
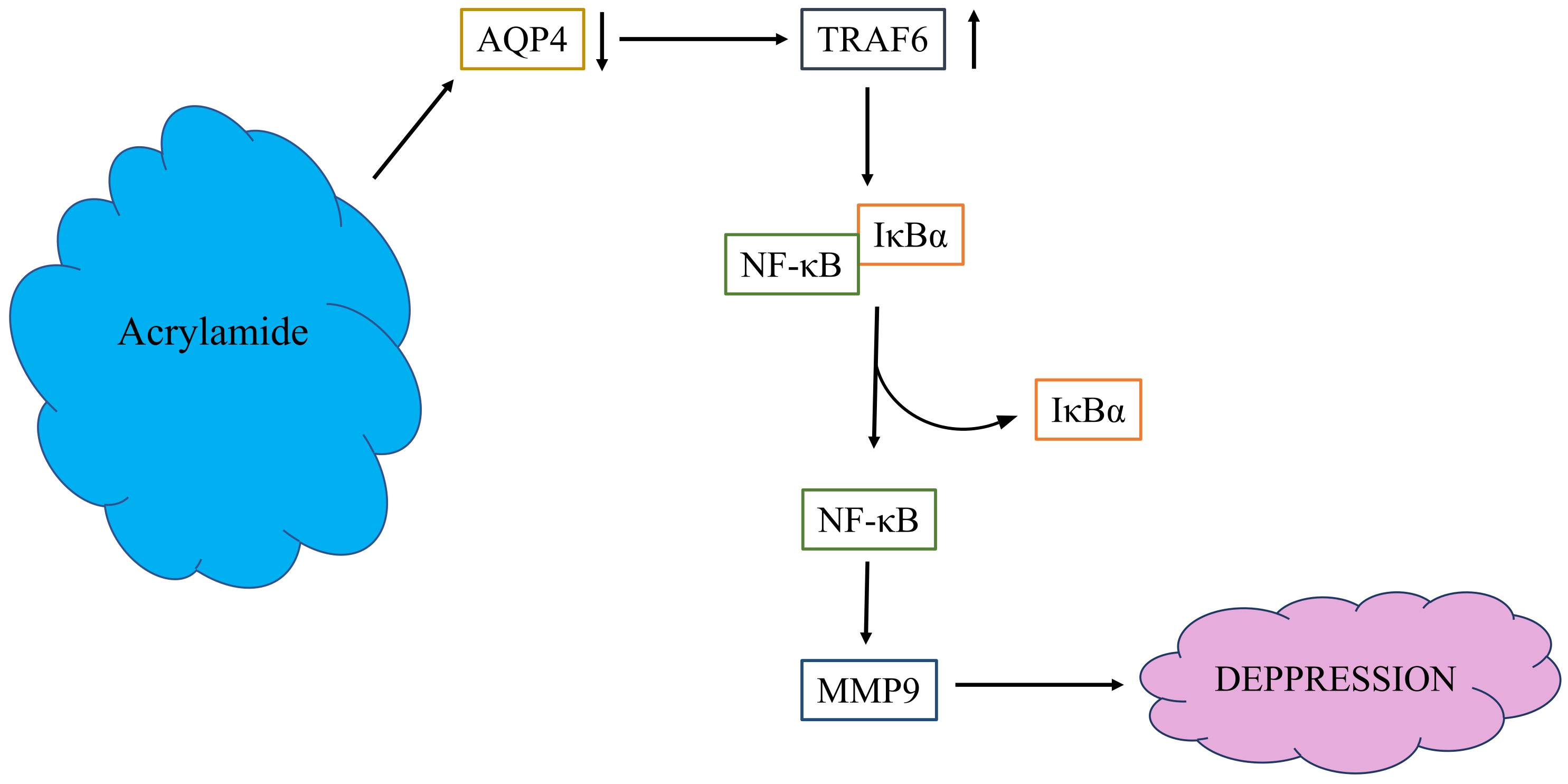
AQP4 Attenuated TRAF6/NFκB Activation in Acrylamide-Induced Neurotoxicity



Abstract
:1. Introduction
2. Results
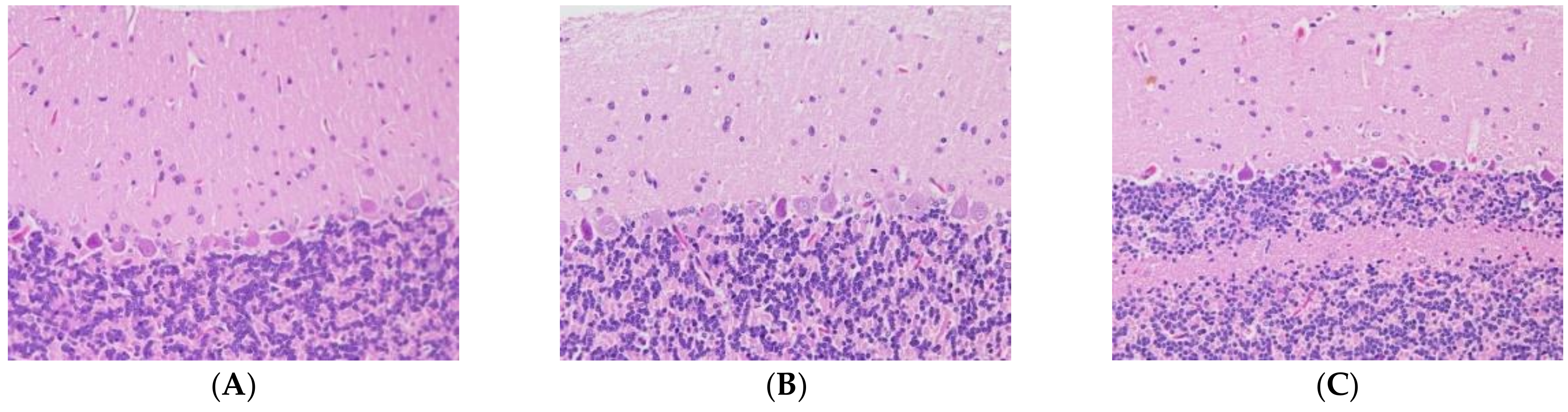
2.1. Acrylamide-Induced Neurotoxicity Symptoms in Rats
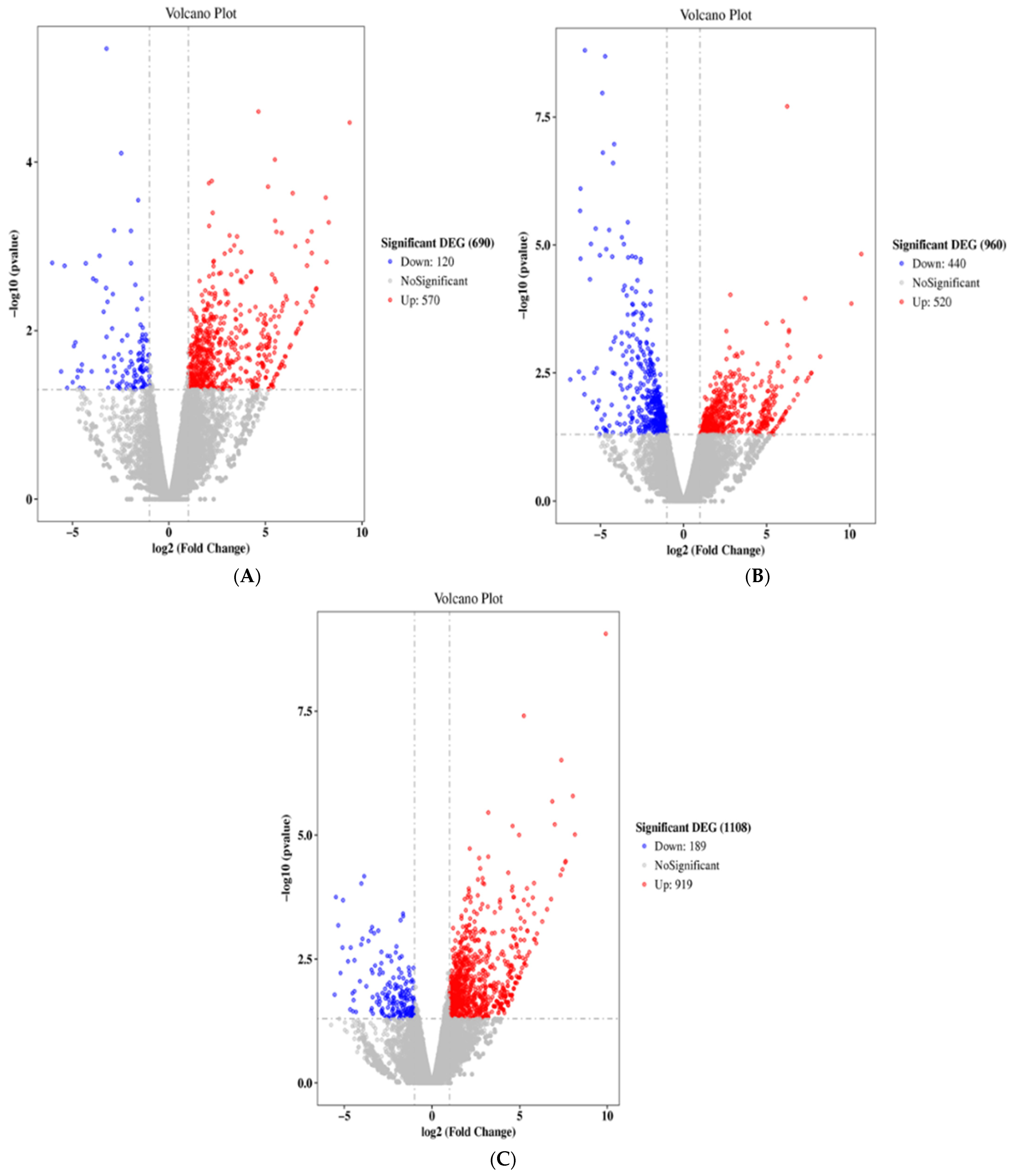
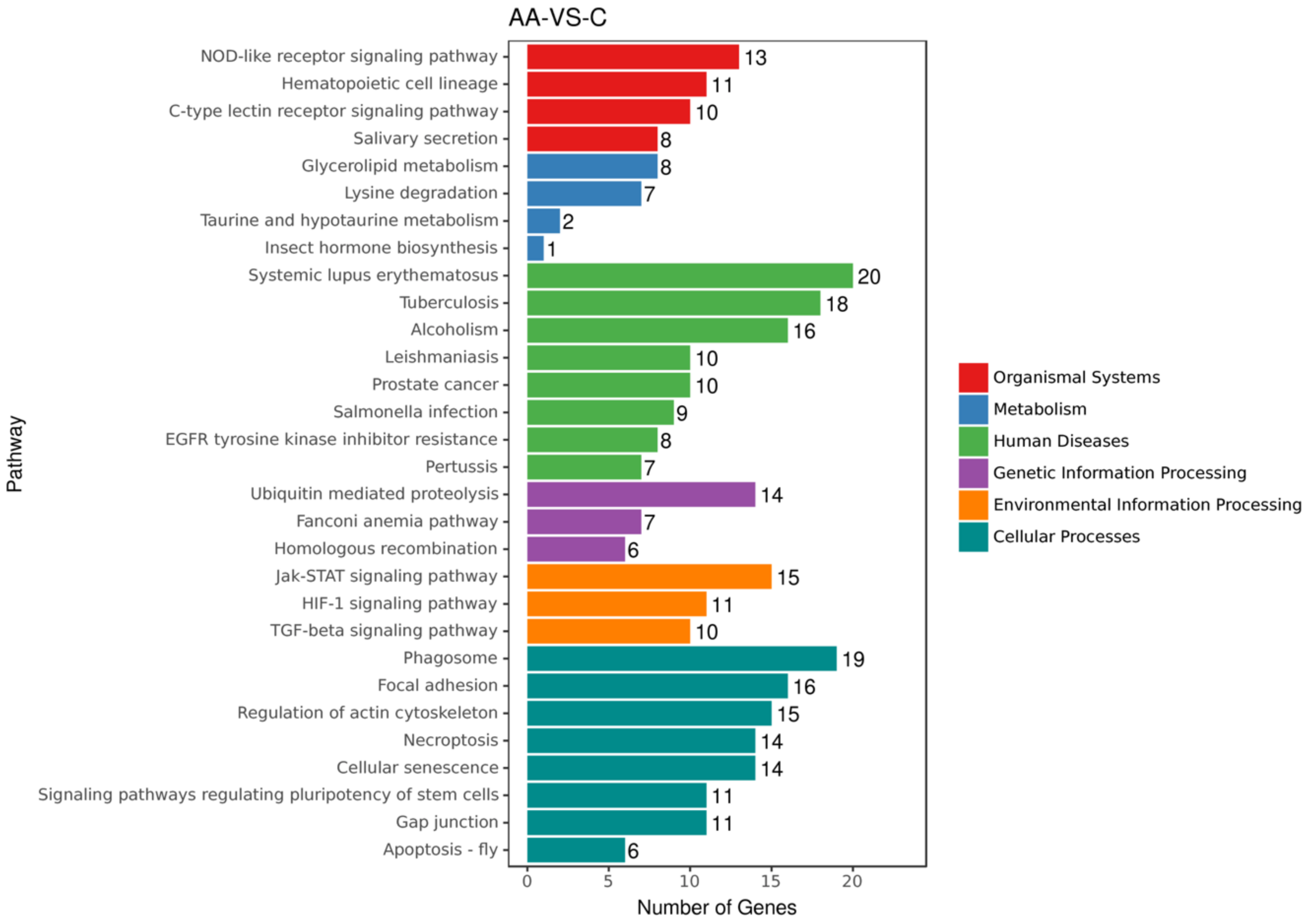
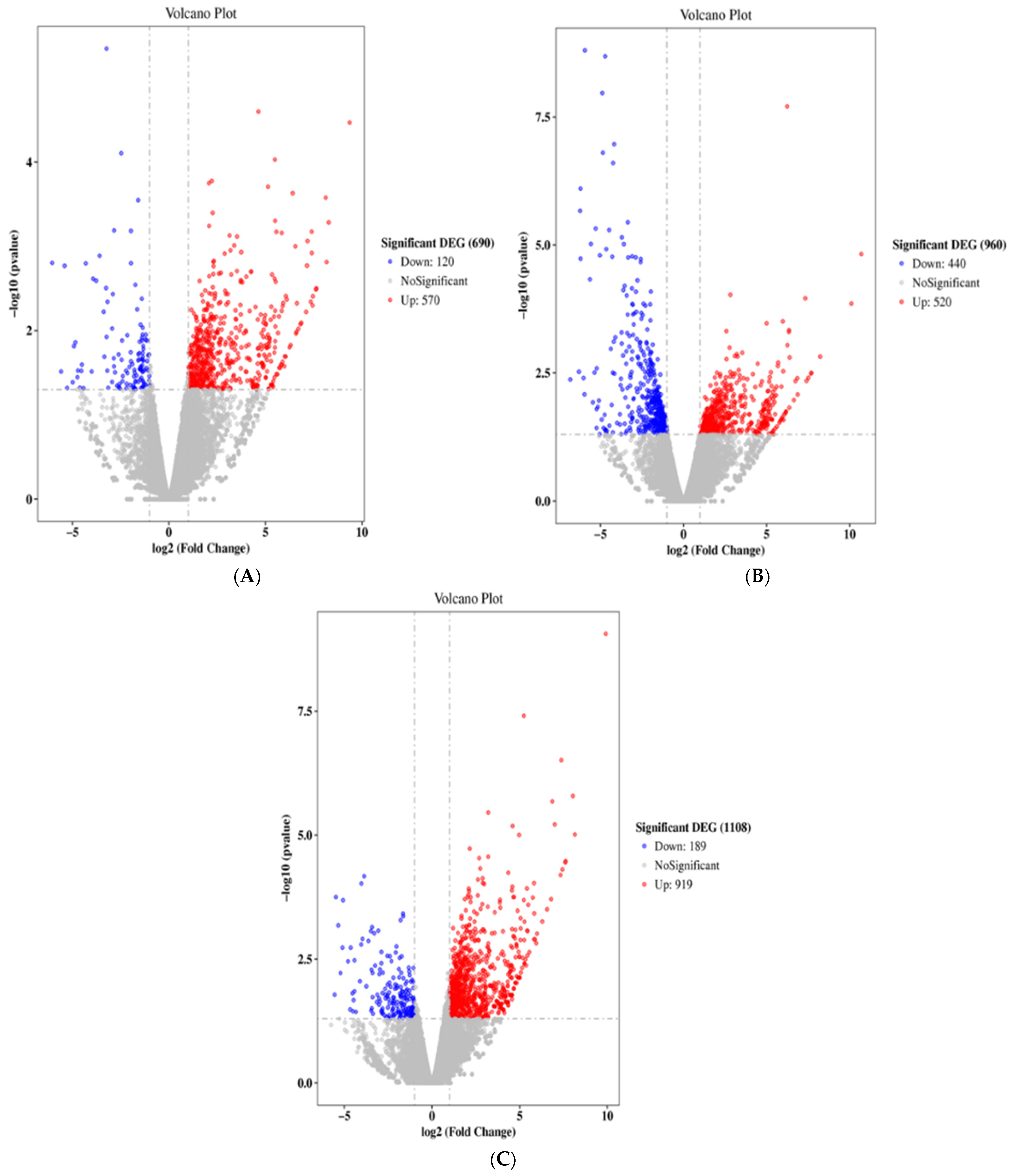
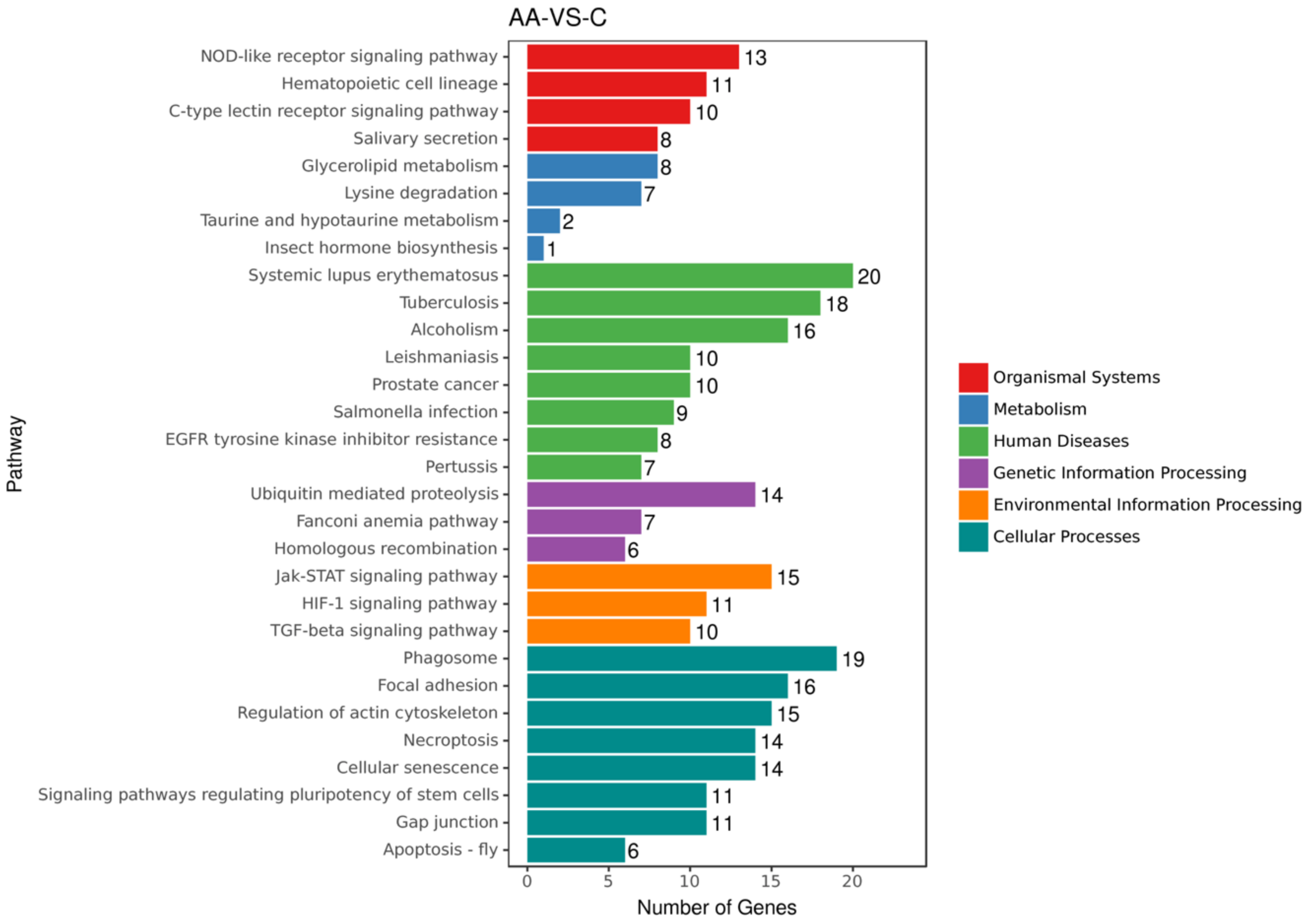
2.2. ACR Alters Genes Expression in Rats
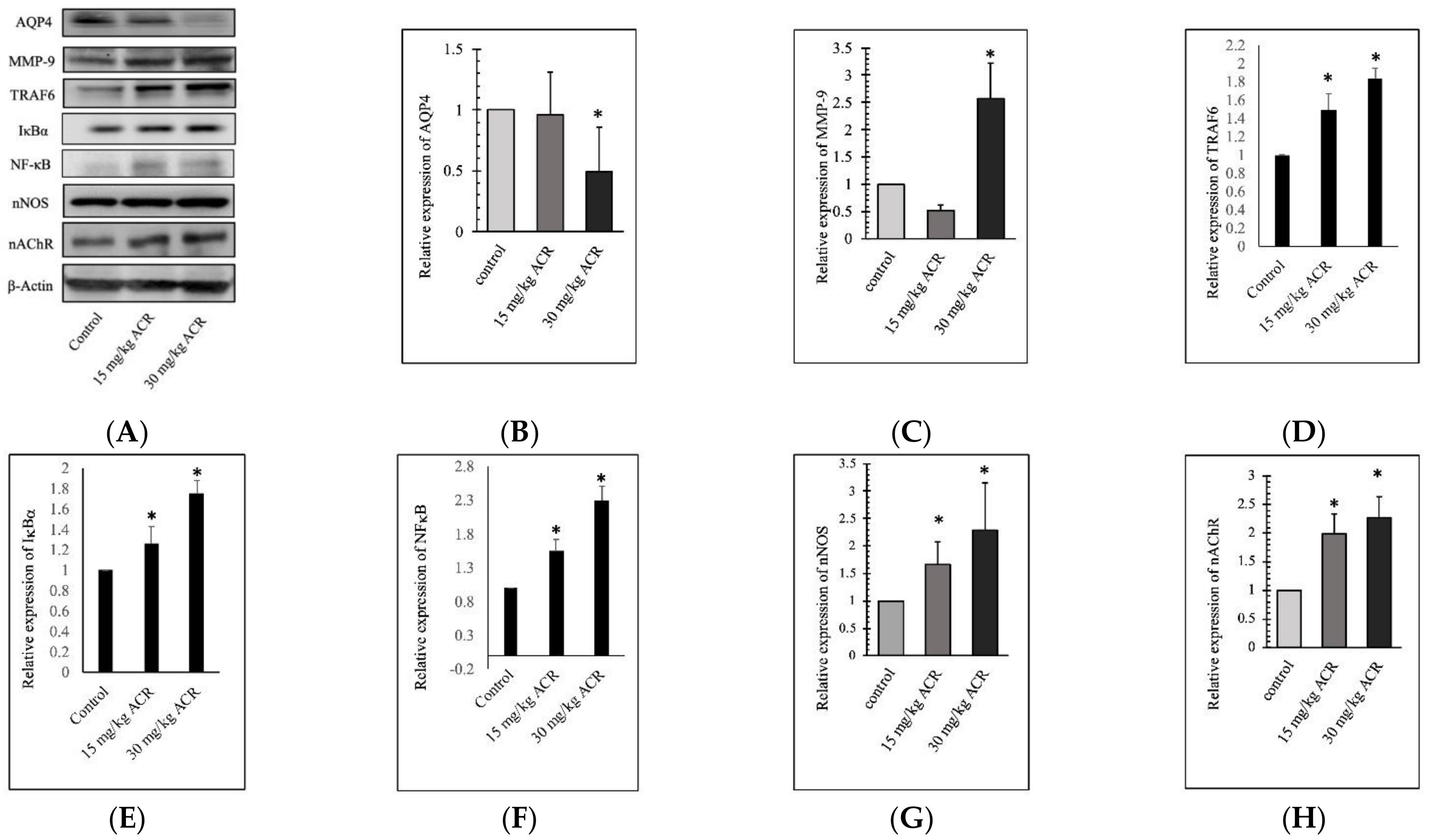
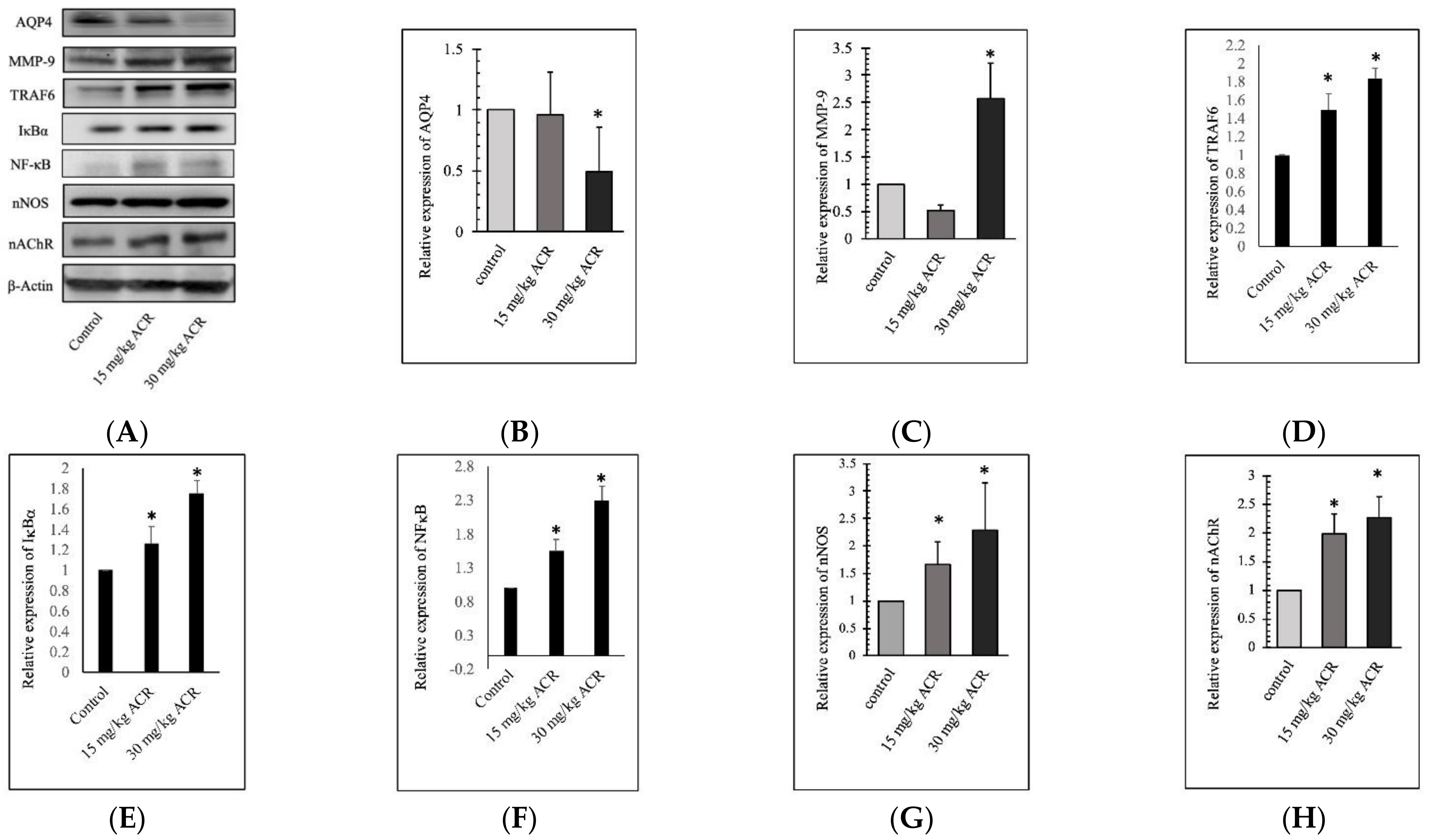
2.3. ACR Induces MMP-9 and AQP4 Expression in the Brain Tissue
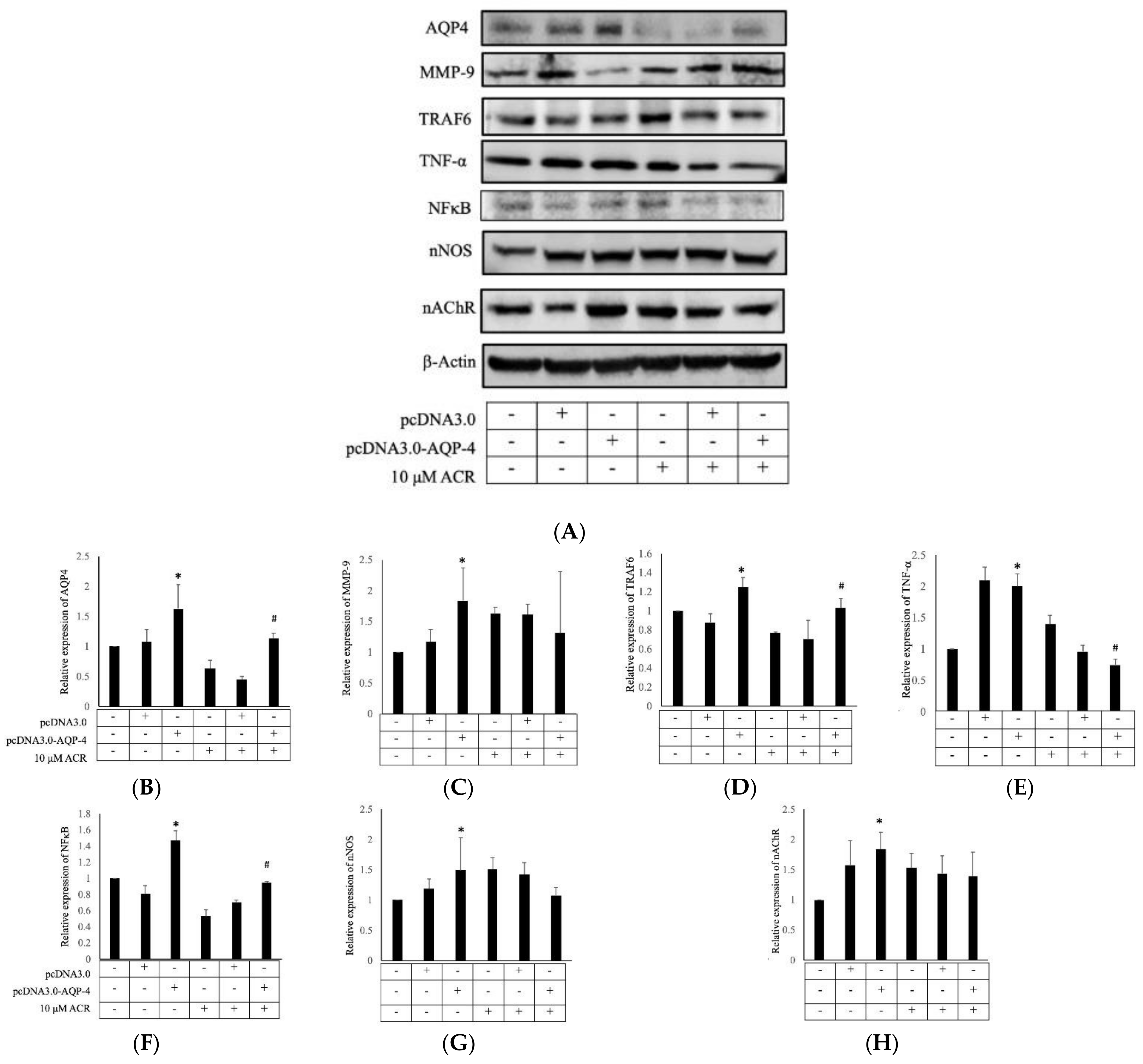
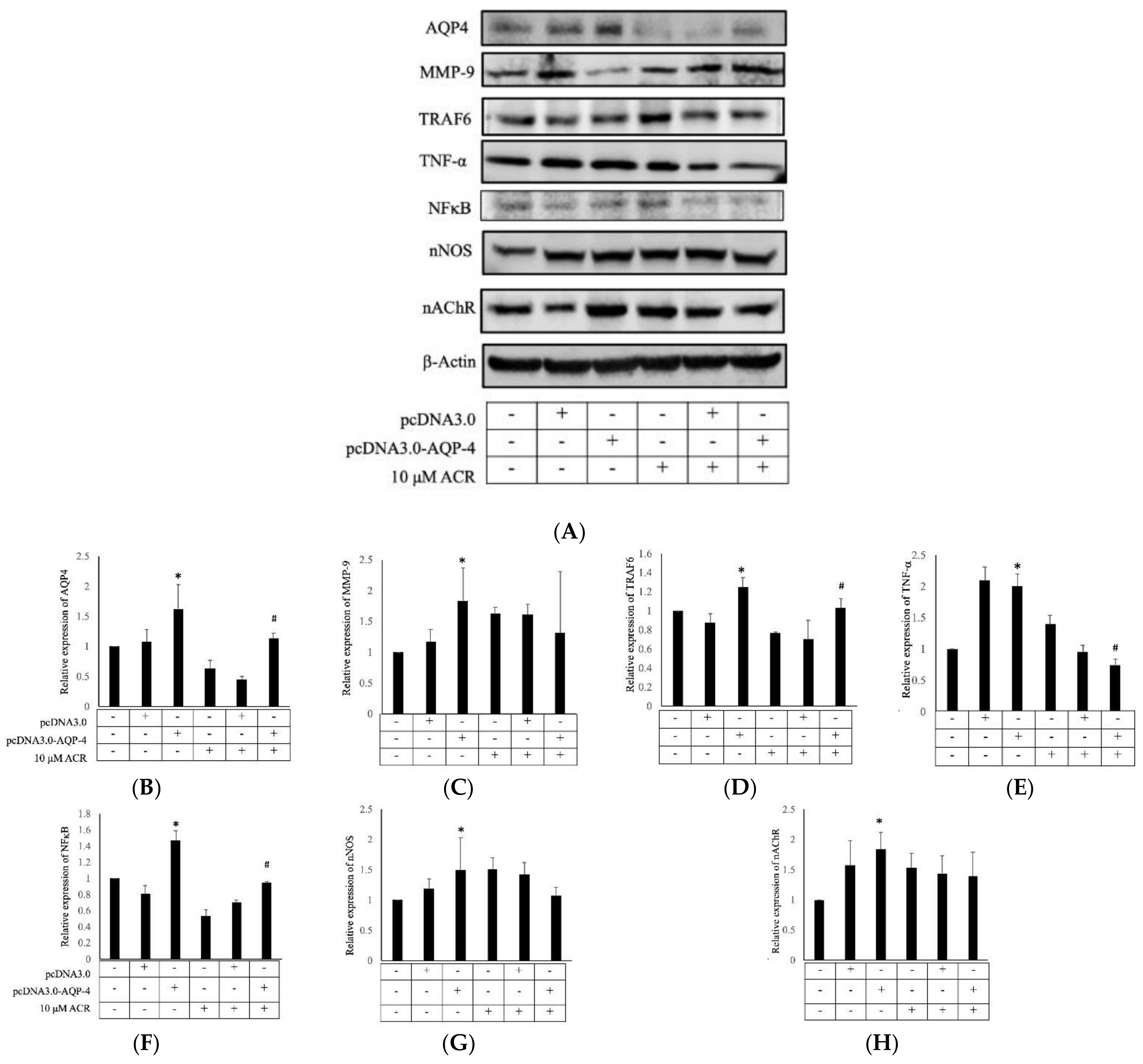
2.4. AQP4 Regulates ACR-Induced MMP-9 Expression
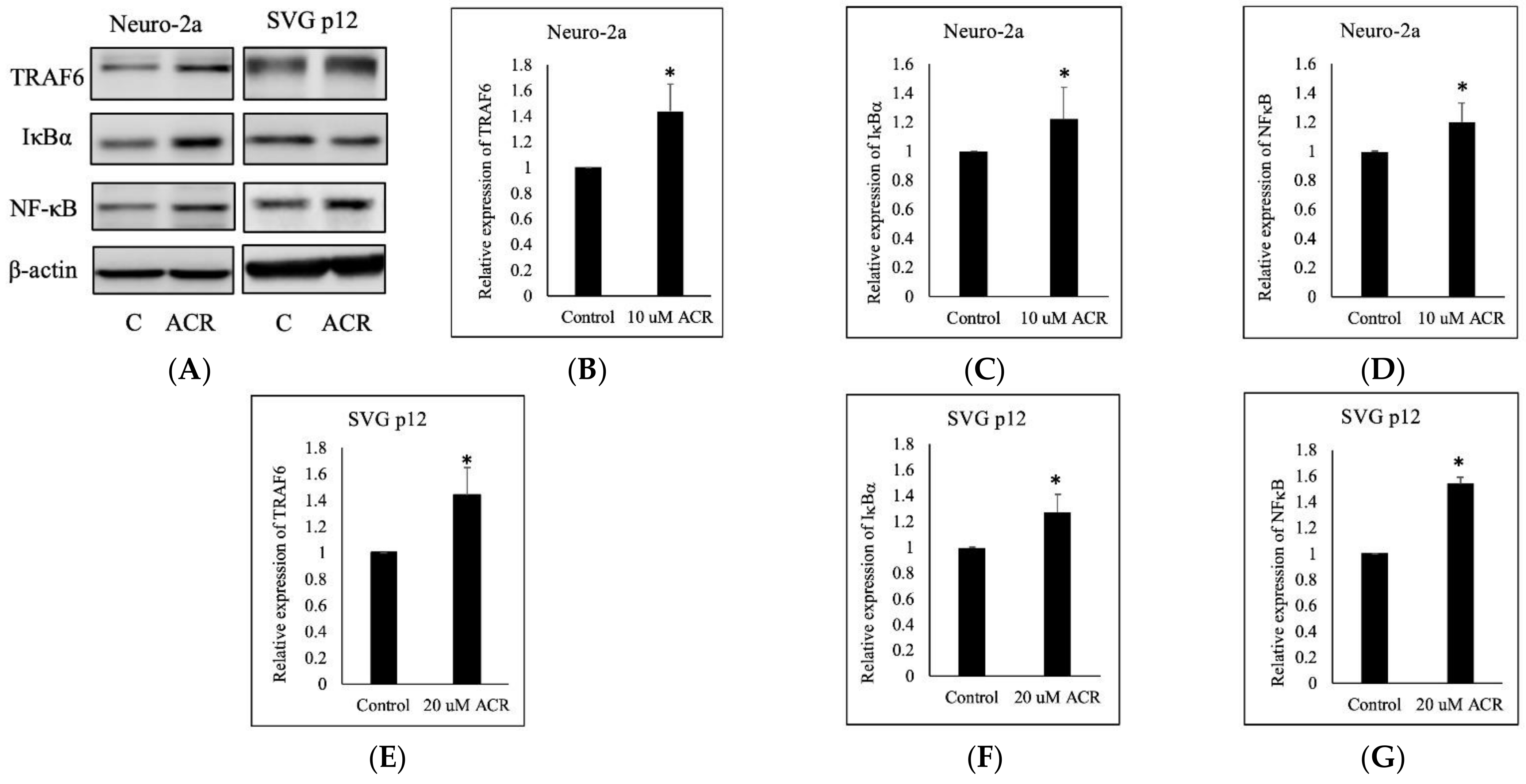
2.5. ACR-Induced TRAF6, IκBα, and NFκB Expression
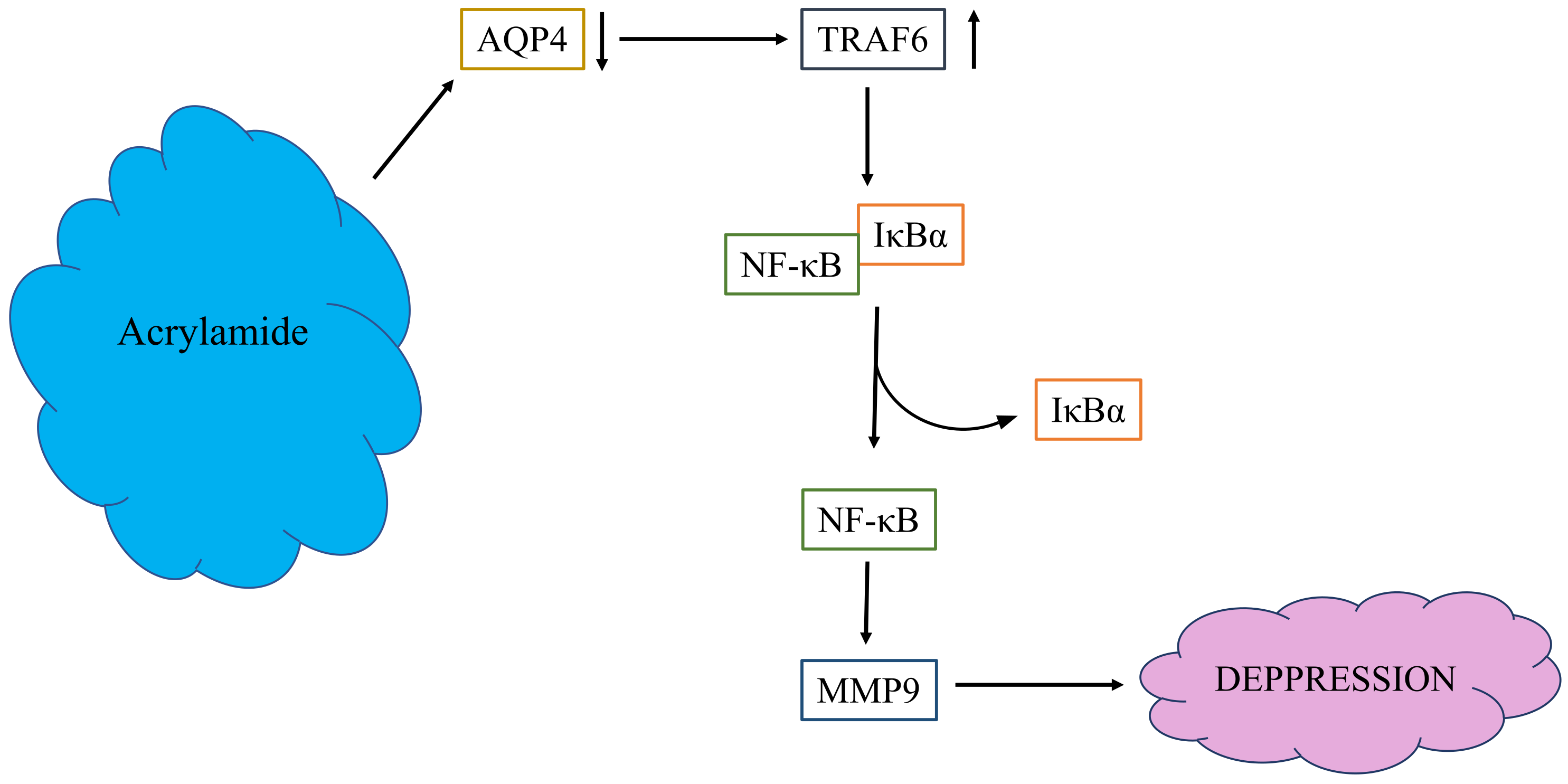
3. Discussion
4. Materials and Methods
4.1. ACR Subacute Oral Toxicity in SD Rats
4.2. Blood Gene Expression Difference by ACR in Rats
4.3. Protein Extraction and Western Blot
4.4. Cell Culture and ACR Dosage Selection
4.5. AQP4 Recombinant Vector Construction and Cell Transient Transfection
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- National Toxicology Program. Toxicology and carcinogenesis studies of acrylamide (CASRN 79-06-1) in F344/N rats and B6C3F1 mice (feed and drinking water studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2012, 575, 1–234. [Google Scholar]
