
Antileukemia Activity and Mechanism of Platinum(II)-Based Metal Complexes

 ,
, 
 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
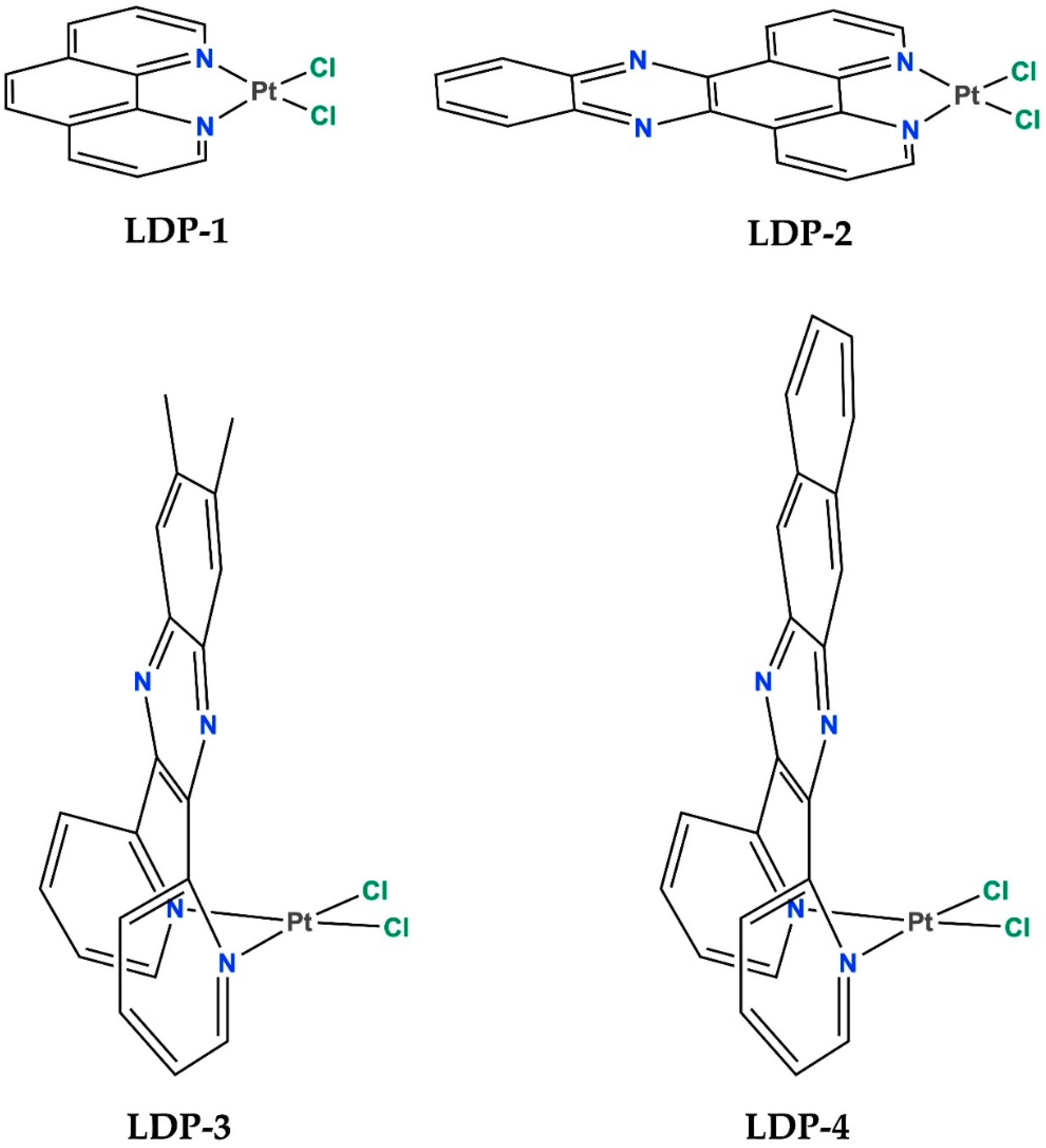
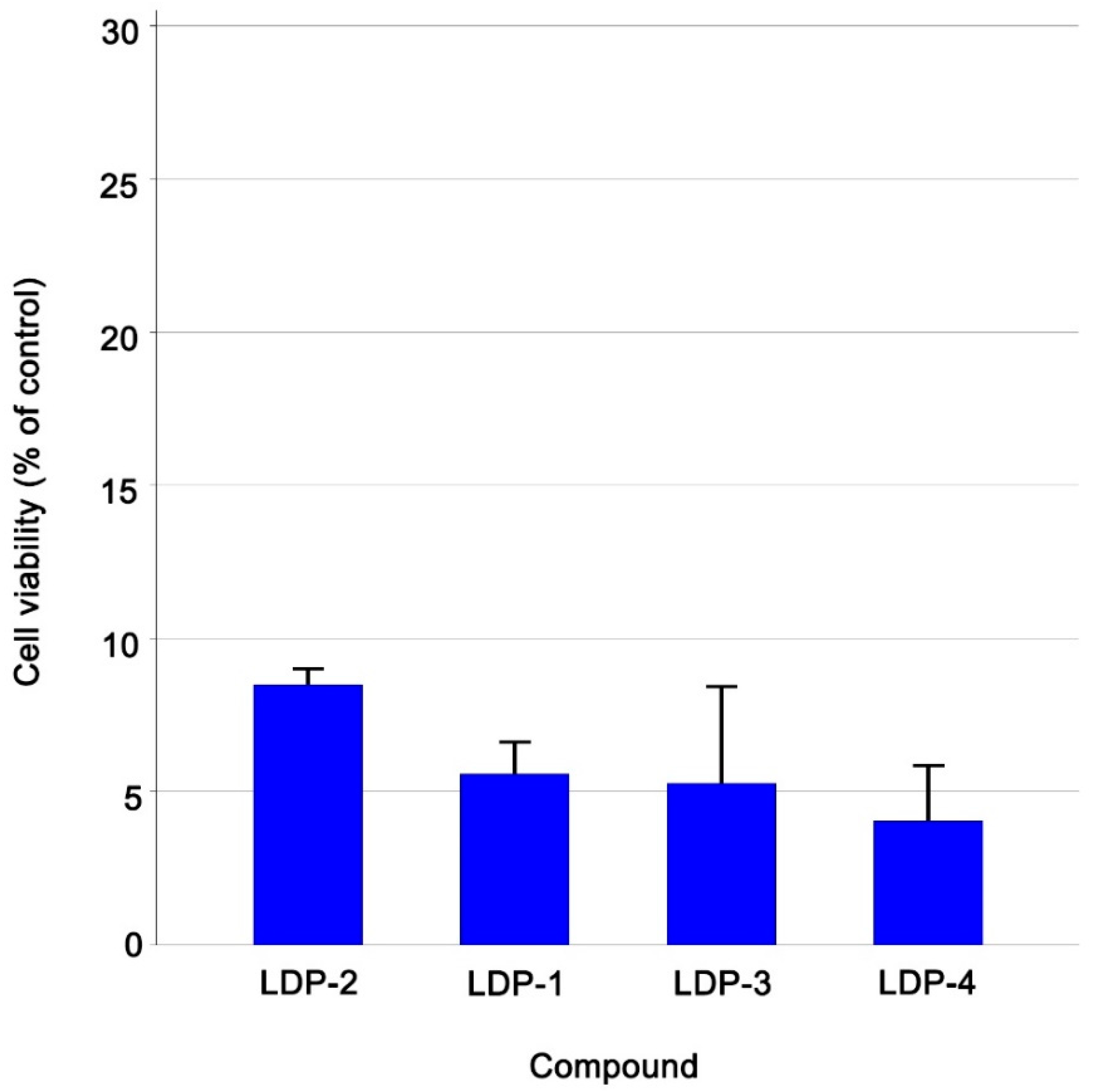
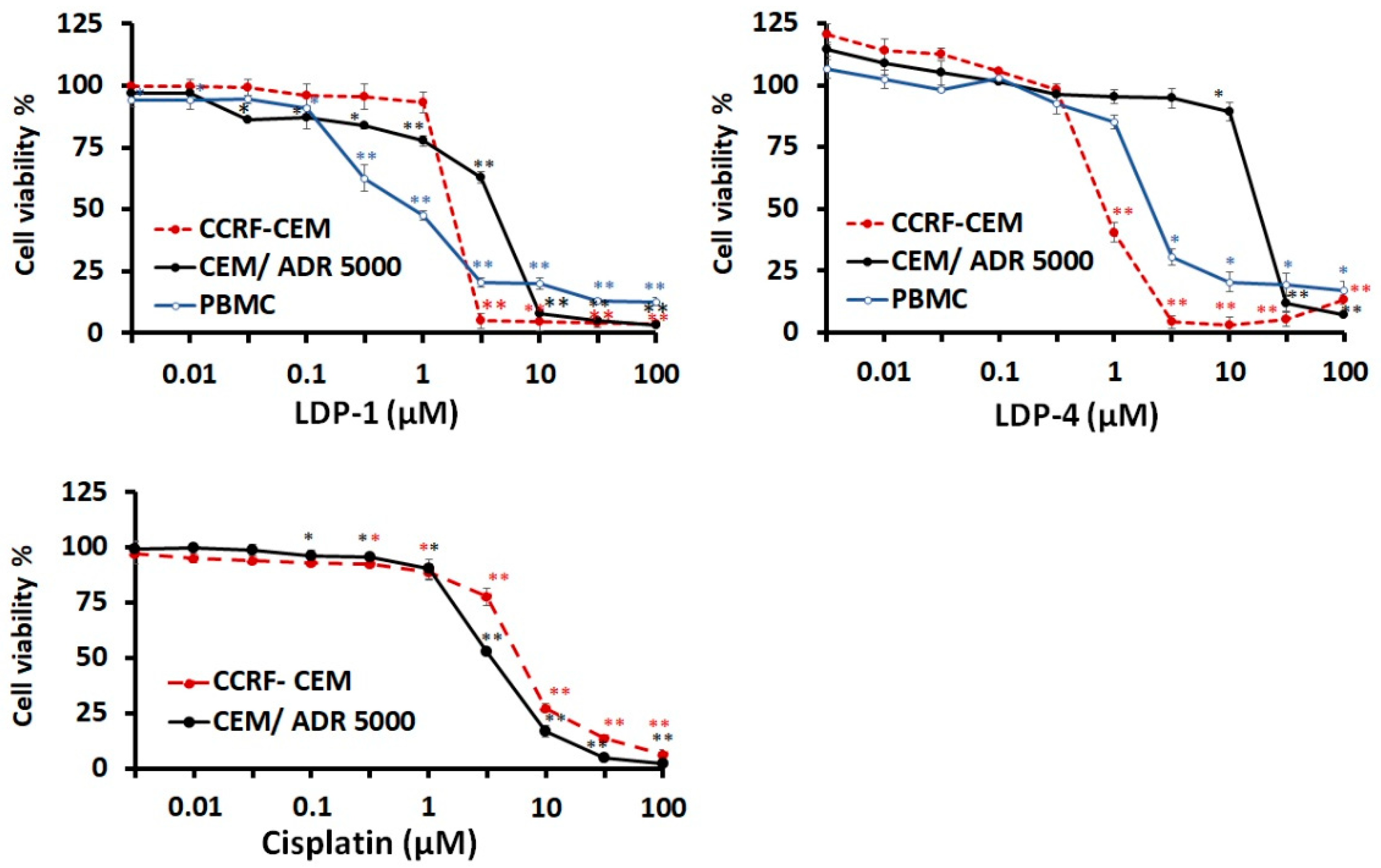
2.1. Biological Assessments
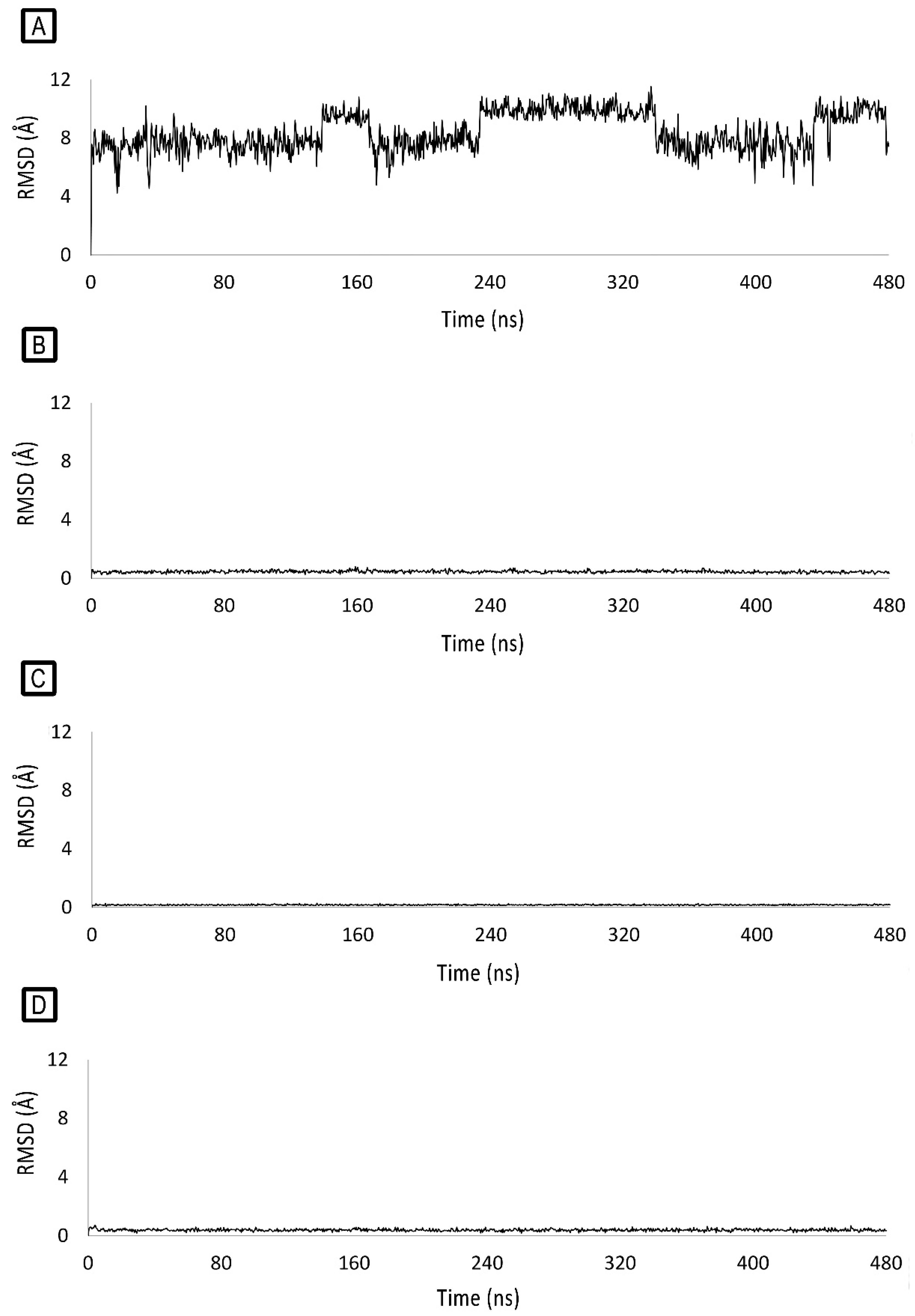
2.2. Molecular-Modelling Studies
3. Materials and Methods
3.1. Cell Lines and Compounds
3.2. Cell Proliferation Inhibition Assay
3.3. Toxicity in Normal Cells
3.4. CAM Assay
3.5. Molecular-Modelling Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, G. DNA Damage Repair Pathways and Repair of Cisplatin-Induced DNA Damage. Chem. Mol. Sci. Chem. Eng. 2018. preprint. [Google Scholar] [CrossRef]
- Lippert, B. (Ed.) Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1999. [Google Scholar]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]
- Gielen, E.R.T.T.M. (Ed.) Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Boulikas, T. Clinical overview on Lipoplatin™: A successful liposomal formulation of cisplatin. Expert Opin. Investig. Drugs 2009, 18, 1197–1218. [Google Scholar] [CrossRef]
- Ma, P.a.; Xiao, H.; Li, C.; Dai, Y.; Cheng, Z.; Hou, Z.; Lin, J. Inorganic nanocarriers for platinum drug delivery. Mater. Today 2015, 18, 554–564. [Google Scholar] [CrossRef]
- Piorecka, K.; Kurjata, J.; Stanczyk, W.A. Nanoarchitectonics: Complexes and Conjugates of Platinum Drugs with Silicon Containing Nanocarriers. An Overview. Int. J. Mol. Sci. 2021, 22, 9264. [Google Scholar] [CrossRef]
- Chaires, J.B. Daunomycin Binding to DNA: From the Macroscopic to the Microscopic; Springer: Dordrecht, The Netherlands, 1990; pp. 123–136. [Google Scholar]
