
On-Site Measurement of Fat and Protein Contents in Milk Using Mobile NMR Technology

 ,
,
Abstract

1. Introduction
2. Results and Discussions
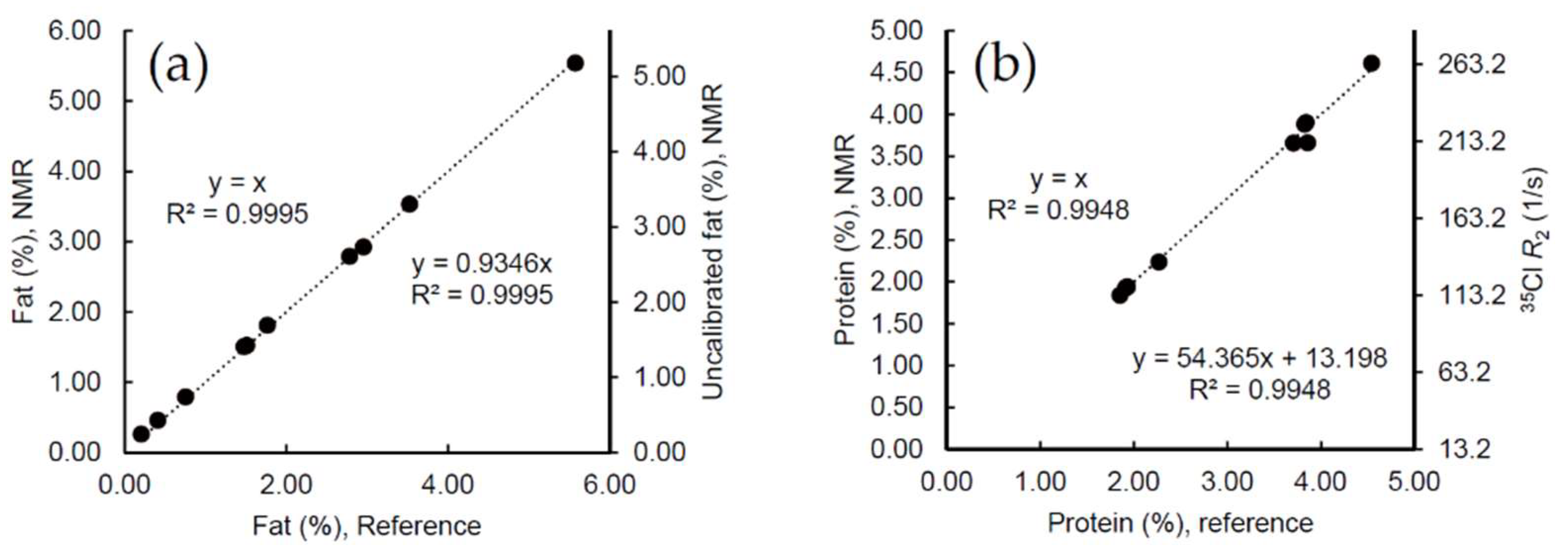
2.1. Calibration Samples
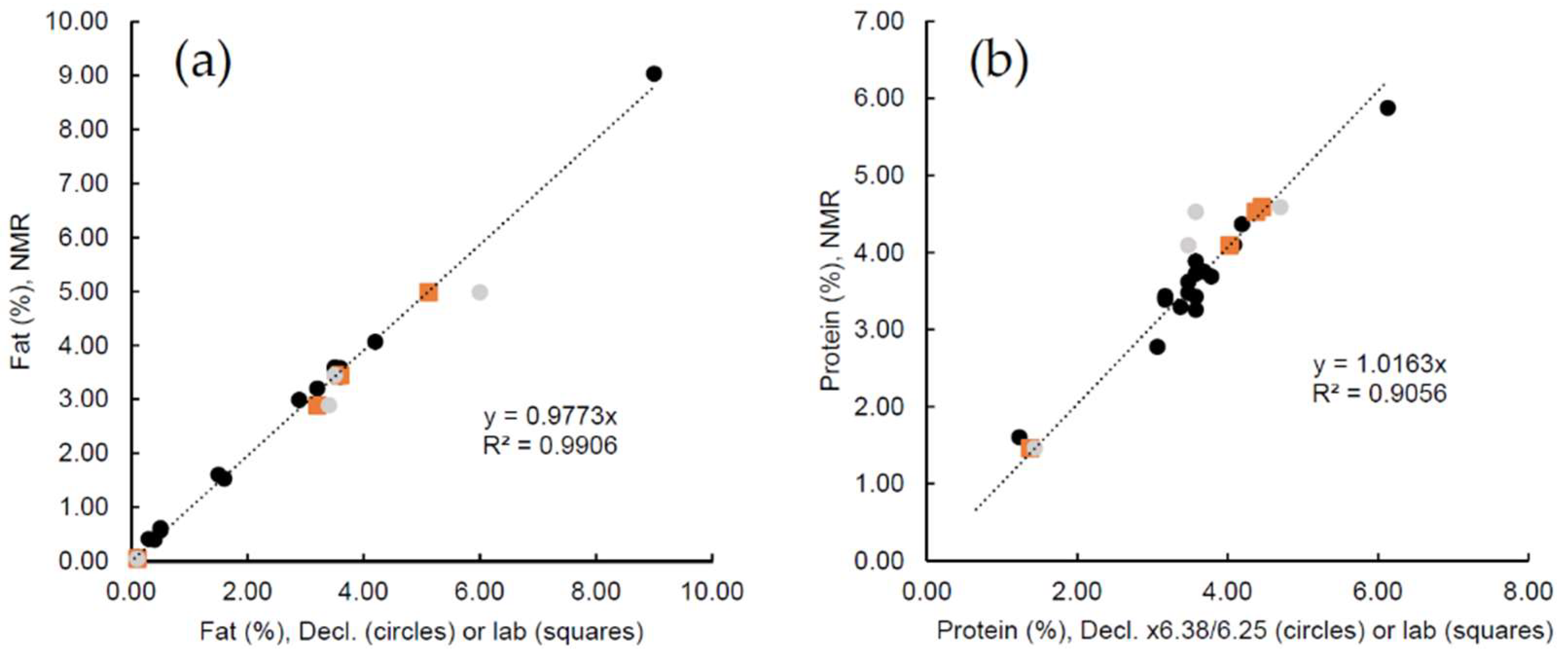
2.2. Supermarket Milk Products
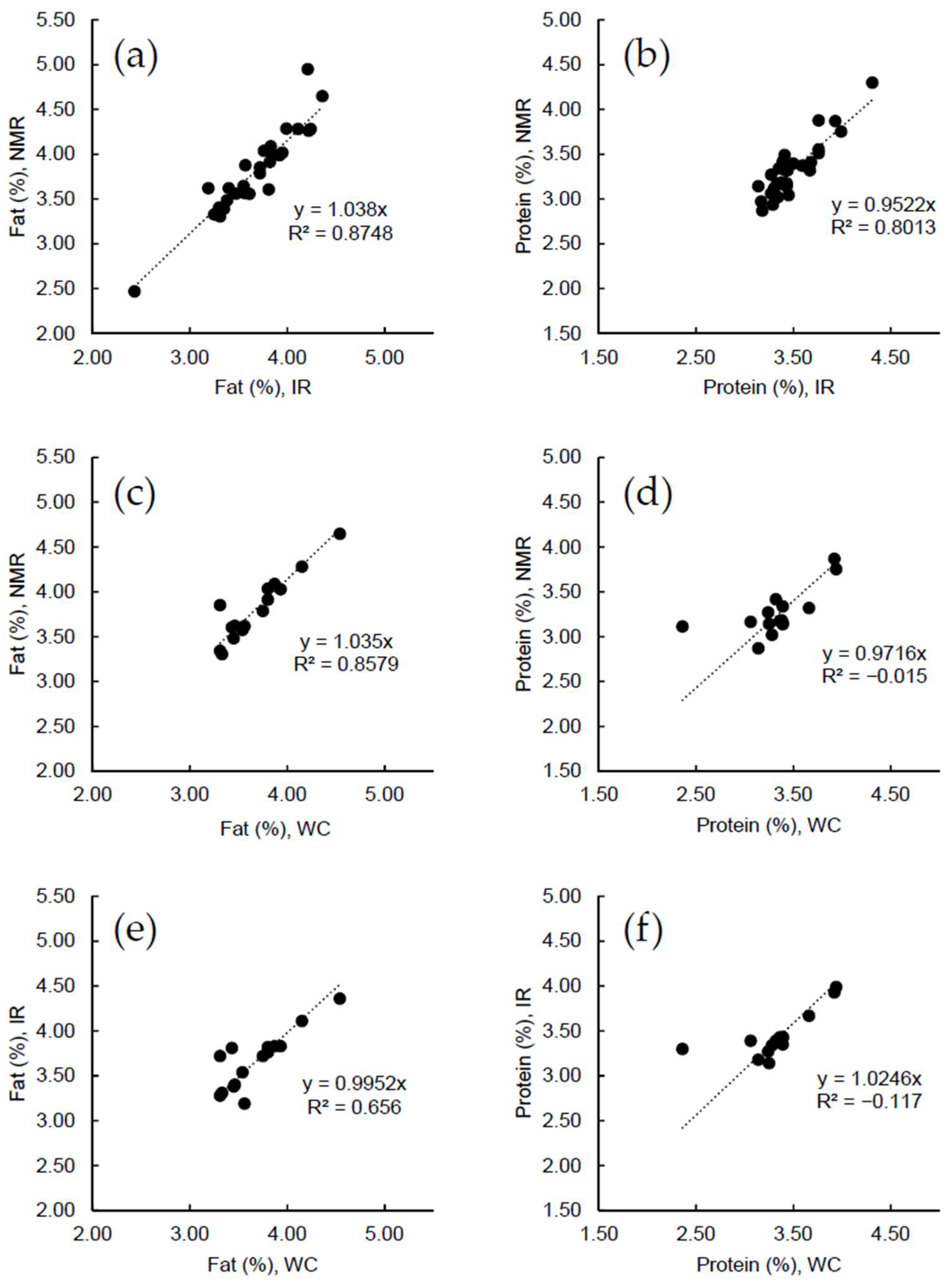
2.3. Raw Milk Samples
2.4. Precision
3. Materials and Methods
3.1. NMR Instrument and Pulse Sequences
3.2. Contrast Solution
3.3. Conduction of Measurements
3.4. Signal Processing
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Džidić, A.; Zamberlin, Š.; Antunac, N.; Šalamon, D. Review on the advances in dairy milk chemistry. J. Cent. Eur. Agric. 2021, 22, 497–509. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, W. Recent developments on rapid detection of main constituents in milk: A review. Crit. Rev. Food Sci. Nutr. 2020, 61, 312–324. [Google Scholar] [CrossRef]
- Kala, R.; Samková, E.; Hanuš, O.; Pecová, L.; Sekmokas, K.; Riaukienė, D. Milk protein analysis: An overview of the methods–Development and application. Acta Univ. Agric. Silvic. Mendel. Brun. 2019, 67, 345–359. [Google Scholar] [CrossRef]
- Mariotti, F.; Tomé, D.; Mirand, P.P. Converting nitrogen into protein--beyond 6.25 and Jones’ factors. Crit. Rev. Food Sci. Nutr. 2008, 48, 177–184. [Google Scholar] [CrossRef]
- Blümich, B. Low-field and benchtop NMR. J. Magn. Reson. 2019, 306, 27–35. [Google Scholar] [CrossRef]
- Sørensen, M.K.; Jensen, O.; Bakharev, O.N.; Nyord, T.; Nielsen, N.C. NPK NMR Sensor: Online Monitoring of Nitrogen, Phosphorus, and Potassium in Animal Slurry. Anal. Chem. 2015, 87, 6446–6450. [Google Scholar] [CrossRef]
