
A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance


Abstract
1. Introduction
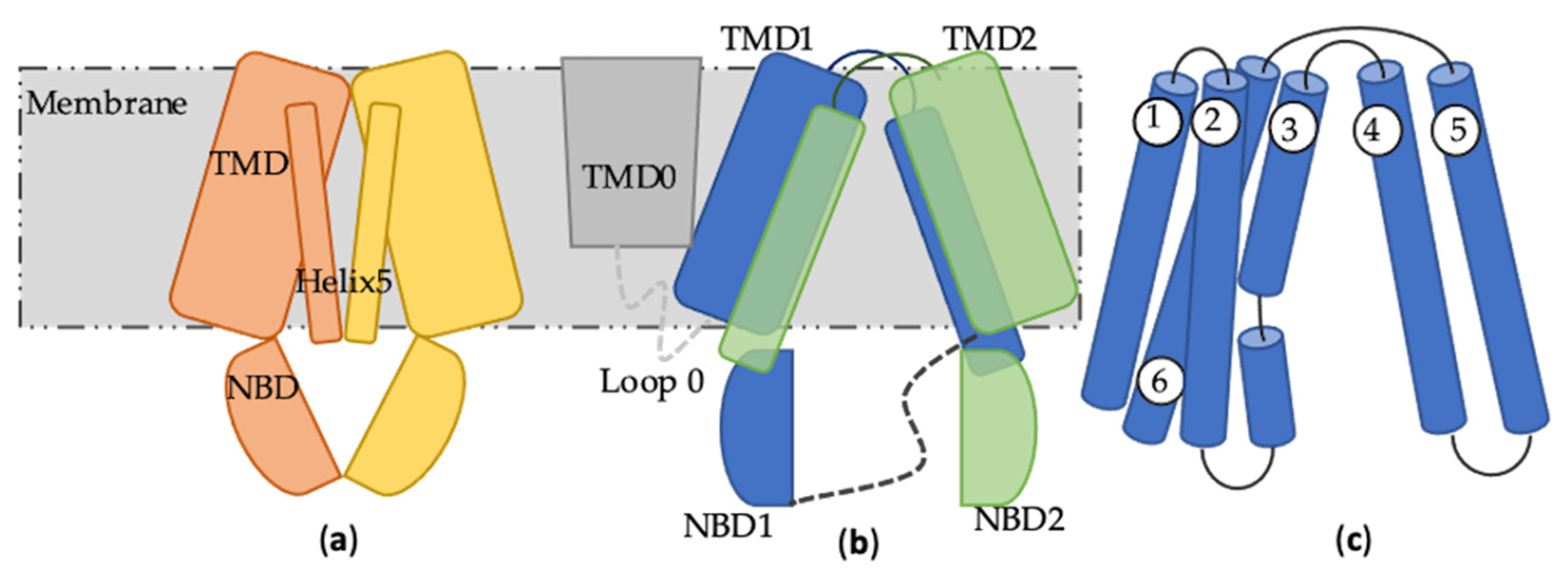
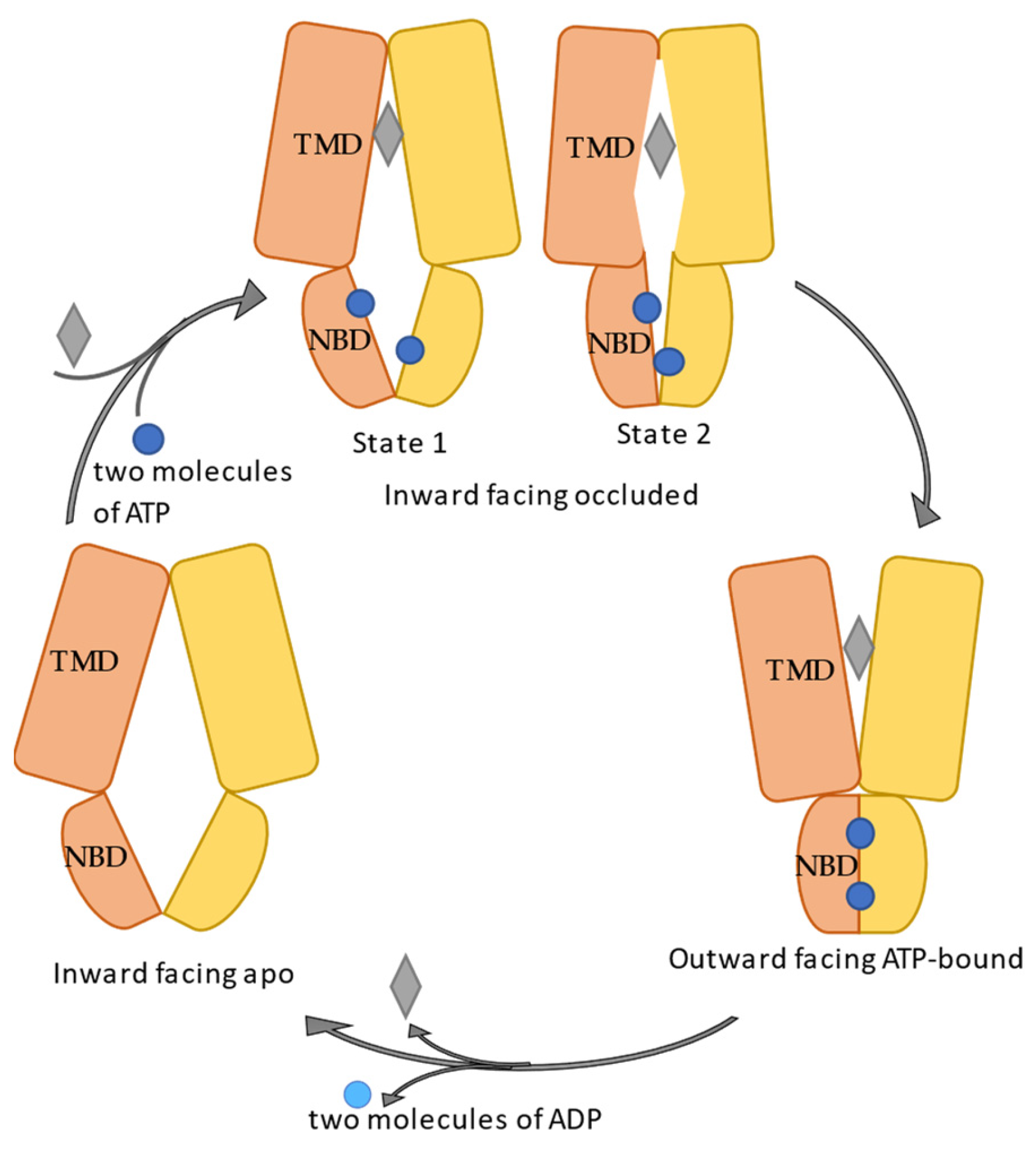
2. ABCB1
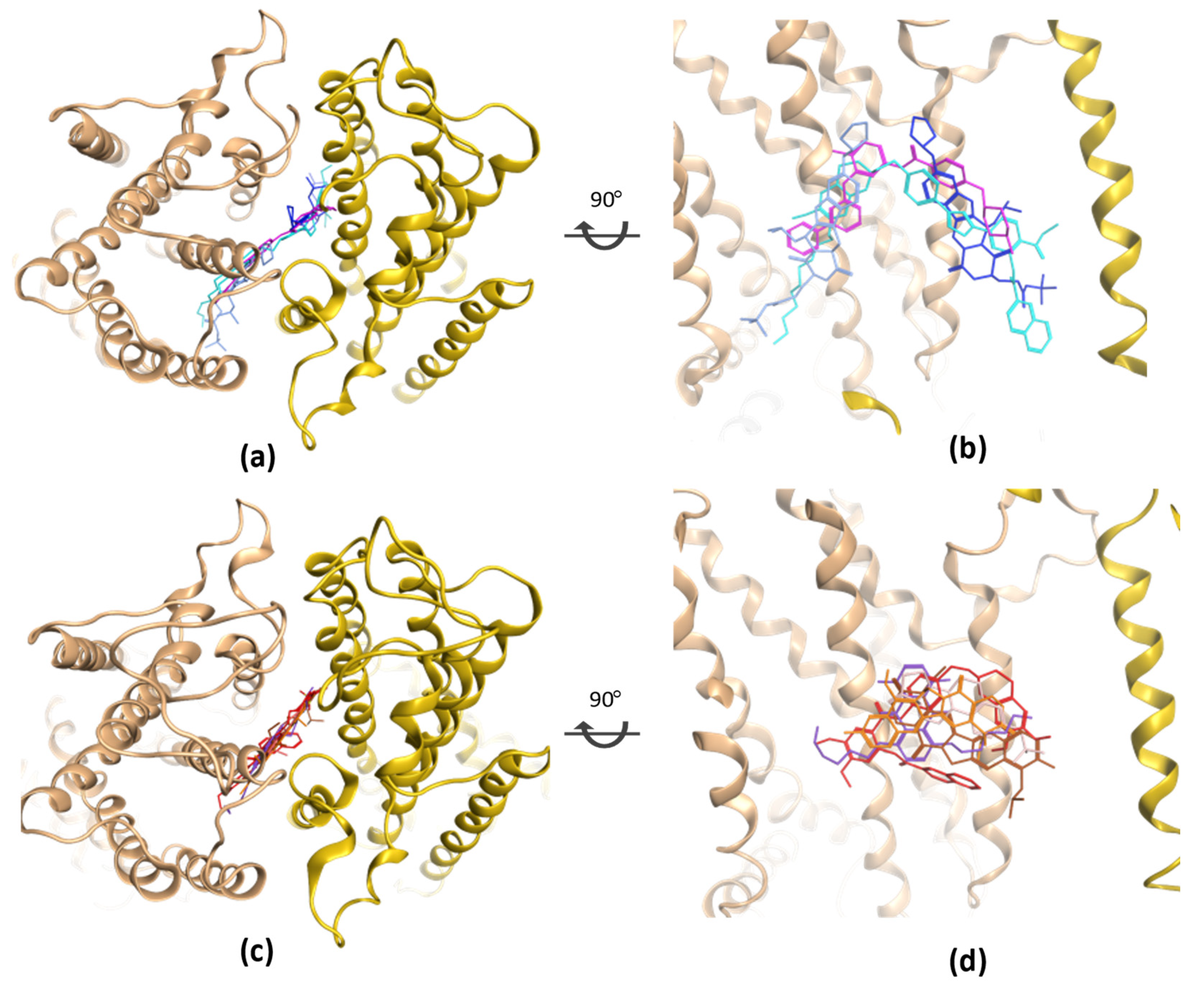
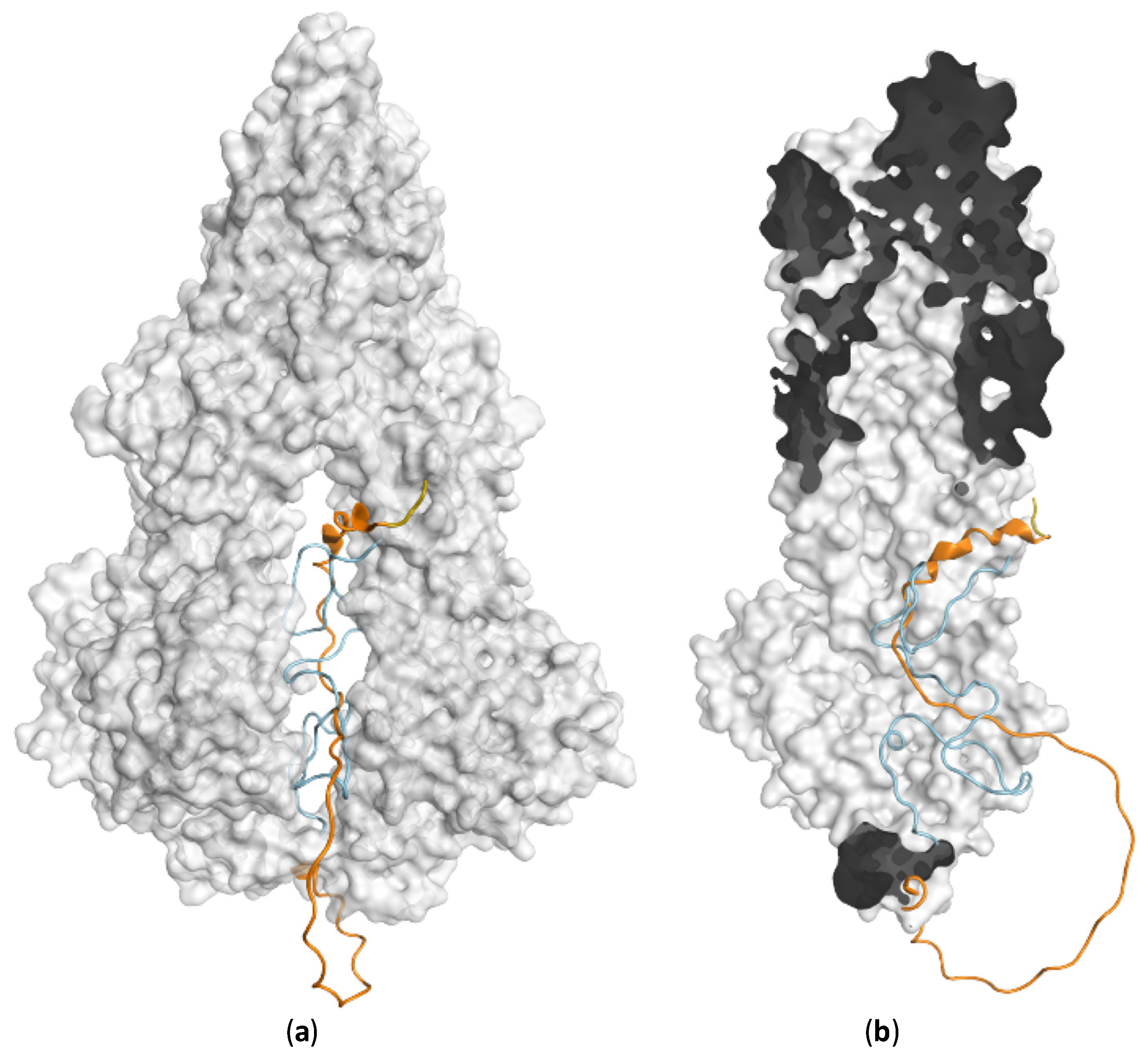
3. ABCC1
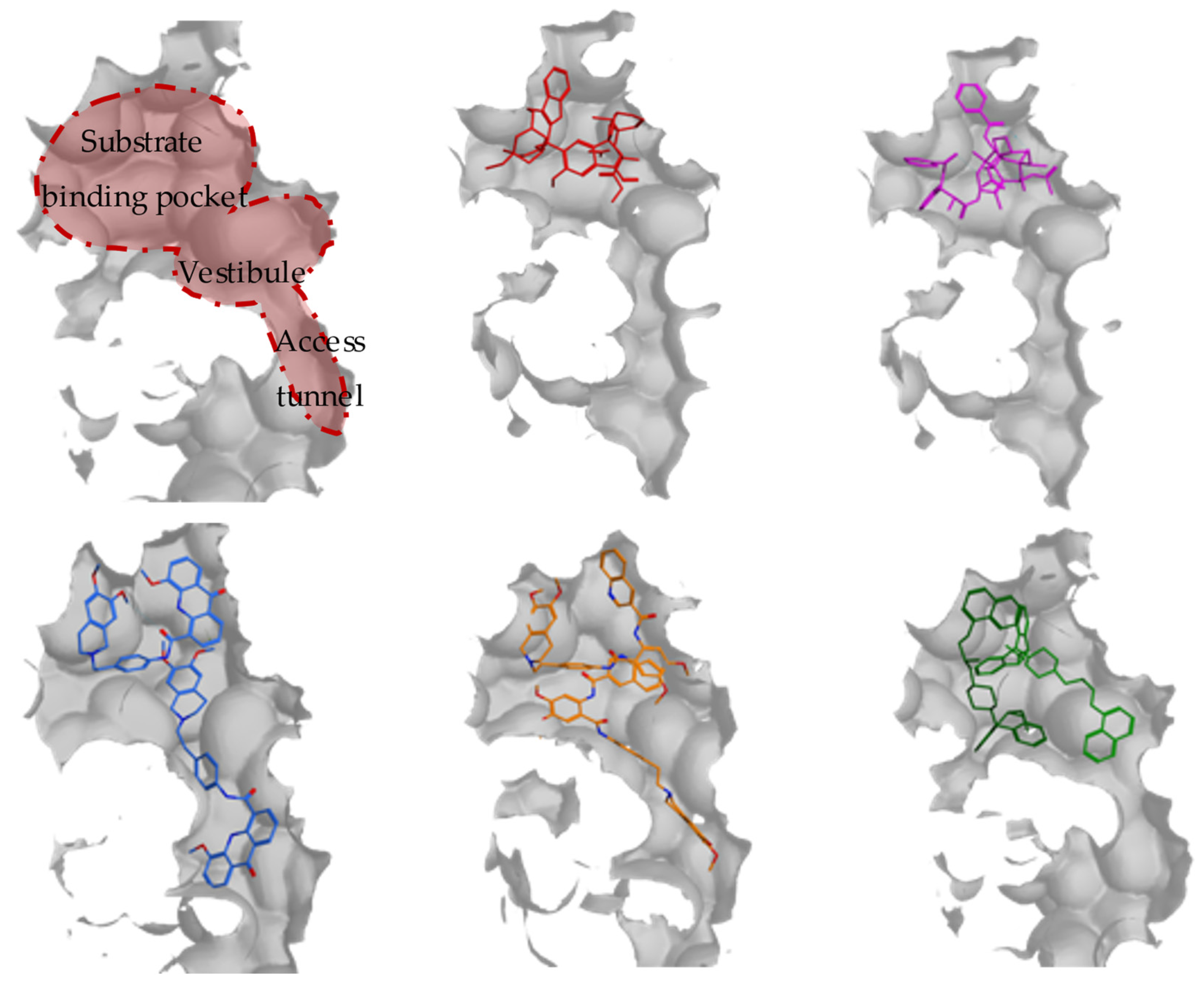
4. ABCG2
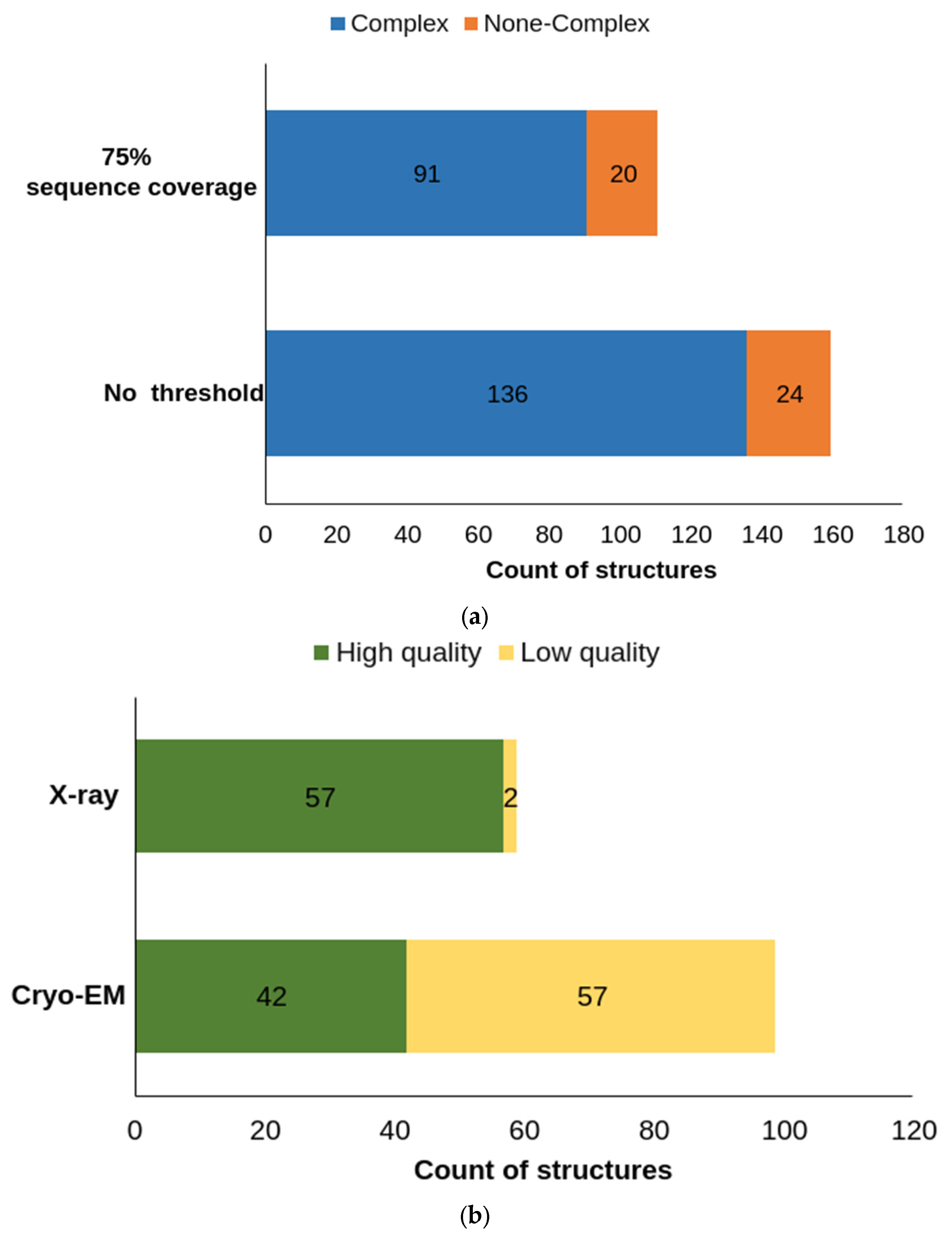
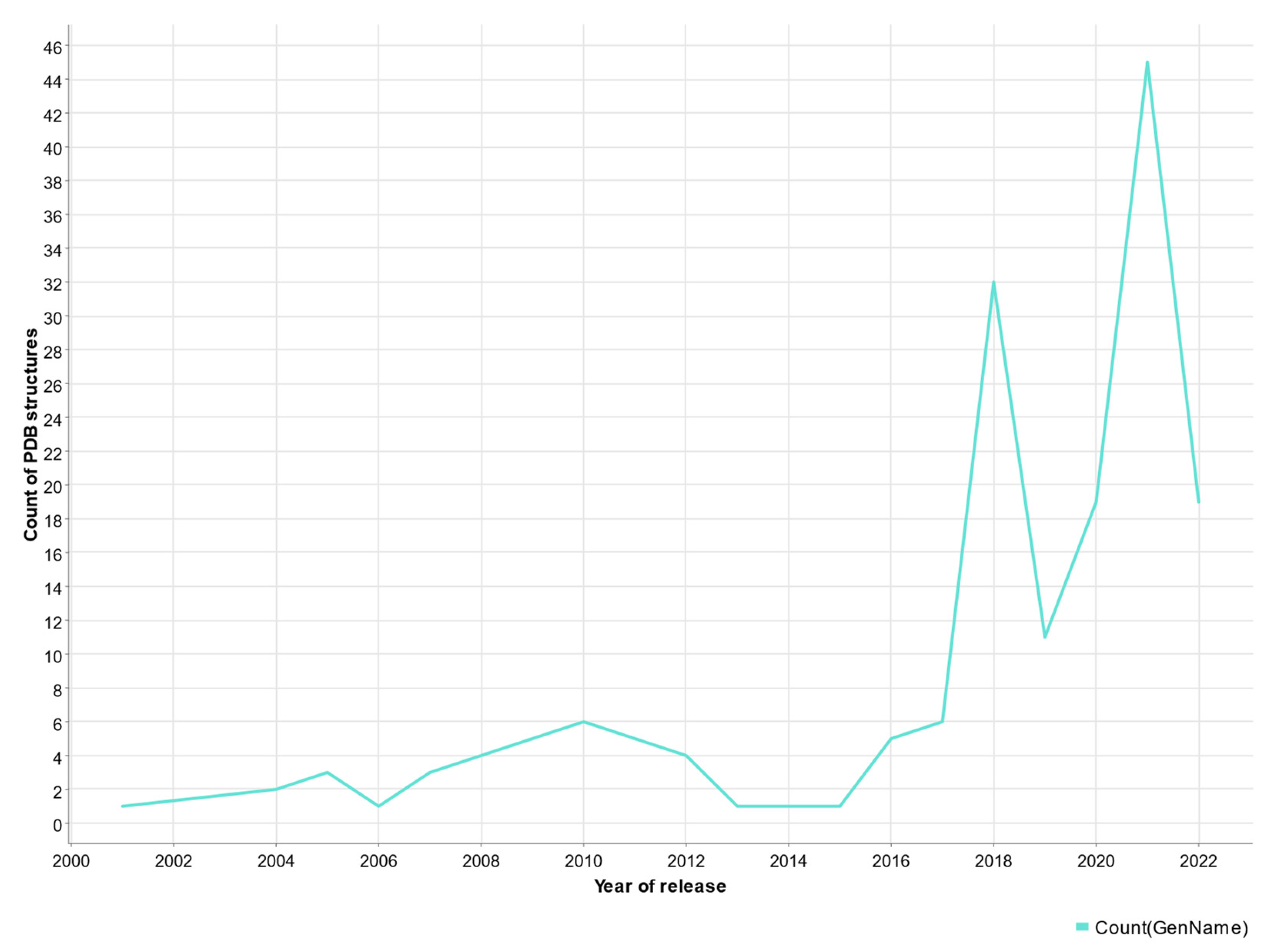
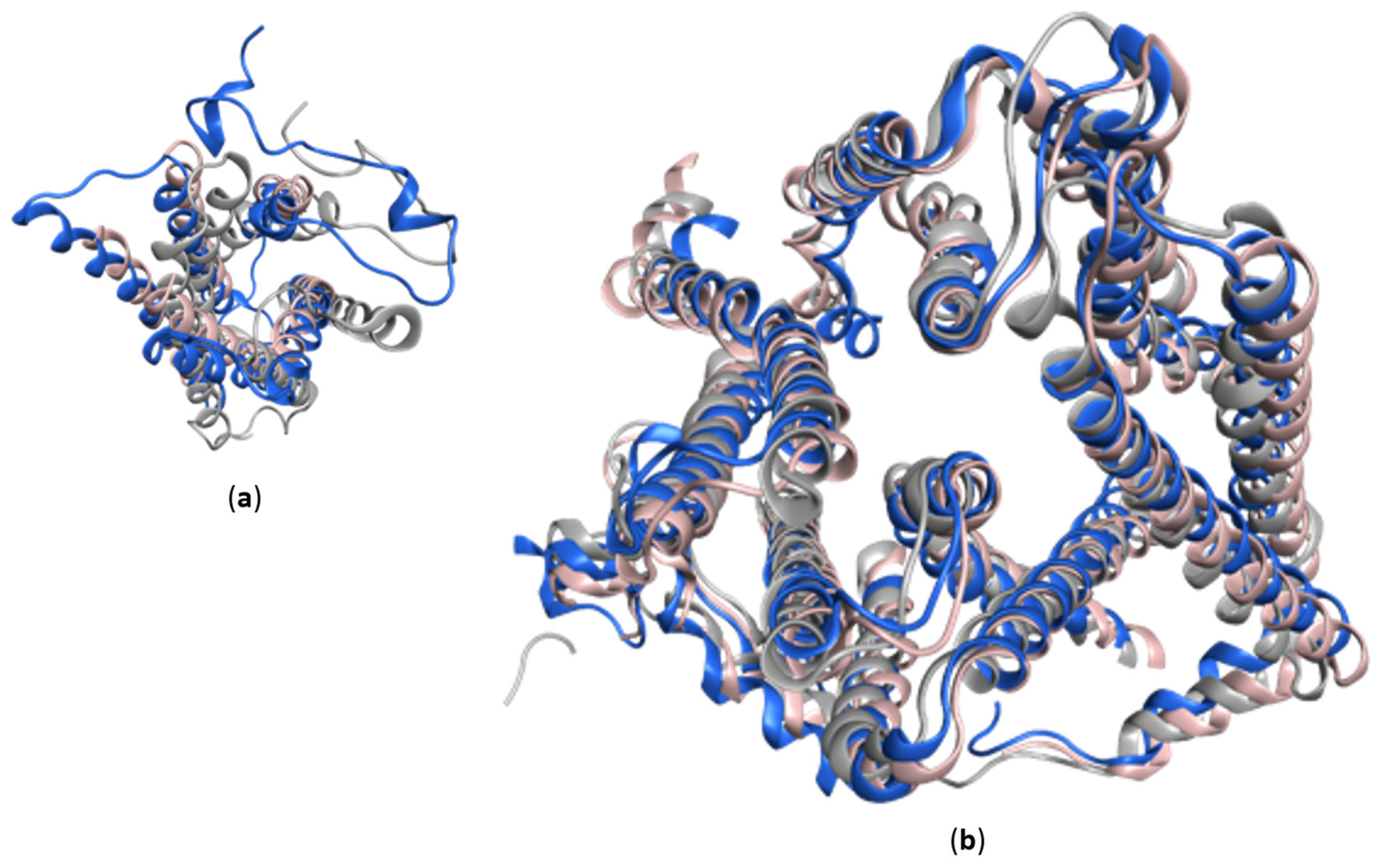
5. Protein Models and AlphaFold-2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- ECIS—European Cancer Information System. Series of Cancer Factsheets in EU-27 Countries. Available online: https://ecis.jrc.ec.europa.eu (accessed on 10 December 2021).
- Ferlay, J.L.M.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/tomorrow (accessed on 10 December 2021).
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta BBA-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Guo, X.-L. Tumor stem cells and drug resistance. Sheng Li Ke Xue Jin Zhan Prog. Physiol. 2007, 38, 115–119. [Google Scholar]
- Cole, S.; Bhardwaj, G.; Gerlach, J.; Mackie, J.; Grant, C.; Almquist, K.; Stewart, A.; Kurz, E.; Duncan, A.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.; Yeh, R.L.; Georges, E.; Ling, V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995, 55, 2029–2034. [Google Scholar] [PubMed]
- Childs, S.; Yeh, R.L.; Hui, D.; Ling, V. Taxol resistance mediated by transfection of the liver-specific sister gene of P-glycoprotein. Cancer Res. 1998, 58, 4160–4167. [Google Scholar]
- Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Chen, Z.-S. Multidrug resistance proteins (MRPs) and cancer therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef]
- Bugde, P.; Biswas, R.; Merien, F.; Lu, J.; Liu, D.-X.; Chen, M.; Zhou, S.; Li, Y. The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 2017, 21, 511–530. [Google Scholar] [CrossRef]
- Kerr, I.D. Structure and association of ATP-binding cassette transporter nucleotide-binding domains. Biochim. Biophys. Acta BBA-Biomembr. 2002, 1561, 47–64. [Google Scholar] [CrossRef]
- Hanekop, N.; Zaitseva, J.; Jenewein, S.; Holland, I.; Schmitt, L. Molecular insights into the mechanism of ATP-hydrolysis by the NBD of the ABC-transporter HlyB. FEBS Lett. 2006, 580, 1036–1041. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha-and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef]
- Linton, K.J.; Higgins, C.F. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 1998, 28, 5–13. [Google Scholar] [CrossRef]
- Diederichs, K.; Diez, J.; Greller, G.; Müller, C.; Breed, J.; Schnell, C.; Vonrhein, C.; Boos, W.; Welte, W. Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. EMBO J. 2000, 19, 5951–5961. [Google Scholar] [CrossRef]
- McMullan, G.; Faruqi, A.; Henderson, R.; Guerrini, N.; Turchetta, R.; Jacobs, A.; Van Hoften, G. Experimental observation of the improvement in MTF from backthinning a CMOS direct electron detector. Ultramicroscopy 2009, 109, 1144–1147. [Google Scholar] [CrossRef]