- Rifai, L.; Saleh, F.A. A Review on Acrylamide in Food: Occurrence, Toxicity, and Mitigation Strategies. Int. J. Toxicol. 2020, 39, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.D.; Ou, C.C.; Hsiao, S.H.; Chang, C.H.; Tsai, F.J.; Liao, J.W.; Chen, Y.T. Effects of acrylamide-induced vasorelaxation and neuromuscular blockage: A rodent study. Toxics 2021, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Abd AI Haleem, E.N.; Hasan, W.Y.S.; Arafa, H.M.M. Therapeutic effects of thymoquinone or capsaicin on acrylamide-induced reproductive toxicity in rats mediated by their effect on oxidative stress, inflammation, and tight junction integrity. Drug Chem. Toxicol. 2021, 7, 1–13. [Google Scholar] [CrossRef]
- De Conti, A.; Tryndyak, V.; Von Tungeln, L.S.; Churchwell, M.I.; Beland, F.A.; Antunes, A.M.M.; Pogribny, I.P. Genotoxic and epigenotoxic alterations in the lung and liver of mice induced by acrylamide: A 28 Day Drinking Water Study. Chem. Res. Toxicol. 2019, 32, 869–877. [Google Scholar] [CrossRef]
- IARC. Acrylamide. Some Industrial Chemicals. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1994; Volume 60, pp. 389–433. [Google Scholar]
- Ngo-Thanh, H.; Thuy, T.D.; Suzue, K.; Kamitani, W.; Yokoo, H.; Isoda, K.; Shimokawa, C.; Hisaeda, H.; Imai, T. Long-term acrylamide exposure exacerbates brain and lung pathology in a mouse malaria model. Food Chem. Toxicol. 2021, 151, 112132. [Google Scholar] [CrossRef]