- Arcamone, F. Doxorubicin: Anticancer Antibiotics; Elsevier: Amsterdam, The Netherlands, 1981; Volume 17. [Google Scholar]
- Marverti, G.; Cusumano, M.; Ligabue, A.; Di Pietro, M.L.; Vainiglia, P.A.; Ferrari, A.; Bergomi, M.; Moruzzi, M.S.; Frassineti, C. Studies on the anti-proliferative effects of novel DNA-intercalating bipyridyl-thiourea-Pt(II) complexes against cisplatin-sensitive and -resistant human ovarian cancer cells. J. Inorg. Biochem. 2008, 102, 699–712. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Montanari, M.; Guerrieri, D.; Cusumano, M.; Di Pietro, M.L.; Troiano, L.; Di Vono, E.; Iotti, S.; Farruggia, G.; et al. Characterization of the cell growth inhibitory effects of a novel DNA-intercalating bipyridyl-thiourea-Pt(II) complex in cisplatin-sensitive and -resistant human ovarian cancer cells. Investig. New Drugs 2011, 29, 73–86. [Google Scholar] [CrossRef]
- Mancuso, A.; Barattucci, A.; Bonaccorsi, P.; Giannetto, A.; La Ganga, G.; Musarra-Pizzo, M.; Salerno, T.M.G.; Santoro, A.; Sciortino, M.T.; Puntoriero, F.; et al. Carbohydrates and Charges on Oligo(phenylenethynylenes): Towards the Design of Cancer Bullets. Chemistry 2018, 24, 16972–16976. [Google Scholar] [CrossRef]
- Saad Hmoud, A.; Awad Abdalla, M. Anticancer Drugs’ Deoxyribonucleic Acid (DNA) Interactions. In Biophysical Chemistry; Mohammed, A.A.K., Ed.; IntechOpen: Rijeka, Croatia, 2019; p. 6. [Google Scholar]
- Di Pietro, M.L.; La Ganga, G.; Nastasi, F.; Puntoriero, F. Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting. Appl. Sci. 2021, 11, 3038. [Google Scholar] [CrossRef]