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-electron microscopy. Cell 2015, 161, 438–449. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and Bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef]
- Sheppard, D.N.; Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar] [CrossRef]
- Philipson, L.H.; Steiner, D.F. Pas de deux or more: The sulfonylurea receptor and K+ channels. Science 1995, 268, 372–374. [Google Scholar] [CrossRef]
- Chan, K.W.; Zhang, H.; Logothetis, D.E. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J. 2003, 22, 3833–3843. [Google Scholar] [CrossRef]
- Leier, I.; Jedlitschky, G.; Buchholz, U.; Cole, S.; Deeley, R.G.; Keppler, D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J. Biol. Chem. 1994, 269, 27807–27810. [Google Scholar] [CrossRef]
- Müller, M.; Meijer, C.; Zaman, G.; Borst, P.; Scheper, R.J.; Mulder, N.H.; De Vries, E.; Jansen, P. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc. Natl. Acad. Sci. USA 1994, 91, 13033–13037. [Google Scholar] [CrossRef]
- Bakos, E.; Evers, R.; Szakács, G.; Tusnády, G.E.; Welker, E.; Szabó, K.; de Haas, M.; van Deemter, L.; Borst, P.; Váradi, A. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J. Biol. Chem. 1998, 273, 32167–32175. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Q.; Zhang, J.-T. Structural and functional consequences of mutating cysteine residues in the amino terminus of human multidrug resistance-associated protein 1. J. Biol. Chem. 2002, 277, 44268–44277. [Google Scholar] [CrossRef]
