

Comparative Investigation of Chemical Constituents of Kernels, Leaves, Husk, and Bark of Juglans regia L., Using HPLC-DAD-ESI-MS/MS Analysis and Evaluation of Their Antioxidant, Antidiabetic, and Anti-Inflammatory Activities

 , , , , ,
, , , , ,  , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plant Material and Extraction
2.3. Determination of Phenolic Contents
2.3.1. Total Phenolic Content
2.3.2. Total Flavonoid Content
2.3.3. Total Tannin Content
2.4. HPLC-DAD-ESI-MS/MS
2.5. Antioxidant Assay
2.5.1. DPPH Method
2.5.2. ABTS Method
2.5.3. FRAP Method
2.6. Anti-Inflammatory Assay
2.6.1. Lipooxidase Inhibition Assay
2.6.2. Tyrosinase Inhibition Assay
2.7. Anti-Diabetic Activity
2.7.1. α-Amylase Inhibition Assay
2.7.2. α-Glucosidase Inhibition Assay
2.8. Statistical Analysis
3. Results and Discussion
3.1. Total Phenolic, Flavonoid, and Tocopherol Contents
3.2. HPLC-DAD-ESI-MS/MS Analysis
3.3. Antioxidant Activity: DPPH, FRAP, and ABTS Assays
3.4. Anti-Inflammatory Activity
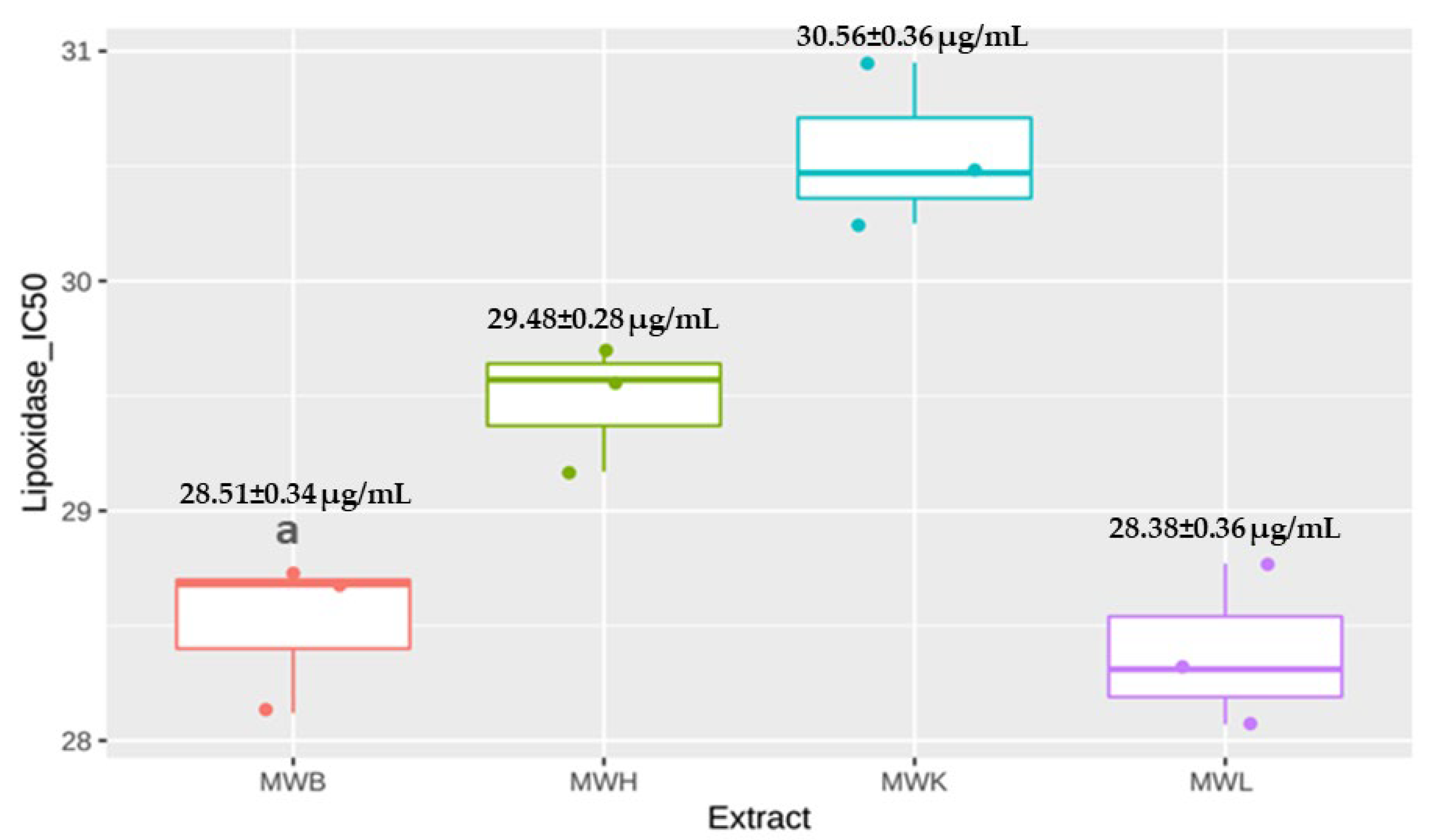
3.4.1. Anti-Lipoxidase Activity
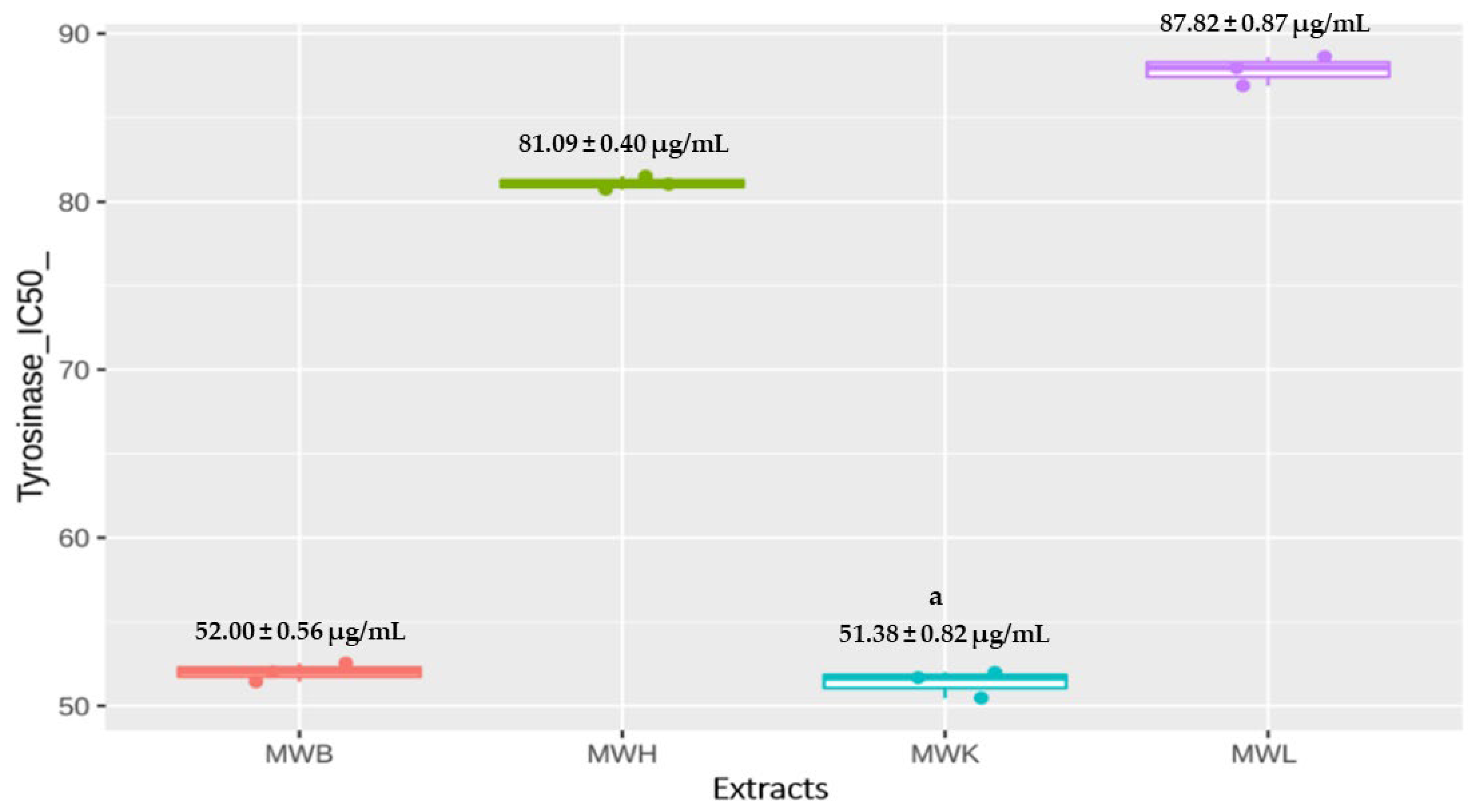
3.4.2. Anti-Tyrosinase Activity
3.5. Antidiabetic Activity
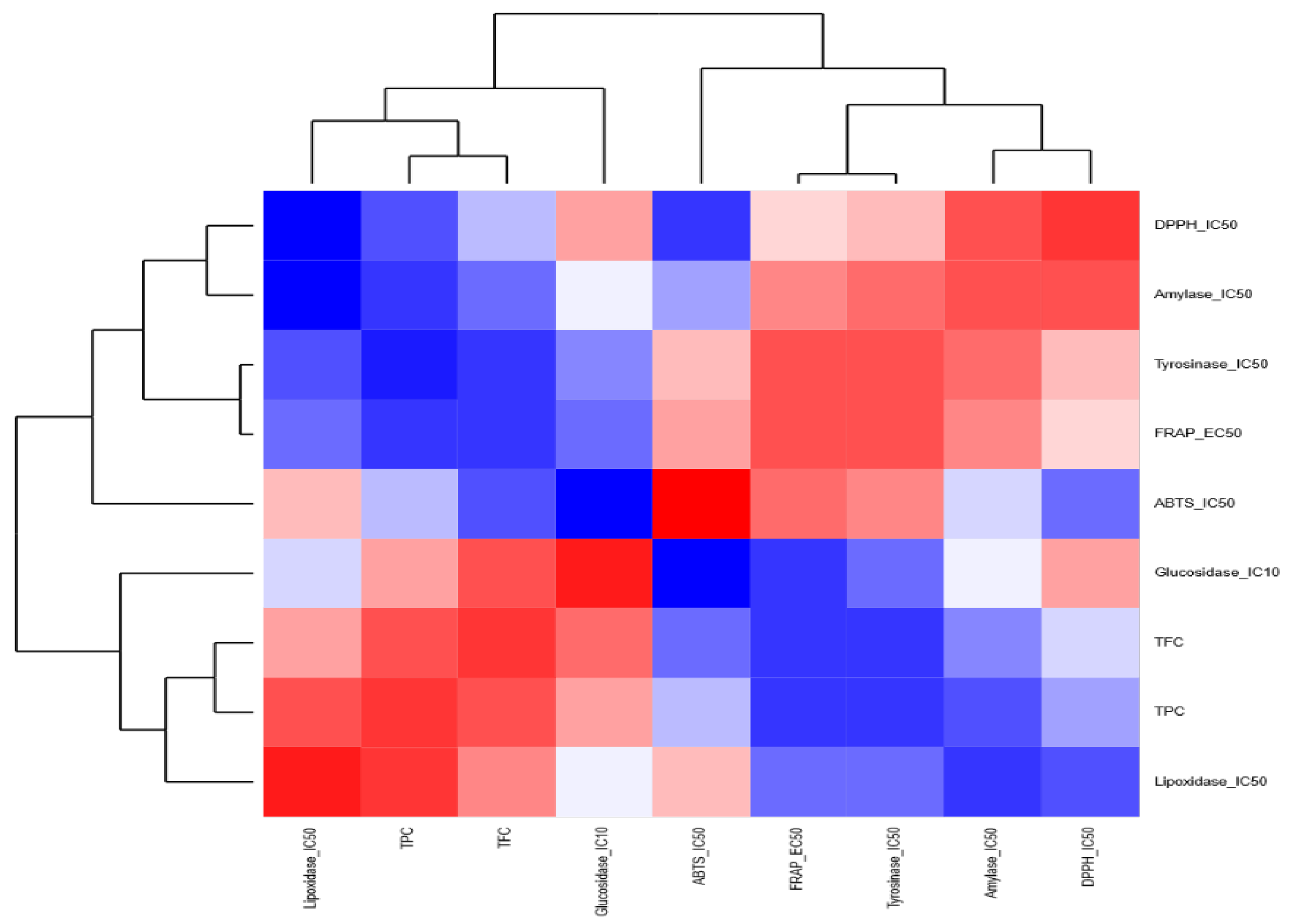
3.6. Correlations among J. regia Biochemical Activities in Different Extracts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABTS | 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) |
| Cat | Catechin |
| CE | Impact Energy |
| CES | Collision Energy Propagation |
| DP | Defusing Potential |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| EMS | Enhanced MS Analysis |
| EP | Input Potential |
| EPIA | Enhanced Product Ion Analysis |