- He, F.S.; Zhang, S.L.; Wang, H.L.; Li, G.; Zhang, Z.M.; Li, F.L.; Dong, X.M.; Hu, F.R. Neurological and electroneuromyographic assessment of the adverse effects of acrylamide on occupationally exposed workers. Scand. J. Work Environ. Health. 1989, 15, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sun, J.; Zhang, D. Association between acrylamide hemoglobin adduct levels and depression symptoms in US adults: NHANES 2013–2016. J. Agric. Food Chem. 2021, 69, 13762–13771. [Google Scholar] [CrossRef]
- Battle, D.E. Diagnostic and Statistical Manual of Mental Disorders (DSM). Codas 2013, 25, 191–192. [Google Scholar]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Ignácio, Z.M.; da Silva, R.S.; Plissari, M.E.; Quevedo, J.; Reus, G.Z. Physical Exercise and Neuroinflammation in Major Depressive Disorder. Mol. Neurobiol. 2019, 56, 8323–8335. [Google Scholar] [CrossRef]
- Kim, Y.K.; Won, E. The influence of stress on neuroinflammation and alterations in brain structure and function in major depressive disorder. Behav. Brain Res. 2017, 329, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139, 91–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kooij, M.A.; Fantin, M.; Rejmak, E.; Grosse, J.; Zanoletti, O.; Fournier, C.; Ganguly, K.; Kalita, K.; Kaczmarek, L.; Sandi, C. Role for MMP-9 in stress-induced downregulation of nectin-3 in hippocampal CA1 and associated behavioural alterations. Nat. Commun. 