- Leslie, E.M.; Bowers, R.J.; Deeley, R.G.; Cole, S.P. Structural requirements for functional interaction of glutathione tripeptide analogs with the human multidrug resistance protein 1 (MRP1). J. Pharmacol. Exp. Ther. 2003, 304, 643–653. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 2016, 167, 1586–1597. [Google Scholar] [CrossRef]
- Westlake, C.J.; Cole, S.P.; Deeley, R.G. Role of the NH2-terminal membrane spanning domain of multidrug resistance protein 1/ABCC1 in protein processing and trafficking. Mol. Biol. Cell 2005, 16, 2483–2492. [Google Scholar] [CrossRef][Green Version]
- Yang, Y.; Liu, Y.; Dong, Z.; Xu, J.; Peng, H.; Liu, Z.; Zhang, J.-T. Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. J. Biol. Chem. 2007, 282, 8821–8830. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. ATP binding enables substrate release from multidrug resistance protein 1. Cell 2018, 172, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Johnson, Z.L.; Wasserman, M.R.; Levring, J.; Chen, J.; Liu, S. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. eLife 2020, 9, e56451. [Google Scholar] [CrossRef] [PubMed]
- Loe, D.W.; Almquist, K.C.; Deeley, R.G.; Cole, S.P. Multidrug Resistance Protein (MRP)-mediated Transport of Leukotriene C4 and Chemotherapeutic Agents in Membrane Vesicles: Demonstration of Glutathione-Dependent Vincristine Transport (∗). J. Biol. Chem. 1996, 271, 9675–9682. [Google Scholar] [CrossRef]