- Jensen, O.N.; Beyer, M.; Sørensen, M.K.; Kreimeyer, M.; Nielsen, N.C. Fast and Accurate Quantification of Nitrogen and Phosphorus Constituents in Animal Slurries Using NMR Sensor Technology. ACS Omega 2021, 6, 17335–17341. [Google Scholar] [CrossRef]
- Sørensen, M.K.; Beyer, M.; Jensen, O.N.; Nielsen, N.C. Quantification of protein and phosphorus in livestock feed using mobile NMR sensor technology. Biosyst. Eng. 2021; submitted (under review). [Google Scholar]
- Sørensen, M.K.; Vinding, M.S.; Bakharev, O.N.; Nesgaard, T.; Jensen, O.; Nielsen, N.C. NMR sensor for onboard ship detection of catalytic fines in marine fuel oils. Anal. Chem. 2014, 86, 7205–7208. [Google Scholar] [CrossRef]
- Sørensen, M.K.; Bakharev, O.N.; Jensen, O.; Nielsen, N.C. Natural abundant 17O NMR in a 1.5-T Halbach magnet. Magn. Reson. Chem. 2016, 54, 510–512. [Google Scholar] [CrossRef]
- Sørensen, M.K.; Balsgart, N.M.; Jensen, O.; Nielsen, N.C.; Vosegaard, T. Fast Wide-Line Solid-State NMR on a Low-Cost Benchtop Spectrometer. ChemPhysChem 2018, 19, 2985–2988. [Google Scholar] [CrossRef]
- De Leõn-Rodríguez, L.M.; Martins, A.F.; Pinho, M.C.; Rofsky, N.M.; Sherry, A.D. Basic MR Relaxation Mechanisms & Contrast Agent Design. J. Magn. Reson. Imaging 2015, 42, 545. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2018, 119, 957–1057. [Google Scholar] [CrossRef]
- Wickramasinghe, N.P.; Kotecha, M.; Samoson, A.; Past, J.; Ishii, Y. Sensitivity Enhancement in 13C Solid-State NMR of Protein Microcrystals by Use of Paramagnetic Metal Ions for Optimizing 1H T1 Relaxation. J. Magn. Reson. 2007, 184, 350. [Google Scholar] [CrossRef][Green Version]
