Abstract
Like other organs, brain functions diminish with age. Furthermore, for a variety of neurological disorders—including Alzheimer’s disease—age is one of the higher-risk factors. Since in many Western countries the average age is increasing, determining approaches for decreasing the effects of aging on brain function is taking on a new urgency. Neuroinflammation and oxidative stress are two convoluted key factors in brain aging and chronic neurodegenerative diseases. The diverseness of factors, causing an age-related decrease in brain functions, requires identifying small molecules that have multiple biological activities that can affect all these factors. One great source of these small molecules is related to polyphenolic flavonoids. Recently, 3,3′,4′,7-tetrahydroxyflavone (fisetin) has been reported as a potent senotherapeutic capable of extending lifespan by reducing peroxidation levels and enhancing antioxidant cell responses. The neuroprotective effects of fisetin have been shown in several in vitro and in vivo models of neurological disorders due to its actions on multiple pathways associated with different neurological disorders. The present work aims to collect the most recent achievements related to the antioxidant and neuroprotective effects of fisetin. Moreover, in silico pharmacokinetics, pharmacodynamics, and toxicity of fisetin are also comprehensively described along with emerging novel drug delivery strategies for the amelioration of this flavonol bioavailability and chemical stability.
1. Introduction
Aging is a highly malleable process that can be modulated in different ways, such as caloric restriction, intermittent fasting, exercise, and a plant-based diet rich in phytochemicals [1]. The use of bioactive compounds to eliminate senescent cells has recently emerged as a promising approach to delay aging and reduce the severity of chronic diseases. Among others, neurodegenerative age-related diseases—including Alzheimer’s disease (AD) and Parkinson’s disease (PD)—currently affect people worldwide. Unfortunately, no treatments are currently available to prevent disease development and progression. Plant-derived flavonoids have a wide range of activities that could make them particularly effective for blocking the age-associated toxicity pathways associated with neurodegenerative diseases [2]. Fisetin has recently emerged as a potential anti-inflammatory, chemopreventive, chemotherapeutic, and senotherapeutic agent, thus making it a good candidate for the treatment of neurodegenerative diseases [3].
Fisetin is a 3,7,3′,4′-tetrahydroxyflavone widely found as glycosides or as aglycone in various edible fruits and vegetables such as strawberries (Fragaria sp. 160 μg/g), apples (Malus sp. 26.9 μg/g), persimmons (Diospyros sp. 10.6 μg/g), Lotus roots (Nelumbo sp. 5.8 μg/g), onions (Allium sp. 4.8 μg/g), grapes (Vitis sp. 3.9 μg/g), kiwi fruits (Actinidia sp. 2.0 μg/g), peaches (Prunus sp. 0.6 μg/g), cucumbers (Cucumis sp. 0.1 μg/g), and tomatoes (Solanum sp. 0.1 μg/g). The average human daily intake is estimated to be around 0.4 mg [4]. It has been recently demonstrated that this natural compound could modulate different pleiotropic pathways (phosphatidylinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) and p38, mitogen-activated protein kinases (MAPK)-dependent nuclear factor kappalight-chain-enhancer of activated B cells (NF-κB)) exerting a large plethora of biological effects including anti-inflammatory, hypolipidemic, hypoglycemic, antioxidant, neuroprotective, antiangiogenic, and antitumor ones [5,6,7].
The present review aims at collecting the most recent achievements related to the antioxidant and neuroprotective effects of fisetin. Moreover, pharmacokinetics and pharmacodynamics of fisetin and fisetin-related metabolites are also comprehensively described.
2. Crossroads of Neuroinflammation and Neurodegenerative Disorders
2.1. Neurodegenerative Disorders
Neurodegenerative disorders are classified by their clinical features, with few patients showing pure syndromes and most having mixed clinical presentations. Most neurodegenerative diseases show common physiological signs characterized by progressive neuronal dysfunction and death triggered by several dysfunctional physiological processes, such as protein abnormalities, proteotoxic stress, and its attendant abnormalities in ubiquitin–proteasomal and autophagosomal/lysosomal systems, oxidative stress, programmed cell death, and neuroinflammation [8]. Among protein abnormalities, amyloidosis represents the most common dysfunction in almost all common neurodegenerative diseases. Amyloids are insoluble fibrous proteins and amyloid-like filamentous aggregates are mostly within the cytoplasm of neurons and glia under neurodegenerative conditions. The most common aggregate is referred to β-amyloid or Aβ which is the main pathological dysfunction leading to Alzheimer’s disease. AD is nowadays the commonest neurodegenerative disease and the most frequent cause of dementia, affecting 50 million people worldwide in 2018 [9]. Currently, there are only two classes of approved drugs to treat AD, including cholinesterase inhibitors and antagonists to N-methyl D-aspartate (NMDA), which are effective only in treating the symptoms but do not cure or prevent the disease.
Neurological disorders are now the leading source of disability in the world, and Parkinson’s disease is the fastest growing of these disorders. Clinically characterized by the cardinal features of rest tremor, bradykinesia, rigidity and postural instability, and a variety of other motor and non-motor symptoms, PD etiopathogenesis includes the accumulation of intracytoplasmic bodies named “Lewy’s bodies” and dopamine deficiency. The underlying key molecular mechanisms include α-synuclein misfolding and aggregation, mitochondrial dysfunction, impairment of protein clearance, neuroinflammation, and oxidative stress [10]. Individuals with mild motor-predominant PD (49–53%) have mild symptoms, a good response to dopaminergic medications (e.g., carbidopa–levodopa, dopamine agonists, monoamine oxidase B inhibitors [11]), and slower disease progression. Dopamine-based therapies initially help to decrease motor symptoms. Contrariwise, non-motor symptoms require non-dopaminergic approaches (e.g., selective serotonin reuptake inhibitors for psychiatric symptoms, cholinesterase inhibitors for cognition). No disease-modifying pharmacologic treatments are currently available [12].