2014, 5, 4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genel, O.; Pariante, C.M.; Borsini, A. The role of AQP4 in the pathogenesis of depression, and possible related mechanisms. Brain Behav. Immun. 2021, 98, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Watson, S.J.; Bunney, W., Jr.; Myers, R.M.; Schatzberg, A.; Barchas, J.; Akil, H.; Thompson, R.C. Evidence for alterations of the glial syncytial function in major depressive disorder. J. Psychiatr. Res. 2016, 72, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Yan, J.; Chen, G.; Hu, G.; Zhou, X.; Zeng, X. AQP4-knockout alleviates the lipopolysaccharide-induced inflammatory response in astrocytes via SPHK1/MAPK/AKT signaling. Int. J. Mol. Med. 2018, 42, 1716–1722. [Google Scholar] [CrossRef] [Green Version]
- Vilcinskas, A. Matrix metalloproteinases and their inhibitors-pleiotropic functions in insect immunity and metamorphosis. FEBS J. 2021. [Google Scholar] [CrossRef]
- Hui, B.; Zhang, L.; Zhou, Q.; Hui, L. Pristimerin inhibits LPS-triggered neurotoxicity in BV-2 Microglia cells through modulating IRAK1/TRAF6/TAK1-Mediated NF-κB and AP-1 signaling pathways in vitro. Neurotox. Res. 2018, 33, 268–283. [Google Scholar] [CrossRef]