- Mao, Q.; Deeley, R.G.; Cole, S.P. Functional reconstitution of substrate transport by purified multidrug resistance protein MRP1 (ABCC1) in phospholipid vesicles. J. Biol. Chem. 2000, 275, 34166–34172. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Bickers, S.C.; Benlekbir, S.; Rubinstein, J.L.; Kanelis, V. Structure of Ycf1p reveals the transmembrane domain TMD0 and the regulatory region of ABCC transporters. Proc. Natl. Acad. Sci. USA 2021, 118, e2025853118. [Google Scholar] [CrossRef]
- Khandelwal, N.K.; Millan, C.R.; Zangari, S.I.; Avila, S.; Williams, D.; Thaker, T.M.; Tomasiak, T.M. The structural basis for regulation of the glutathione transporter Ycf1 by regulatory domain phosphorylation. Nat. Commun. 2022, 13, 1278. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular structure of the human CFTR ion channel. Cell 2017, 169, 85–95.e88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Schriml, L.M.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar] [PubMed]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Dezi, M.; Fribourg, P.-F.; Di Cicco, A.; Arnaud, O.; Marco, S.; Falson, P.; Di Pietro, A.; Lévy, D. The multidrug resistance half-transporter ABCG2 is purified as a tetramer upon selective extraction from membranes. Biochim. Biophys. Acta BBA-Biomembr. 2010, 1798, 2094–2101. [Google Scholar] [CrossRef]