In this light, alternative and innovative therapeutic approaches for neurodegenerative diseases are urgently needed. Since neuroinflammation is a common feature in most pathologies leading to neurodegeneration, a valid approach to adjuvate traditional treatments must counteract the molecular mechanisms underlying inflammation in the CNS.
2.2. The Interplay between Neuroinflammation and Neurodegeneration
The term neuroinflammation defines an inflammatory cell or tissue response within the brain or spinal cord. Resident central nervous system (CNS) glia (microglia and astrocytes), endothelial cells, and immune cells produce cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers to mediate neuroinflammation processes (Figure 1). As for general inflammation, the degree of neuroinflammation is strongly influenced by the cell context, duration, and course of the primary stimulus or insult [13]. Microglia contains the main cell types implicated in neuroinflammation. Indeed, most of the CNS innate immune capacity resides there and thus microglia has an active role in immune surveillance [14].
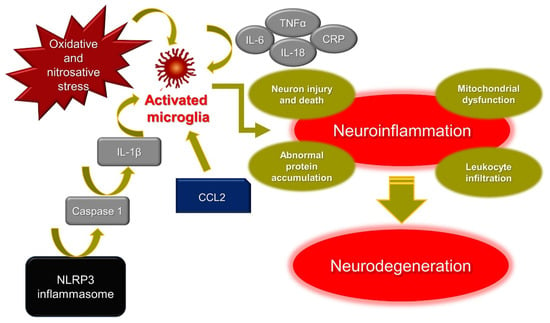
Figure 1.
Mechanisms of neuroinflammation. IL-6: interleukin-6; IL-18: interleukin-18; IL-1β: interleukin-1β; TNFα: tumor necrosis factor α; CRP: C-reactive protein; CCL2: C-C motif chemokine ligand 2; NLRP3: NOD-like receptor protein 3.
Under infection or disease, microglia become “activated” and resident cells function as inflammatory mediators. Activated cells in microglia rapidly produce pro-inflammatory cytokines and chemokines, facilitating the recruitment of leukocytes in the brain [15]. Neuroinflammation is mediated by several key cytokines, interleukin (IL)-1β, IL-18, IL-6, tumor necrosis factor (TNF)α, chemokines (CCL2, CCL5, CXCL1), secondary messengers (NO and prostaglandins), and ROS. In general, CNS benefits from fine and tight modulation of microglial activation which is intended to protect the host organism. Nonetheless, chronic microglial activation due to amplified or exaggerated cytokine production can lead to robust pathological changes and neurodegeneration [14].
IL-1β and IL-18 have prominent functions in the CNS, and therefore many nervous cell types express their cognate receptors which mediate inflammatory signaling cascades that may contribute to neuronal injury and cell death. Increased levels of IL-1β and IL-18 are often observed upon CNS infection, brain injury, and neurodegenerative diseases [16]. It has been reported that pro-inflammatory markers, including IL-1β, IL-6, TNF-α, and C-Reactive Protein (CRP), are significantly higher in the elderly with depression and AD [17]. NOD-like receptor protein 3 (NLRP3) inflammasomes play a crucial role in PD via caspase 1 activation, inducing IL-1β maturation and leading to neuronal pyroptosis. Furthermore, the abnormal aggregation of α-synuclein, which is the main pathogenesis factor of PD, also activates NLRP3 inflammasomes [18].
Recently, chemokines have become a focus in the context of the development and treatment of brain diseases. Among them, chemokine CCL2 and its main receptor CCR2 have become candidate mediators of abnormal brain–immune-system dialogue in depression [19]. It has also been reported that an atypical chemokine receptor CCL2 binding, supports sustained inflammation and increased AD risk [20].
Mitochondrial dysfunction and oxidative and nitroxidative stress have emerged as major contributors to degeneration of dopaminergic neurons in PD [21], AD [22] and in general in neurodegenerative diseases. One of the histopathological hallmarks of AD is the formation of extracellular senile plaques of the amyloid-β (Aβ) peptide aggregated with metal ions such as copper, iron, or zinc. Redox-active metal ions, such as copper, for example, can catalyze the production of ROS when bound to the Aβ. It has been reported that the ROS produced thus may contribute to oxidative damage on both the Aβ peptide itself and on the surrounding molecule [23].
To date, available therapy options for neurodegenerative diseases have not shown fully satisfactory results in finding a curative or disease-modifying treatment and many drug candidates have failed in clinical development. Thus, novel approaches in the prevention of neurodegenerative processes could be necessary. One possible solution could be a combination of easily and cheaply available food-derived substances (so-called “nutraceuticals”) with a low profile of adverse reactions, aiming at supporting canonical therapies, targeting the multiple involved pro-inflammatory mediators implicated [24]. Natural compounds, such as flavonoids, own neuroprotective potential related to their ability to modulate the inflammatory responses involved in neurodegenerative diseases in the microglia [25]. Fisetin is one of the most common and bioactive flavonoids which possesses potential neuroprotective effects. It has been reported that fisetin enhances learning and memory, decreases neuronal aging-related cell death, and suppresses oxidative stress [26]. Most important for our discussion, it has been reported that, on ten natural flavonoids tested in vivo, fisetin was the most potent senolytic compound, reducing senescence markers in progeroid and old mice [27]. In this work, it has been therefore demonstrated that fisetin reduced senescence in a subset of cells in murine and human adipose tissue, demonstrating cell-type specificity. Moreover, the fisetin administration to wild-type mice late in life restored tissue homeostasis, reduced age-related pathology, and extended median and maximum lifespan, disclosing that fisetin is a promising senotherapeutic compound both in mice and humans. Since aging is deemed one of the major risk factors which leads to various chronic diseases and disabilities such as neurodegenerative diseases [28,29], in the next paragraphs the crossroads between senescence and neurodegeneration are highlighted, as well as the molecular mechanisms underlying the effects of fisetin as a senolytic drug for neuroprotection.