- Pan, X.; Guo, X.; Xiong, F.; Cheng, G.; Lu, Q.; Yan, H. Acrylamide increases dopamine levels by affecting dopamine transport and metabolism related genes in the striatal dopaminergic system. Toxicol. Lett. 2015, 236, 60–68. [Google Scholar] [CrossRef]
- Zhang, L.; Hara, S.; Ichinose, H.; Nagashima, D.; Morita, K.; Sakurai, T.; Ichihara, S.; Ichihara, G. Exposure to acrylamide decreases noradrenergic axons in rat brain. Neurotoxicology 2020, 78, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, S.; Lundberg, A.K.; Jonasson, L. Overexpression of MMP-9 and its inhibitors in blood mononuclear cells after myocardial infarction - is it associated with depressive symptomatology? PLoS ONE 2014, 9, e105572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibasaki, C.; Takebayashi, M.; Itagaki, K.; Abe, H.; Kajitani, N.; Okada-Tsuchioka, M.; Yamawaki, S. Altered serum levels of matrix metalloproteinase-2, -9 in response to electroconvulsive therapy for mood disorders. Int. J. Neuropsychopharmacol. 2016, 19, pyw019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Mihaylova, I.; Kubera, M.; Ringel, K. Activation of cell-mediated immunity in depression: Association with inflammation, melancholia, clinical staging and the fatigue and somatic symptom cluster of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 36, 169–175. [Google Scholar] [CrossRef]