- McDevitt, C.A.; Collins, R.F.; Conway, M.; Modok, S.; Storm, J.; Kerr, I.D.; Ford, R.C.; Callaghan, R. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure 2006, 14, 1623–1632. [Google Scholar] [CrossRef]
- Taylor, N.M.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural basis of drug recognition by the multidrug transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Van Loevezijn, A.; Lakhai, J.M.; Van Der Valk, M.; Van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.-J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar]
- Puentes, C.O.; Höcherl, P.; Kühnle, M.; Bauer, S.; Bürger, K.; Bernhardt, G.; Buschauer, A.; König, B. Solid phase synthesis of tariquidar-related modulators of ABC transporters preferring breast cancer resistance protein (ABCG2). Bioorg. Med. Chem. Lett. 2011, 21, 3654–3657. [Google Scholar] [CrossRef]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef]
- Eadie, L.; Hughes, T.; White, D. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin. Pharmacol. Ther. 2014, 95, 294–306. [Google Scholar] [CrossRef]
- Houghton, P.J.; Germain, G.S.; Harwood, F.C.; Schuetz, J.D.; Stewart, C.F.; Buchdunger, E.; Traxler, P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 In Vitro. Cancer Res. 2004, 64, 2333–2337. [Google Scholar] [CrossRef]