3. Fisetin as a Senolytic Drug: Crosstalk between Neuroprotection and Senescence
One of the characteristics of normal aging is a decrease in both cognitive and motor functions, resulting in changes in learning and memory as well as deficits in balance and coordination. Furthermore, for a variety of neurological disorders (including AD), age is one of the higher risk factors. Since in many Western countries the average age is increasing, determining approaches to decrease the effects of aging on brain function is taking on a new urgency. However, to choose among possible approaches, it is first necessary to identify the factors which contribute to the decrease in brain function with age. Recent evidence suggests an association between cellular senescence and age-related diseases. Senescent cells are characterized by an irreversible replicative arrest, resistance to apoptosis, increased ROS generation, metabolic shifts, and acquisition of a pro-inflammatory senescence-associated secretory phenotype (SASP). The latter can accumulate in various tissue sites such as the CNS, suggesting that it can contribute significantly to the cognitive decline characteristic of neurological disease.
In order, focusing attention and therapeutic interventions on the biological mechanisms underlying aging and particularly on the reduction of senescent cells using senolytic drugs could be important to alleviate age-related diseases such as neurodegenerative diseases.
3.1. Senescence Mechanisms
Cellular senescence occurrence is widely reported as the first sign of aging [30]. In 1961 the first experimental evidence was highlighted for cellular aging in vitro. Leonard Hayflick and Paul Moorhead established in a ground-breaking study that normal diploid human fibroblasts have a restricted ability to replicate before they enter a stage known as replicative senescence [31]. Subsequently, this specific form of senescence (known as replicative senescence) was connected to telomere attrition, which promotes tumorigenesis and chromosomal instability, consistent with the original hypothesis that uncontrolled cell growth of damaged cells is counteracted via senescence [32]. Permanent cell cycle arrest in senescent cells plays a fundamental role in anticancer machinery [33]. Several stressors can induce senescence; for instance, ROS, DNA damage, strong mitogenic/oncogenic signaling, depletion of specific tumor suppressors, arrested DNA replication, and chromatin disruption [34,35,36,37,38,39,40].
However, the senescent cell cycle arrest is governed by tumor-suppressive pathways, and it is organized through p53/p21 and p16INK4A/retinoblastoma (RB) [41]. The DNA damage response (DDR) implies the occurrence of DNA double-strand breaks and uncapped telomeres, which results in post-translational phosphorylation events via ATM and ATR serine/threonine protein kinases. These events lead to p53-dependent DNA stabilization or to p14ARF-mediated inhibition of the ubiquitin ligase MDM2 upon hindering degradation of p53 [42,43]. The transcription of the cyclin-dependent kinase inhibitor (CDKi) p21 takes place once p53 is stabilized, triggering transient cell cycle arrest [44]. Next, a permanent arrest is maintained by the transcriptional upregulation of p16INK4A mediated by p38 [45] and/or ERK signaling [46]. Once established, a permanent blockage of the S phase entry occurs, due to the inhibition of the activity of both CDK4 and CDK6 via p16INK4A, thus leading to RB hyperphosphorylation. Substantially, the expression of p16INK4A increases with aging in different types of tissues and it is considered as a biomarker of natural aging [47].
The number of senescent cells increases with age in vivo, and one of the most important methods to detect them is measuring the senescence-associated β-galactosidase activity (SA β-gal) at pH 6.0 [48]. Senescent cells are considered to apply their adverse effects with aging and in age-related diseases to a certain extent via the SASP, which consists of multiple growth factors, proteases, and inflammatory cytokines [49].
Cell fate depends on various incentives, such as impairment of specific tumor suppressors or oncogene activation. As a strategy for preventing neoplastic transformation, cells might undergo senescence or apoptosis. p53 is a major key regulator of this cell decision based on the level of its activation. Excessive p53 signaling leads to the overexpression of proapoptotic modulators including NOXA and PUMA, which causes cell death. Milder “senescence-inducing” stressors stimulate p53-mediated transcription of the CDKi p21 and BCL-2 anti-apoptotic family members, including BCL-W, BCL-2, and BCL-XL [50]. These proteins bind to proapoptotic proteins with BH3 domains, preventing mitochondrial outer membrane permeabilization and apoptosis [51]. While transient low-grade p53 activation aids repair, persistent activation can lead to cell senescence. These situations cause antiapoptotic proteins to be produced to promote survival, which might explain why senescent cells grow in vitro with passaging and in vivo with increasing age [52].
3.2. CNS Senescence
Aging drives chronic brain inflammation in a gradual manner and influences the homeostasis of all brain cell types, including neurons, microglia, astrocytes, oligodendrocytes, and endothelial cells [53]. The accumulation of inflammatory cytokines occurs without clear evidence of the pathogen. This leads to decreased pre- and postsynaptic densities alongside a total decrease in synapses and dendritic spines, which induce memory loss and cognitive deficits in old individuals. However, a typical feature of many neurodegenerative disorders is chronic inflammation levels associated with aging, which indicates that the normal aging process and pathological modification have mutual proinflammatory pathways [52].