- Trovato, E.; Di Pietro, M.L.; Giannetto, A.; Dupeyre, G.; Lainé, P.P.; Nastasi, F.; Puntoriero, F.; Campagna, S. Designing expanded bipyridinium as redox and optical probes for DNA. Photochem. Photobiol. Sci. 2020, 19, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A. DNA interaction of platinum(II) complexes with 1,10-phenanthroline and extended phenanthrolines. Inorg. Chem. 2006, 45, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Nicolo, F.; Norden, B.; Lincoln, P. Ambivalent intercalators for DNA: L-shaped platinum(II) complexes. Inorg. Chem. 2004, 43, 2416–2421. [Google Scholar] [CrossRef] [PubMed]
- Bostancıoğlu, R.B.; Işık, K.; Genç, H.; Benkli, K.; Koparal, A.T. Studies on the cytotoxic, apoptotic and antitumoral effects of Au(III) and Pt(II) complexes of 1, 10-phenanthroline on V79 379A and A549 cell lines. J. Enzyme Inhib. Med. Chem. 2012, 27, 458–466. [Google Scholar] [CrossRef][Green Version]
- Roy, S.; Hagen, K.D.; Maheswari, P.U.; Lutz, M.; Spek, A.L.; Reedijk, J.; van Wezel, G.P. Phenanthroline Derivatives with Improved Selectivity as DNA-Targeting Anticancer or Antimicrobial Drugs. ChemMedChem 2008, 3, 1427–1434. [Google Scholar] [CrossRef]
- Nicolò, F.; Cusumano, M.; Di Pietro, M.L.; Scopelliti, R.; Bruno, G. Dichloro[6,7-dimethyl-2,3-bis(2-pyridyl-N)quinoxaline]palladium(II) monohydrate. Acta Crystallogr. Sect. C 1998, 54, 485–487. [Google Scholar] [CrossRef]
- Rotondo, E.; Rotondo, A.; Nicolo, F.; Di Pietro, M.L.; Messina, M.A.; Cusumano, M. NMR study of L-shaped (quinoxaline)platinum(II) complexes—Crystal structure of [Pt(DMeDPQ)(bipy)](PF6)(2). Eur. J. Inorg. Chem. 2004, 2004, 4710–4717. [Google Scholar] [CrossRef]