- Yamamoto, K.; Vivekanandan, S.; Ramamoorthy, A. Fast NMR Data Acquisition From Bicelles Containing a Membrane-Associated Peptide at Natural-Abundance. J. Phys. Chem. B 2011, 115, 12448. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Larsen, F.H.; Jakobsen, H.J.; Ellis, P.D.; Nielsen, N.C. Sensitivity-enhanced quadrupolar-echo NMR of half-integer quadrupolar nuclei. Magnitudes and relative orientation of chemical shielding and quadrupolar coupling tensors. J. Phys. Chem. A 1997, 101, 8597–8606. [Google Scholar] [CrossRef]
- Nascimento, P.A.M.; Barsanelli, P.L.; Rebellato, A.P.; Pallone, J.A.L.; Pereira, F.M.V. Time-Domain Nuclear Magnetic Resonance (TD-NMR) and Chemometrics for Determination of Fat Content in Commercial Products of Milk Powder. J. AOAC Int. 2017, 100, 330–334. [Google Scholar] [CrossRef]
- Todt, H.; Guthausen, G.; Burk, W.; Schmalbein, D.; Kamlowski, A. Water/moisture and fat analysis by time-domain NMR. Food Chem. 2006, 96, 436–440. [Google Scholar] [CrossRef]
- Mariette, F. NMR Relaxation of Dairy Products. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer: Dordrecht, The Netherlands, 2008; pp. 1697–1701. [Google Scholar] [CrossRef]
- Hester, R.E.; Quine, D.E.C. Quantitative analysis of food products by pulsed nuclear magnetic resonance: II. Simultaneous analysis of water and fat in milk powder and cottage cheese. J. Dairy Res. 1977, 44, 125–130. [Google Scholar] [CrossRef]
- Synek, V. Evaluation of the standard deviation from duplicate results. Accredit. Qual. Assur. 2008, 13, 335–337. [Google Scholar] [CrossRef]