3.2.1. Neurons
In vivo, terminally differentiated neurons may show signs of senescence correlated with advanced age. Additionally, it has been demonstrated that in neurodegenerative diseases, for instance in AD patients, nervous cells have shortened telomeres [54]. On the contrary, PD patients do not exhibit these alterations [55]. Furthermore, in 2012, Jurk et al. illustrated more extensive and comprehensive evidence for neuronal senescence [56]. For the first time, this study demonstrated that Purkinje and cortical neurons in 32-month-old mice have various cell senescent hallmarks, including the most common senescent-driven pathways in a p21/CDKN1A dependent manner [56]. These findings demonstrated that neuronal cells have similar senescent features compared to proliferating cells. In 2015, a study on human brains verified neuronal senescence in mice and rats, demonstrating p16/CDKN2A expression in pyramidal neurons in the brains of old individuals (above 77 years) [57]. Furthermore, senescent neurons secrete pro-inflammatory cytokines which may play a key role at the beginning of neurodegeneration. It has been demonstrated that neurons in AD patients have senescent features such as augmented p38/MAPK activity, TGF mRNA expression, IL-6 expression, and p16/CDKN2A expression and that higher SASP activity can trigger senescence in surrounding cells in a paracrine way [45].
3.2.2. Microglia
Microglia, which originated during development in the bone marrow, are the fundamental immune cells of the brain and are functionally macrophage-like [58]. Normally quiescent microglia become activated as a reaction to invading microorganisms or injury, release different growth factors and cytokines, and induce phagocytosis [59]. Senescence-upregulated proinflammatory cytokines, including IL-6, IL-1β, and TNF-α, were highly expressed in microglia from aged mice [49,60,61]. Furthermore, shorter telomeres are present in naturally aged microglia [62]. Chronically activated microglia demonstrate different features of senescence, including growth arrest, heterochromatic foci formation, and SA β-gal activity, among others [63]. Interestingly, TGF-β levels in plasma and cerebrospinal fluid were observed to be increased in Alzheimer’s patients. However, unpaired TGF-β1-SMAD3 signaling was expressed in senescent microglia, which involved aberrant activation and decrease of phagocytic Aβ peptide activity [64].
3.2.3. Astrocytes
The brain is one of the richest body organs with astrocytes, which were formerly thought to be non-functional neural network fillers. Nevertheless, as time passes and technology improves, the significance of these cells in a variety of biological processes has been established. Astrocytes are crucial for maintaining homeostasis by balancing osmotic balance [65]. Furthermore, astrocytes have been shown to have a role in synaptic transmission by being adjacent to neurons, which is a component of the tripartite synapse and enhances neurotransmission between pre- and postsynaptic parts. Moreover, because of their participation in innate immunity, astrocytes play a role in the defense against brain inflammation such as neurodegeneration, trauma, and infection [66].
3.3. Senolytic Drugs
To our knowledge, during pathological conditions senescent cells may accumulate with age in the brain. Currently, there are no methods or strategies for identifying, isolating, and eliminating senescent cells, and distinguishing their mechanistic involvement in neuropathology is exceedingly challenging. Until now, identifying senescent cells has relied on a mixed group of markers that are not specific, to verify that the alterations are not caused by other factors. It was observed that the use of genetically modified mice, through genetic inactivation of p16INK4A or the elimination of senescent cells, may impede cells from becoming senescent, which causes neuropathology. However, few relevant researchers have applied these techniques to neurodegenerative pathologies, and p16INK4A mice with genetic inactivation are extremely tumor prone and die before cognitive impairment is noticed. To deal with this problem, two genetically engineered mice models were established. The INK-ATTAC transgenic mouse uses a 2.6-kb p16INK4A promoter region to induce drug-responsive caspase-8 expression when the drug AP20187 is introduced to mice [67]. Another useful model, the p16-3MR transgenic model, expresses a trimodal reporter construct under the control of a bacterial artificial chromosome that contains the whole p16INK4A promoter. Monomeric red fluorescent protein (mRFP), synthetic Renilla luciferase, and a shortened herpes simplex virus thymidine kinase make up this reporter (HSV-TK). When ganciclovir is given to p16INK4A-expressing cells, HSV-TK turns it into a hazardous DNA chain terminator, causing senescent cells to die through mitochondrial-dependent cell death [68,69]. These models helped to improve our understanding of senescent cell role in various age-related diseases. Senescent cells (SCs) might be removed for therapeutic benefit without causing detrimental side effects. This is a major finding that has paved the way for the creation of drugs and strategies that target SNC, particularly for the prevention and finding a cure for age-related diseases [67,70,71]. Several approaches have been developed aiming at the clearance of senescent cells and the reduction of SASP, e.g., the development of genetic clearance models (transgenic animals) and the identification and development of drugs referred to as “senotherapeutics”. Currently, the identification of senotherapeutic drugs represents a promising area of research for new therapies. Even if the knowledge of their potential molecular targets and their mechanisms of action are still limited, senotherapeutics are classified as (i) senolytics agents selectively targeting senescent cells to eliminate them; (ii) senomorphics agents, which create a controlled environment by suppressing markers of senescence and targeting a specific component of SASP. As a consequence, the functions and morphology of senescence cell are modulated by reverting it or by slowing the progression of cells towards the senescent phenotype; (iii) immune-system mediators of the clearance of SCs. Tests conducted to investigate the mechanism of action of different senotherapeutic agents have revealed that they can be classified as dual agents (senolytic and senomorphic), in a cell-specific and concentration-dependent manner, obviously in correlation to the heterogenicity of senescent cells [72].
Senolysis, rather than SASP suppression, has the most therapeutic potential for two main reasons. First, the permanent removal of SCs results in the long-term elimination of harmful SASP components. Second, there is no probability of tumorigenic “escape” from senescence once a SC has been removed [73].