- Bobińska, K.; Szemraj, J.; Czarny, P.; Gałecki, P. Role of MMP-2, MMP-7, MMP-9 and TIMP-2 in the development of recurrent depressive disorder. J. Affect. Disord. 2016, 205, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblat, J.D.; Cha, D.S.; Mansur, R.B.; McIntyre, R.S. Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 23–34. [Google Scholar] [CrossRef]
- Bobińska, K.; Szemraj, J.; Czarny, P.; Gałecki, P. Expression and Activity of metalloproteinases in depression. Med. Sci. Monit. 2016, 22, 1334–1341. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, A.; Ferrari, C.; Turner, L.; Mariani, N.; Enache, D.; Hastings, C.; Kose, M.; Lombardo, G.; McLaughlin, A.P.; Nettis, M.A.; et al. Whole-blood expression of infammasome- and glucocorticoid-related mRNAs correctly separates treatment-resistant depressed patients from drug-free and responsive patients in the BIODEP study. Transl. Psychiatry 2020, 10, 232. [Google Scholar] [CrossRef]
- Wallensten, J.; Nager, A.; Åsberg, M.; Borg, K.; Beser, A.; Wilczek, A.; Mobarrez, F. Leakage of astrocyte-derived extracellular vesicles in stress-induced exhaustion disorder: A cross-sectional study. Sci. Rep. 2021, 11, 2009. [Google Scholar] [CrossRef]
- Mikita, J.; Dubourdieu-Cassagno, N.; Deloire, M.S.; Vekris, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.H.; Franconi, J.M.; Boiziau, C.; et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. 