- Hodges, K.D.; Rund, J.V. Oxidative addition of halogens and pseudohalogens to dihalo (1,10-phenanthroline) platinum(II). Inorg. Chem. 1975, 14, 525–528. [Google Scholar] [CrossRef]
- Askari, B.; Amiri Rudbari, H.; Micale, N.; Schirmeister, T.; Efferth, T.; Seo, E.J.; Bruno, G.; Schwickert, K. Ruthenium(ii) and palladium(ii) homo- and heterobimetallic complexes: Synthesis, crystal structures, theoretical calculations and biological studies. Dalton Trans. 2019, 48, 15869–15887. [Google Scholar] [CrossRef] [PubMed]
- Cirri, D.; Schirmeister, T.; Seo, E.J.; Efferth, T.; Massai, L.; Messori, L.; Micale, N. Antiproliferative Properties of a Few Auranofin-Related Gold(I) and Silver(I) Complexes in Leukemia Cells and their Interferences with the Ubiquitin Proteasome System. Molecules 2020, 25, 4454. [Google Scholar] [CrossRef]
- Rudbari, H.A.; Kordestani, N.; Cuevas-Vicario, J.V.; Zhou, M.; Efferth, T.; Correia, I.; Schirmeister, T.; Barthels, F.; Enamullah, M.; Fernandes, A.R.; et al. Investigation of the influence of chirality and halogen atoms on the anticancer activity of enantiopure palladium(ii) complexes derived from chiral amino-alcohol Schiff bases and 2-picolylamine. New J. Chem. 2022, 46, 6470–6483. [Google Scholar] [CrossRef]
- Whiteley, A.E.; Price, T.T.; Cantelli, G.; Sipkins, D.A. Leukaemia: A model metastatic disease. Nat. Rev. Cancer 2021, 21, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Carmeliet, P. Angiogenesis: A Target in Solid Tumors, Also in Leukemia? Hematol. Am. Soc. Hematol. Educ. Progr. 2011, 2011, 1–8. [Google Scholar] [CrossRef]