Senolytics were the first senotherapeutic agents to be effectively evaluated in preclinical in vivo models, and there are already multiple senolytic drugs available. Many of these drug targets increased anti-apoptosis mechanisms in SCs, including signaling via the BCL2 protein family (BCL2, BCLXL, and BCLW). These proteins bind to pro-apoptotic BCL2 family members and functionally deactivate them [51,74]. BCL2 proteins that are pro-apoptotic, activate the BAX and BCL2 homologous antagonist/killer (BAK) proteins, inducing cytochrome c release and programmed cell death [75].
3.4. Fisetin as Senolytic Drug in the CNS
Fisetin is a senolytic flavonoid that is found in nature. It functions by suppressing BCL-2 family members such as BCL-xL, as well as HIF-α and other components of the SCAP network [27]. Moreover, Singh and colleagues evaluated the neuroprotection properties of fisetin in induced, accelerated, and natural aging models of rat. Interestingly, it was found that fisetin significantly reduced the peroxidation levels and increased antioxidants responses. Furthermore, mitochondrial membrane depolarization and apoptotic cell death were attenuated in aging rat brain. At the genetic level, fisetin increased the expression of autophagy genes (Atg-3 and Beclin-1) and NSE and Ngb sirtuin-1 neuronal markers and decreased the expression of IL-1β, TNF-α, and Sirt-2 inflammatory genes in the aging brain [76]. Interestingly, Cho et and coworkers [77] found that fisetin significantly decreased neuronal defects at days 5 and 10 in a Caenorhabditis elegans model. Indeed, worms exposed to fisetin exhibited decreased anterior lateral microtubule (ALM) and posterior lateral microtubule (PLM) morphological defects at day 5 compared to the DMSO control.
It has been furthermore reported that treatment with fisetin attenuated brain edema and cell apoptosis in intracerebral hemorrhage (ICH) mice, which exhibited a significant increase in the modified neurologic severity scores (mNSS). Fisetin attenuated severe brain deficit by decreasing levels of proinflammatory cytokines. Moreover, diminution of NF-κB signaling was reported after fisetin treatment [78]. However, a recent study demonstrated an increase in p16Ink4a senescent cells in an age-dependent manner, which was more noticeable in microglia and oligodendrocyte progenitor cells. In aged mice, with p16Ink4a-positive senescent cells, they were genetically eliminated upon treatment with two specific drugs or with the senolytic cocktail (dasatinib and quercetin). It was demonstrated that both strategies resulted in a decrease in p16Ink4a particularly in the microglial population, resulting in reduced microglial activation and reduced expression of SASP factors. Substantially, these two methods significantly improved cognitive function in aged mice, suggesting that senolytic interventions might be a potential therapeutic approach to mitigate age-associated cognitive impairment [79].
One of the physiological brain age indicators is the electroencephalograph (EEG). Various sensory–cognitive inputs modify brain oscillatory waves from neural tissue. Furthermore, fast-wave oscillations (e.g., 8–12 Hz or 12–28 Hz) are linked to coordination and deal with difficult behavioral tasks. In a recent study, Das and colleagues [80] tested fisetin effects in vivo through cortical spectral power oscillations and multi-unit activity (MUA), finding that in aged rats the relative spectral power of α and β decreased along with the MUA count compared to young ones.
Intriguingly, in vivo study on aging senescence-accelerated prone 8 (SAMP8) mice demonstrated that fisetin prevents cognitive and locomotor deficits. Additionally, three proteins linked to synaptic function were reduced in aged mice compared to young mice, and fisetin treatment imped their reduction almost completely [81]. Moreover, in another study on brain mice injected with D-galactose (D-gal) to accelerate senescence, fisetin significantly reduced ROS generation induced by D-galactose, along with neuroinflammation-related pathways and pro-apoptotic markers [82].
4. Fisetin: In Silico Evaluation of Pharmacodynamics, Pharmacokinetics, and Toxicity
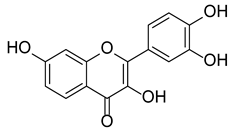
Fisetin, chemically known as 2-(3,4-dihydroxy)-3,7-dihydroxy-4H-1-benzopyran-4-one, has a peculiar flavone scaffold like that of the more famous quercetin (see paragraph 3.4). Like other flavonoids, it displays poor aqueous solubility (9.55–10.45 µg/mL at 37 °C) [83] and high affinity to polar organic solvents (2.89 mg/mL in ethanol:water 50/50 at 27 °C), pKa1 = 7.27 ± 0.09 and pKa2 = 9.44 ± 0.07 [84], and experimental LogP of 2.20 [85]; contrariwise, its antioxidant activity is not so efficient despite the presence of a reactive catechol portion [3]. This feature led to the discovery of low stability or bioavailability of this secondary metabolite when tested in cell culture conditions or after in vivo administration, respectively [86]. These unfavorable characteristics can be partially overcome by lipid-based delivery systems such as liposomes, nanoemulsions, nanocochelates, and spherulites [87].
Keeping under consideration that the appraisal of a natural compound as a promising drug is a long and uncertain process, we aimed to evaluate this small molecule by means of fourteen commercially available online machine-learning platforms in terms of pharmacodynamics, pharmacokinetics, and toxicity (Table 1, Table 2 and Table 3). Some data were initially predicted through a tool and then further validated by others if available. We aim to provide new information to better ascertain the biological potential and drug likeness of this widely spread flavonoid, which has been involved till Phase 2 of clinical trials according to the ZINC database (code ZINC39111) (http://zinc15.docking.org, accessed on 30 December 2021) and ChEMBL (https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL31574/, accessed on 30 December 2021) for the treatment of osteoarthritis of the knee, childhood cancer, and severe acute respiratory syndrome in COVID-19.