- Özvegy-Laczka, C.; Köblös, G.; Sarkadi, B.; Váradi, A. Single amino acid (482) variants of the ABCG2 multidrug transporter: Major differences in transport capacity and substrate recognition. Biochim. Biophys. Acta BBA-Biomembr. 2005, 1668, 53–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- David, A.; Islam, S.; Tankhilevich, E.; Sternberg, M.J. The AlphaFold Database of Protein Structures: A Biologist’s Guide. J. Mol. Biol. 2022, 434, 167336. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. AcM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Hrycyna, C.A.; Airan, L.E.; Germann, U.A.; Ambudkar, S.V.; Pastan, I.; Gottesman, M.M. Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry 1998, 37, 13660–13673. [Google Scholar] [CrossRef]
- Sato, T.; Kodan, A.; Kimura, Y.; Ueda, K.; Nakatsu, T.; Kato, H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009, 276, 3504–3516. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; Dos Santos, D.J. Insights on P-glycoprotein’s efflux mechanism obtained by molecular dynamics simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; Dos Santos, D.J. Assessing the Stabilization of P-Glycoprotein’s Nucleotide-Binding Domains by the Linker, Using Molecular Dynamics. Mol. Inform. 2013, 32, 529–540. [Google Scholar] [CrossRef]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Gillet, J.-P.; Cordeiro, M.N.D.; Dos Santos, D.J. Theoretical insights on helix repacking as the origin of P-glycoprotein promiscuity. Sci. Rep. 2020, 10, 9823. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), 1010 Sherbooke St. West, Suite 2017, 910. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 3 October 2022).