- Trujillo, A.; McGee, C.; Cogle, C.R. Angiogenesis in acute myeloid leukemia and opportunities for novel therapies. J. Oncol. 2012, 2012, 128608. [Google Scholar] [CrossRef]
- Tufan, A.C.; Satiroglu-Tufan, N.L. The Chick Embryo Chorioallantoic Membrane as a Model System for the Study of Tumor Angiogenesis, Invasion and Development of Anti-Angiogenic Agents. Curr. Cancer Drug Targets 2005, 5, 249–266. [Google Scholar] [CrossRef]
- Kue, C.S.; Tan, K.Y.; Lam, M.L.; Lee, H.B. Chick embryo chorioallantoic membrane (CAM): An alternative predictive model in acute toxicological studies for anti-cancer drugs. Exp. Anim. 2015, 64, 129–138. [Google Scholar] [CrossRef]
- DeBord, L.C.; Pathak, R.R.; Villaneuva, M.; Liu, H.C.; Harrington, D.A.; Yu, W.; Lewis, M.T.; Sikora, A.G. The chick chorioallantoic membrane (CAM) as a versatile patient-derived xenograft (PDX) platform for precision medicine and preclinical research. Am. J. Cancer Res. 2018, 8, 1642–1660. [Google Scholar]
- Taizi, M.; Deutsch, V.R.; Leitner, A.; Ohana, A.; Goldstein, R.S. A novel and rapid in vivo system for testing therapeutics on human leukemias. Exp. Hematol. 2006, 34, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem. Cell Biol. 2008, 130, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Lokman, N.A.; Elder, A.S.F.; Ricciardelli, C.; Oehler, M.K. Chick Chorioallantoic Membrane (CAM) Assay as an In Vivo Model to Study the Effect of Newly Identified Molecules on Ovarian Cancer Invasion and Metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kaiser, J.T.; Barton, J.K. Crystal structure of Δ-[Ru(bpy)2dppz]2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nat. Chem. 2012, 4, 615–620. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Structure of duplex DNA containing the cisplatin 1,2-{Pt(NH3)2}2+-d(GpG) cross-link at 1.77 A resolution. J. Inorg. Biochem. 2010, 104, 902–908. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Kuete, V.; Mbaveng, A.T.; Nono, E.C.N.; Simo, C.C.; Zeino, M.; Nkengfack, A.E.; Efferth, T. Cytotoxicity of seven naturally occurring phenolic compounds towards multi-factorial drug-resistant cancer cells. Phytomedicine 2016, 23, 856–863. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Mahmoud, N.; Sugimoto, Y.; Efferth, T.; Abdel-Aziz, H. Molecular Determinants of Sensitivity or Resistance of Cancer Cells Toward Sanguinarine. Front. Pharmacol. 2018, 9, 136. [Google Scholar] [CrossRef]
- Certo, G.; Costa, R.; D’Angelo, V.; Russo, M.; Albergamo, A.; Dugo, G.; Germanò, M.P. Anti-angiogenic activity and phytochemical screening of fruit fractions from Vitex agnus castus. Nat. Prod. Res. 2017, 31, 2850–2856. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-3; Maestro, Schrödinger, LLC: New York, NY, USA, 2021; Available online: https://www.schrodinger.com/citations#Maestro (accessed on 17 November 2022).
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossváry, I.; Moraes, M.; Sacerdoti, F.; et al. Molecular Dynamics—Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Alhindi, T.; Zhang, Z.; Ruelens, P.; Coenen, H.; Degroote, H.; Iraci, N.; Geuten, K. Protein interaction evolution from promiscuity to specificity with reduced flexibility in an increasingly complex network. Sci. Rep. 2017, 7, 44948. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Cell Viability % (±Sd) Ccrf-Cem Cells |
|---|---|
| LDP-1 | 5.57 ± 1.08 |