Table 1.
In silico prediction of putative pharmacodynamics for fisetin (threshold: probability score ≥ 0.6).
Table 2.
Molecular properties and ADME (absorption, distribution, metabolism, excretion) profile of fisetin.
Table 3.
Prediction of toxicity for fisetin.
Starting with the web-service tool SwissTargetPrediction implemented with SwissSimilarity, we collected data and robust predictive models in Table 1 regarding the highly putative human targets for fisetin with a probability score ≥0.6 [88] (Table 1). Then, we checked if ZINC and ChEMBL could corroborate these data. Moreover, fisetin had no structural similarity to other FDA-approved drugs.
Epigenetic Target Profiler [89] and ChEMBL also proposed the strong affinity for Cyclin-dependent kinase 2 and to a lesser extent to other epigenetic targets (Cyclin-dependent kinase 1, Bromodomain-containing protein 2, Histone acetyltransferase p300, Histone deacetylase 2, Protein kinase 1, Serine-protein kinase ATM).
Lastly, PASS (Prediction of Activity Spectra for Substances, http://www.pharmaexpert.ru/PASSonline/index.php, accessed on 30 December 2021) was developed to predict biological activities with 90% accuracy. Each outcome is characterized by Pa (probability to be active, high if ≥ 0.7) or Pi (probability to be inactive, good if Pa is much higher than Pi). Fisetin displayed discrete cytotoxicity expressed as Pa value of 0.523 and Pi = 0.049 against human Hs 683 cell line (oligodendroglioma).
Drug likeness of fisetin was predicted by determining whether it had properties consistent with being an orally active drug and important in the drug design and development to prevent drug candidates from failing. Calculations were made using different web-service tools such as SwissADME and BOILED-Egg [90,91], pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction, accessed on 30 December 2021) [92], FAF-Drugs4 (https://fafdrugs4.rpbs.univ-paris-diderot.fr/ [93], accessed on 30 December 2021), OSIRIS Property Explorer (http://www.cheminfo.org/Chemistry/Cheminformatics/Property_explorer/index.html, accessed on 30 December 2021), and PreADMET (v. 2.0, Yonsei University, Seoul, Korea, https://preadmet.bmdrc.kr/adme/, accessed on 30 December 2021) (Table 2). The molecule respected Lipinski’s, Ghose’s, Veber’s, Egans’, and Muegge’s filters with no violation of the drug likeness according to SwissADME and PreADMET. Moreover, it displayed an Abbott bioavailability score of 0.55 and synthetic accessibility score of 3.16 (but it can be also obtained by natural sources).
The recommended range for Caco2 permeability is: <25 poor, >500 great or if Papp is higher than 8 × 10−6 cm/s; for HIA is: >80% high, <25% poor; for BBB: optimum range between −3.0 and −1.2, whereas logBBB >0.3 highly permeable and <−1 poorly permeable; for skin permeability: optimum range between −8.0 and −1.0; for log PS > −2 possible penetration into the CNS, for log PS < −3 not able to penetrate the CNS.
The main metabolites (and their percentages) of fisetin predicted by GLORYx software are 7- (98%), 4′- (97%), and 3′-glucuronides (90%) along with 3′-OCH3 (90%) and 3′-sulfated (87%) compounds [94].
Finally, to conclude an exhaustive analysis on fisetin, preliminary data on toxicity influencing the fate of drug candidates were also calculated by ProTox-II [95], CarcinoPred-EL [96], OSIRIS Property Explorer, PreADMET, pkCSM and presented in Table 3. These in silico toxicity models can substitute animal testing, avoiding timewasting and unnecessary cost.
Data collected in silico described fisetin as a good lead compound to be further investigated for specific biological activities, despite the limited aqueous solubility and moderate BBB permeation. This information further justified the loading of fisetin into more complex structures capable of enhancing the delivery and targeting of this bioactive flavonol [96,97,98,99].
5. Conclusions
Pathophysiological determinants leading to aging and age-related neurodegenerative diseases are still unknown. The lack of understanding of the mechanisms underlying the onset of neuronal aging and related pathologies represents an obstacle to the development of targeted therapeutic strategies and delays the initiation of clinical translation for a wide range of applications. Recent evidence suggests accumulation of senescent cells with aging may actively contribute to chronic and age-related diseases and conditions such as neurodegenerative diseases. Their detrimental effects appear to be determined by metabolic shifts, increased generation of reactive oxygen species (ROS), and SASP factors. However, the overall significance and mechanistic contribution of these cells to neurodegenerative diseases have not yet been clarified due to the lack of identification and isolation tools. Scientific evidence, showing the efficacy through the elimination of senescent cells in the positive modulation of inflammatory diseases, has aroused an interest in the development of therapeutic strategies for the elimination of senescent cells, indicated as “senotherapy” in the absence of gene modifications. Recently, fisetin was shown to act as a senotherapeutic agent capable of extending lifespan, reducing ROS levels, and enhancing antioxidant cell responses. This neuroprotection has been detected in in vitro and in vivo models associated with different neurological disorders. The present review collected the most recent findings of the antioxidant and neuroprotective effects of fisetin. Furthermore, in silico pharmacokinetics, pharmacodynamics, and toxicity of fisetin were assessed by means of several commercially available web tools. Although senolytics provide a promising avenue for exploration as a treatment for age-related and neurodegenerative diseases, further clinical studies are needed to determine the safety and efficacy of these drugs before routine clinical use can be considered, and then possibly inclusion as agents that are available over the counter as dietary supplements. Determining high-risk populations, reliably identifying early signs of these diseases, recruiting patients at higher risk of developing the disease, and identifying the right time to start treatment are key parameters to consider for the effectiveness of the treatment. Diversifying and exploring new therapeutic ideas is essential, especially for these inherently complex and not very well-understood pathologies.