- MATLAB, R2019a; The Mathworks Inc.: Natick, MA, USA, 2019.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR vs. IR | NMR vs. WC | IR vs. WC | |
|---|---|---|---|
| Fat (%) | 0.15 | 0.14 | 0.19 |
| Protein (%) | 0.13 | 0.27 | 0.25 |
| Fat (%) | Protein (%) | |||
|---|---|---|---|---|
| Repeats | Duplicates | Repeats | Duplicates | |
| Homogenized calibration samples | 0.02 | 0.01 | 0.12 | 0.07 |
| Unhomogenized calibration samples | 0.03 | 0.05 | 0.09 | 0.05 |
| Homogenized supermarket milk | 0.04 | 0.03 | 0.10 | 0.07 |
| Unhomogenized supermarket milk | 0.03 | 0.08 | 0.19 | 0.15 |
| Raw milk (unhomogenized) | 0.02 | 0.21 | 0.09 | 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sørensen, M.K.; Balsgart, N.M.; Beyer, M.; Jensen, O.N.; Nielsen, N.C. On-Site Measurement of Fat and Protein Contents in Milk Using Mobile NMR Technology. Molecules 2022, 27, 583. https://doi.org/10.3390/molecules27030583
Sørensen MK, Balsgart NM, Beyer M, Jensen ON, Nielsen NC. On-Site Measurement of Fat and Protein Contents in Milk Using Mobile NMR Technology. Molecules. 2022; 27(3):583. https://doi.org/10.3390/molecules27030583
Chicago/Turabian StyleSørensen, Morten K., Nicholas M. Balsgart, Michael Beyer, Ole N. Jensen, and Niels Chr. Nielsen. 2022. "On-Site Measurement of Fat and Protein Contents in Milk Using Mobile NMR Technology" Molecules 27, no. 3: 583. https://doi.org/10.3390/molecules27030583
APA StyleSørensen, M. K., Balsgart, N. M., Beyer, M., Jensen, O. N., & Nielsen, N. C. (2022). On-Site Measurement of Fat and Protein Contents in Milk Using Mobile NMR Technology. Molecules, 27(3), 583. https://doi.org/10.3390/molecules27030583