| LDP-2 | 8.49 ± 0.59 |
| LDP-3 | 5.28 ± 3.18 |
| LDP-4 | 4.05 ± 1.84 |
| Compound Name | CCRF-CEM | CEM/ADR5000 | PBMC | Degree of Resistance | |||
|---|---|---|---|---|---|---|---|
| IC50 (µM) | SD | IC50 (µM) | SD | IC50 (µM) | SD | ||
| LDP-1 | 1.71 | 0.05 | 3.97 | 0.16 | 0.82 | 0.1 | 2.32 |
| LDP-4 | 0.82 | 0.06 | 17.46 | 0.96 | 2.03 | 0.06 | 21.29 |
| Cisplatin (µM) | 5.82 | 0.16 | 3.28 | 0.41 | - | - | 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, M.L.; Stagno, C.; Efferth, T.; Omer, E.A.; D’Angelo, V.; Germanò, M.P.; Cacciola, A.; De Gaetano, F.; Iraci, N.; Micale, N. Antileukemia Activity and Mechanism of Platinum(II)-Based Metal Complexes. Molecules 2022, 27, 9000. https://doi.org/10.3390/molecules27249000
Di Pietro ML, Stagno C, Efferth T, Omer EA, D’Angelo V, Germanò MP, Cacciola A, De Gaetano F, Iraci N, Micale N. Antileukemia Activity and Mechanism of Platinum(II)-Based Metal Complexes. Molecules. 2022; 27(24):9000. https://doi.org/10.3390/molecules27249000
Chicago/Turabian StyleDi Pietro, Maria Letizia, Claudio Stagno, Thomas Efferth, Ejlal A. Omer, Valeria D’Angelo, Maria Paola Germanò, Anna Cacciola, Federica De Gaetano, Nunzio Iraci, and Nicola Micale. 2022. "Antileukemia Activity and Mechanism of Platinum(II)-Based Metal Complexes" Molecules 27, no. 24: 9000. https://doi.org/10.3390/molecules27249000
APA StyleDi Pietro, M. L., Stagno, C., Efferth, T., Omer, E. A., D’Angelo, V., Germanò, M. P., Cacciola, A., De Gaetano, F., Iraci, N., & Micale, N. (2022). Antileukemia Activity and Mechanism of Platinum(II)-Based Metal Complexes. Molecules, 27(24), 9000. https://doi.org/10.3390/molecules27249000