Author Contributions
Conceptualization, S.C. and M.G.; methodology, O.E., A.P., S.C. and M.G.; data curation, S.C.; writing—original draft preparation, O.E., A.P., S.C. and M.G.; writing—review and editing, A.P., M.P., A.C., S.C. and M.G.; supervision, S.C. and M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martel, J.; Ojcius, D.; Ko, Y.-F.; Ke, P.-Y.; Wu, C.-Y.; Peng, H.-H.; Young, J.D. Hormetic Effects of Phytochemicals on Health and Longevity. Trends Endocrinol. Metab. 2019, 30, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynkiewicz, G.; Demchuk, O.M. New Perspectives for Fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Sharma, A.; Sak, K.; Tuli, H.S.; Buttar, H.S.; Bishayee, A. Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. 2018, 194, 75–87. [Google Scholar] [CrossRef]
- Ravula, A.R.; Teegala, S.B.; Kalakotla, S.; Pasangulapati, J.P.; Perumal, V.; Boyina, H.K. Fisetin, potential flavonoid with multifarious targets for treating neurological disorders: An updated review. Eur. J. Pharmacol. 2021, 910, 174492. [Google Scholar] [CrossRef]
- Maher, P. Preventing and Treating Neurological Disorders with the Flavonol Fisetin. Brain Plast. 2020, 6, 155–166. [Google Scholar] [CrossRef]
- Yen, F.S.; Qin, C.S.; Xuan, S.T.S.; Ying, P.J.; Le, H.Y.; Darmarajan, T.; Gunasekaran, B.; Salvamani, S. Hypoglycemic Effects of Plant Flavonoids: A Review. Evid. -Based Complement. Altern. Med. 2021, 2021, 2057333. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer′s disease. Neural Regen. Res. 2022, 17, 1666. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Carradori, S.; D’Ascenzio, M.; Chimenti, P.; Secci, D.; Bolasco, A. Selective MAO-B inhibitors: A lesson from natural products. Mol. Divers. 2014, 18, 219–243. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Zengeler, K.E.; Lukens, J.R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 2021, 21, 454–468. [Google Scholar] [CrossRef]
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 2018, 23, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Yan, Y.-Q.; Fang, Y.; Zheng, R.; Pu, J.-L.; Zhang, B.-R. NLRP3 Inflammasomes in Parkinson’s disease and their Regulation by Parkin. Neuroscience 2020, 446, 323–334. [Google Scholar] [CrossRef]
- Curzytek, K.; Leśkiewicz, M. Targeting the CCL2-CCR2 axis in depressive disorders. Pharmacol. Rep. 2021, 73, 1052–1062. [Google Scholar] [CrossRef]
- Murcia, J.D.G.; Weinert, A.; Freitas, C.M.T.; Arens, D.K.; Ferrel, M.N.; Grose, J.H.; Ridge, P.G.; Wilson, E.; Kauwe, J.S.K.; Weber, K.S. Atypical chemokine receptor ACKR2-V41A has decreased CCL2 binding, scavenging, and activation, supporting sustained inflammation and increased Alzheimer’s disease risk. Sci. Rep. 2020, 10, 8019. [Google Scholar] [CrossRef]
- Nunes, C.; Laranjinha, J. Nitric oxide and dopamine metabolism converge via mitochondrial dysfunction in the mechanisms of neurodegeneration in Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108877. [Google Scholar] [CrossRef]
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer’s Disease: A Contextual Link with Nitric Oxide Synthase. Curr. Mol. Med. 2020, 20, 505–515. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Gruendler, R.; Hippe, B.; Jengic, V.S.; Peterlin, B.; Haslberger, A.G. Nutraceutical Approaches of Autophagy and Neuroinflammation in Alzheimer’s Disease: A Systematic Review. Molecules 2020, 25, 6018. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Sureda, A.; Manayi, A.; Nabavi, S.M. Neuroprotective Effects of Fisetin in Alzheimer’s and Parkinson’s Diseases: From Chemistry to Medicine. Curr. Top. Med. Chem. 2016, 16, 1910–1915. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L. Translating advances from the basic biology of aging into clinical application. Exp. Gerontol. 2013, 48, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A. Extending Life: Scientific Prospects and Political Obstacles. Milbank Q. 2002, 80, 155–174. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, E.O.W.; Zhu, Y.; Tchkonia, T.; Kirkland, J.L. Discovery, development, and future application of senolytics: Theories and predictions. FEBS J. 2020, 287, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederländer, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Romanov, V.S.; Abramova, M.V.; Svetlikova, S.B.; Bykova, T.V.; Zubova, S.G.; Aksenov, N.D.; Fornace, A.J.; Pospelova, T.V.; Pospelov, V.A. p21Waf1is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010, 9, 3945–3955. [Google Scholar] [CrossRef] [Green Version]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M.; Lozano, G. The Mdm network and its regulation of p53 activities: A rheostat of cancer risk. Hum. Mutat. 2014, 35, 728–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Wei, W.; Dutriaux, A.; Jobling, W.A.; Sedivy, J.M. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging Cell 2003, 2, 295–304. [Google Scholar] [CrossRef]
