0.056 |
| ν2 (C-H asymmetrical stretching) | 3413.7 | 0.038 |
| ν3 (C≡C stretching) | 2011.3 | 0.045 |
| ν4 and ν5 (symmetrical bending) | 746.8 | 0.122 |
| ν6 and ν7 (asymmetrical bending) | 613.4 | 0.441 |
| Ec | Vibrational Excitation | Total Energy | ftrana | frota | frot (HCCO) a | fvib (HCCO) a |
|---|---|---|---|---|---|---|
| 6.00 | - | 6.00 | 41.53% (41.7% b) | 0.43% | 13.39% | 44.65% |
| 8.22 | - | 8.22 | 39.54% (42% c) | 0.47% | 13.98% | 46.01% |
| 13.05 | - | 13.05 | 36.75% (35% c) | 0.54% | 17.39% | 45.33% |
| 8.22 | ν1 = 1 | 18.22 | 34.88% | 0.44% | 11.57% | 53.11% |
| 8.22 | ν2 = 1 | 17.98 | 34.55% | 0.44% | 11.74% | 53.27% |
| 8.22 | ν3 = 1 | 13.97 | 36.91% | 0.46% | 13.02% | 49.61% |
| 8.22 | ν4 = 1/ν5 = 1 | 10.36 | 38.65% | 0.48% | 13.30% | 47.57% |
| 8.22 | ν6 = 1/ν7 = 1 | 9.97 | 38.95% | 0.46% | 13.81% | 46.78% |
| Ec | Vibrational Excitation | Total Energy | ftrana | frota | frot (CO) a | fvib (CO) a | frot (CH2) a | fvib (CH2) a |
|---|---|---|---|---|---|---|---|---|
| 6.00 | - | 6.00 | 40.36% (41.3% b) | 0.47% | 13.68% | 6.47% | 19.86% | 19.17% |
| 8.22 | - | 8.22 | 39.40% (42% c) | 0.48% | 14.26% | 4.92% | 16.99% | 23.95% |
| 13.05 | - | 13.05 | 37.96% (42% c) | 0.59% | 14.36% | 5.04% | 17.34% | 24.72% |
| 8.22 | ν1 = 1 | 18.22 | 35.88% | 0.45% | 13.52% | 5.83% | 16.96% | 27.35% |
| 8.22 | ν2 = 1 | 17.98 | 36.30% | 0.48% | 14.67% | 6.05% | 17.24% | 25.27% |
| 8.22 | ν3 = 1 | 13.97 | 37.45% | 0.51% | 14.06% | 5.88% | 17.27% | 24.82% |
| 8.22 | ν4 = 1/ν5 = 1 | 10.36 | 39.03% | 0.49% | 14.29% | 4.65% | 16.89% | 24.65% |
| 8.22 | ν6 = 1/ν7 = 1 | 9.97 | 38.10% | 0.44% | 14.41% | 5.67% | 17.24% | 24.15% |
| - | - | - | - | - | - | (7.4% d) | - | - |
| - | - | - | - | - | - | 6.1 ± 1.3% e) | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Chen, Q.; Zuo, J.; Hu, X.; Xie, D. Dissection of the Multichannel Reaction O(3P) + C2H2: Differential Cross-Sections and Product Energy Distributions. Molecules 2022, 27, 754. https://doi.org/10.3390/molecules27030754
Zhang S, Chen Q, Zuo J, Hu X, Xie D. Dissection of the Multichannel Reaction O(3P) + C2H2: Differential Cross-Sections and Product Energy Distributions. Molecules. 2022; 27(3):754. https://doi.org/10.3390/molecules27030754
Chicago/Turabian StyleZhang, Shuwen, Qixin Chen, Junxiang Zuo, Xixi Hu, and Daiqian Xie. 2022. "Dissection of the Multichannel Reaction O(3P) + C2H2: Differential Cross-Sections and Product Energy Distributions" Molecules 27, no. 3: 754. https://doi.org/10.3390/molecules27030754
APA StyleZhang, S., Chen, Q., Zuo, J., Hu, X., & Xie, D. (2022). Dissection of the Multichannel Reaction O(3P) + C2H2: Differential Cross-Sections and Product Energy Distributions. Molecules, 27(3), 754. https://doi.org/10.3390/molecules27030754