2011, 17, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-κB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Muto, T.; Kleppe, M.; Bolanos, L.C.; Hueneman, K.M.; Walker, C.S.; Sampson, L.; Wellendorf, A.M.; Chetal, K.; Choi, K.; et al. TRAF6 mediates basal activation of NF-κB necessary for hematopoietic stem cell homeostasis. Cell Rep. 2018, 22, 1250–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.; Kim, M.J.; Lee, S.; Chun, E.; Lee, K.Y. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy 2018, 14, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, S.; Wang, S.; Liu, Q.; Xu, S. Cadmium-induced oxidative stress promotes apoptosis and necrosis through the regulation of the miR-216a-PI3K/AKT axis in common carp lymphocytes and antagonized by selenium. Chemosphere 2020, 258, 127341. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.M.; Ma, Y.X.; Shi, J.; Lou, T.Y.; Liu, S.S.; Li, G.Y. Acrylamide neurotoxicity on the cerebrum of weaning rats. Neural. Regen. Res. 2015, 10, 938–943. [Google Scholar]
- Shi, J.; Ma, Y.; Zhang, M.; Ruan, Z.; Liu, J.; Tian, S.; Zhang, D.; He, X.; Li, G. Effect of sub-acute exposure to acrylamide on GABAergic neurons and astrocytes in weaning rat cerebellum. Toxicol. Ind. Health 2012, 28, 10–20. [Google Scholar] [CrossRef]
- Shackelford, C.; Long, G.; Wolf, J.; Okerberg, C.; Herbert, R. Qualitative and quantitative analysis of nonneoplastic lesions in toxicology studies. Toxicol. Pathol. 2002, 30, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Log2FC | p-Value |