- Wong, E.S.M.; Le Guezennec, X.; Demidov, O.; Marshall, N.T.; Wang, S.T.; Krishnamurthy, J.; Sharpless, N.; Dunn, N.R.; Bulavin, D.V. p38MAPK Controls Expression of Multiple Cell Cycle Inhibitors and Islet Proliferation with Advancing Age. Dev. Cell 2009, 17, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.G.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Tavana, O.; Benjamin, C.L.; Puebla-Osorio, N.; Sang, M.; Ullrich, S.E.; Ananthaswamy, H.; Zhu, C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010, 9, 3348–3356. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero, D.A.; González-Giraldo, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Meta-analysis of Telomere Length in Alzheimer’s Disease. J. Gerontol. Ser. A 2016, 71, 1069–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero, D.A.; Gonzalez, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Telomere length in Parkinson’s disease: A meta-analysis. Exp. Gerontol. 2016, 75, 53–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Doorn, K.J.; Lucassen, P.J.; Boddeke, H.W.; Prins, M.; Berendse, H.W.; Drukarch, B.; van Dam, A.-M. Emerging roles of microglial activation and non-motor symptoms in Parkinson’s disease. Prog. Neurobiol. 2012, 98, 222–238. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation 2011, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence That Aging and Amyloid Promote Microglial Cell Senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Luo, X.M.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tichauer, J.E.; Flores, B.; Soler, B.; Bernhardi, L.E.-V.; Ramírez, G.; Von Bernhardi, R. Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.-M.; Adler, D.; DeMaria, M.; Mechtouf, N.; Teachenor, R.; Cardin, G.B.; Desprez, P.-Y.; Campisi, J.; Rodier, F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013, 4, e727. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; DeMaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.; Robbins, P.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Luna-Vargas, M.P.; Chipuk, J.E. Physiological and Pharmacological Control of BAK, BAX, and Beyond. Trends Cell Biol. 2016, 26, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, A.K.; Garg, G.; Rizvi, S.I. Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. 2018, 193, 171–179. [Google Scholar] [CrossRef]
- Cho, I.; Song, H.; Cho, J.H. Flavonoids mitigate neurodegeneration in agedCaenorhabditis elegansby mitochondrial uncoupling. Food Sci. Nutr. 2020, 8, 6633–6642. [Google Scholar] [CrossRef]
- Chen, C.; Yao, L.; Cui, J.; Liu, B. Fisetin Protects against Intracerebral Hemorrhage-Induced Neuroinflammation in Aged Mice. Cerebrovasc. Dis. 2018, 45, 154–161. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Das, J.; Singh, R.; Ladol, S.; Nayak, S.K.; Sharma, D. Fisetin prevents the aging-associated decline in relative spectral power of α, β and linked MUA in the cortex and behavioral alterations. Exp. Gerontol. 2020, 138, 111006. [Google Scholar] [CrossRef]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin Reduces the Impact of Aging on Behavior and Physiology in the Rapidly Aging SAMP8 Mouse. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2018, 73, 299–307. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin Rescues the Mice Brains Against D-Galactose-Induced Oxidative Stress, Neuroinflammation and Memory Impairment. Front. Pharmacol. 2021, 12, 57. [Google Scholar] [CrossRef]
- Mohite, R.; Mehta, P.; Arulmozhi, S.; Kamble, R.; Pawar, A.; Bothiraja, C. Synthesis of fisetin co-crystals with caffeine and nicotinamide using the cooling crystallization technique: Biopharmaceutical studies. New J. Chem. 2019, 43, 13471–13479. [Google Scholar] [CrossRef]
- Herrero-Martinez, J.M.; Sanmartin, M.; Roses, M.; Bosch, E.; Ràfols, C. Determination of dissociation constants of flavonoids by capillary electrophoresis. Electrophoresis 2005, 26, 1886–1895. [Google Scholar] [CrossRef]
- Moridani, M.Y.; Galati, G.; O’Brien, P.J. Comparative quantitative structure toxicity relationships for flavonoids evaluated in isolated rat hepatocytes and HeLa tumor cells. Chem.-Biol. Interact. 2002, 139, 251–264. [Google Scholar] [CrossRef]
- Xiao, J.; Högger, P. Stability of Dietary Polyphenols under the Cell Culture Conditions: Avoiding Erroneous Conclusions. J. Agric. Food Chem. 2015, 63, 1547–1557. [Google Scholar] [CrossRef]
- Bothiraja, C.; Yojana, B.D.; Pawar, A.P.; Shaikh, K.; Thorat, U.H. Fisetin-loaded nanocochleates: Formulation, characterisation, in vitroanticancer testing, bioavailability and biodistribution study. Expert Opin. Drug Deliv. 2013, 11, 17–29. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cruz, N.; Medina-Franco, J.L. Epigenetic Target Profiler: A Web Server to Predict Epigenetic Targets of Small Molecules. J. Chem. Inf. Model. 2021, 61, 1550–1554. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kops, C.D.B.; Šícho, M.; Mazzolari, A.; Kirchmair, J. GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chem. Res. Toxicol. 2021, 34, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ai, H.; Chen, W.; Yin, Z.; Hu, H.; Zhu, J.; Zhao, J.; Zhao, Q.; Liu, H. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef] [PubMed]
- Moolakkadath, T.; Aqil, M.; Ahad, A.; Imam, S.S.; Praveen, A.; Sultana, Y.; Mujeeb, M. Preparation and optimization of fisetin loaded glycerol based soft nanovesicles by Box-Behnken design. Int. J. Pharm. 2020, 578, 119125. [Google Scholar] [CrossRef]
- Sengupta, B.; Coleman, J.; Johnson, J.; Feng, M. Graphene oxide as selective transporter of flavonols for physiological target DNA: A two-color fluorescence approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 214, 192–198. [Google Scholar] [CrossRef]
- Pawar, A.; Singh, S.; Rajalakshmi, S.; Shaikh, K.; Bothiraja, C. Development of fisetin-loaded folate functionalized pluronic micelles for breast cancer targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 347–361. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).