- Ruff, K.M.; Pappu, R.V. AlphaFold and implications for intrinsically disordered proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Kage, K.; Tsukahara, S.; Sugiyama, T.; Asada, S.; Ishikawa, E.; Tsuruo, T.; Sugimoto, Y. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int. J. Cancer 2002, 97, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Jensen, U.; Hansen, A.; Covitz, K.-M.; Zhan, Z.; Fetsch, P.; Abati, A.; Hansen, P.R.; Horn, T.; Skovsgaard, T. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim. Biophys. Acta BBA-Biomembr. 2002, 1565, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Yang, Y.; Bates, S.; Zhang, J.-T. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 2004, 279, 19781–19789. [Google Scholar] [CrossRef] [PubMed]
- László, L.; Sarkadi, B.; Hegedűs, T. Jump into a new fold—A homology based model for the ABCG2/BCRP multidrug transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Qi, J.; Peng, H.; Zhang, J.-T. Effect of cysteine mutagenesis on the function and disulfide bond formation of human ABCG2. J. Pharmacol. Exp. Ther. 2008, 326, 33–40. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Walzthoeni, T.; Leitner, A.; Stengel, F.; Aebersold, R. Mass spectrometry supported determination of protein complex structure. Curr. Opin. Struct. Biol. 2013, 23, 252–260. [Google Scholar] [CrossRef]
- Rossmann, M.G. Molecular replacement–historical background. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1360–1366. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Conformation | Cocrystal Molecules | Property | Resolution (Å) | References |
|---|---|---|---|---|---|
| 5NJ3 | IF (NBDs modeled) | - | - | 3.78 | Taylor et al., 2017 [52] |
| 5NJG | IF (Only TMDs) | - | - | 3.78 | Taylor et al., 2017 [52] |
| 6FFC | IF | MZ29 × 2 | Inhibitor | 3.56 | Jackson et al., 2018 [53] |
| 6ETI | IF | MZ29 × 2 (Fab) | Inhibitor | 3.10 | Jackson et al., 2018 [53] |
| 6FEQ | IF | MB136 (Fab) | Inhibitor | 3.6 | Jackson et al., 2018 [53] |
| 6HIJ | IF | MZ29 × 2 (Cholesterol and phospholipid surrounded) | Inhibitor | 3.56 | Jackson et al., 2018 [53] |
| 6HCO | IF | E1S | Substrate | 3.58 | Manolaridis et al., 2018 [54] |
| 6HZM | OF | ATP × 2 + Mg2+ (Alternative placement) | - | 3.09 | Manolaridis et al., 2018 [54] |
| 6HBU | OF | ATP × 2 + Mg2+ | - | 3.09 | Manolaridis et al., 2018 [54] |
| 6VXF | Apo-closed | - | - | 3.50 | Orlando et al., 2020 [55] |
| 6VXH | IF | Imatinib | Inhibitor | 4.00 | Orlando et al., 2020 [55] |
| 6VXI | IF | Mitoxantrone | Substrate | 3.70 | Orlando et al., 2020 [55] |
| 6VXJ | IF | SN38 | Substrate | 4.10 | Orlando et al., 2020 [55] |
| 7NEZ | IF | Topotecan (Fab) | Substrate | 3.39 | Kowal et al., 2021 [56] |
| 7NFD | IF | Mitoxantrone (Fab) | Substrate | 3.51 | Kowal et al., 2021 [56] |
| 7NEQ | IF | Tariquidar (Fab) | Substrate | 3.12 | Kowal et al., 2021 [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Ecker, G.F. A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules 2023, 28, 495. https://doi.org/10.3390/molecules28020495
Huang J, Ecker GF. A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules. 2023; 28(2):495. https://doi.org/10.3390/molecules28020495
Chicago/Turabian StyleHuang, Jiahui, and Gerhard F. Ecker. 2023. "A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance" Molecules 28, no. 2: 495. https://doi.org/10.3390/molecules28020495
APA StyleHuang, J., & Ecker, G. F. (2023). A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules, 28(2), 495. https://doi.org/10.3390/molecules28020495