|---|---|---|---|
| ENSRNOG00000016043 | AQP4 | N/A | N/A |
| ENSRNOG00000017539 | MMP-9 | 1.65759 | 0.088937 |
| ENSRNOG00000016606 | TRAF6 | 1.227902 | 0.028015 |
| ENSRNOG00000023258 | NFκB | 0.13844 | 0.649575 |
| ENSRNOG00000007390 | IκBα | 0.32284 | 0.411419 |
| ENSRNOG00000003773 | HO-1 | 0.231468 | 0.712061 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, C.-Y.; Chang, C.-H.; Lin, T.-J.; Yi, H.-H.; Tsai, N.-Z.; Chen, Y.-R.; Chen, Y.-T. AQP4 Attenuated TRAF6/NFκB Activation in Acrylamide-Induced Neurotoxicity. Molecules 2022, 27, 1066. https://doi.org/10.3390/molecules27031066
Hung C-Y, Chang C-H, Lin T-J, Yi H-H, Tsai N-Z, Chen Y-R, Chen Y-T. AQP4 Attenuated TRAF6/NFκB Activation in Acrylamide-Induced Neurotoxicity. Molecules. 2022; 27(3):1066. https://doi.org/10.3390/molecules27031066
Chicago/Turabian StyleHung, Chia-Yu, Chih-Han Chang, Tzu-Jung Lin, Hsin-Hui Yi, Nian-Zhen Tsai, Yu-Ru Chen, and Yng-Tay Chen. 2022. "AQP4 Attenuated TRAF6/NFκB Activation in Acrylamide-Induced Neurotoxicity" Molecules 27, no. 3: 1066. https://doi.org/10.3390/molecules27031066
APA StyleHung, C.-Y., Chang, C.-H., Lin, T.-J., Yi, H.-H., Tsai, N.-Z., Chen, Y.-R., & Chen, Y.-T. (2022). AQP4 Attenuated TRAF6/NFκB Activation in Acrylamide-Induced Neurotoxicity. Molecules, 27(3), 1066. https://doi.org/10.3390/molecules27031066