| FRAP | Ferric Reducing Antioxidant Power |
| GA | Gallic acid |
| GLUT2 | Glucose transporter 2 |
| HCl | Hydrochloric acid |
| HPLC | High-Performance Liquid Chromatography |
| J. regia | Juglans regia |
| LOX | Lipoxygenase |
| MS | Mass Spectrometer |
| MWB | Methanolic Extract of Bark |
| MWH | Methanolic extract of Husk |
| MWK | Methanolic Extract of Kernels |
| MWL | Methanolic Extract of Leaves |
| ROS | Reactive Oxygen Species |
| RU | Rutin |
| TE | Trolox Equivalent |
| TFC | Total Flavonoid Content |
| TPC | Total Phenolic Content |
| TTC | Total Tocopherol Content |
References
- Bhatti, J.S.; Sehrawat, A.; Mishra, J.; Sidhu, I.S.; Navik, U.; Khullar, N.; Kumar, S.; Bhatti, G.K.; Reddy, P.H. Oxidative Stress in the Pathophysiology of Type 2 Diabetes and Related Complications: Current Therapeutics Strategies and Future Perspectives. Free Radic. Biol. Med. 2022, 184, 114–134. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef] [PubMed]
- Bakhouche, I.; Aliat, T.; Boubellouta, T.; Gali, L.; Şen, A.; Bellik, Y. Phenolic Contents and In Vitro Antioxidant, Anti-Tyrosinase, and Anti-Inflammatory Effects of Leaves and Roots Extracts of the Halophyte Limonium Delicatulum. South Afr. J. Bot. 2021, 139, 42–49. [Google Scholar] [CrossRef]
- Bouyahya, A.; Omari, N.E.; El Hachlafi, N.; Jemly, M.E.; Hakkour, M.; Balahbib, A.; El Menyiy, N.; Bakrim, S.; Naceiri Mrabti, H.; Khouchlaa, A. Chemical Compounds of Berry-Derived Polyphenols and Their Effects on Gut Microbiota, Inflammation, and Cancer. Molecules 2022, 27, 3286. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Shi, S.; Liu, B.; Shan, M.; Tang, D.; Zhang, W.; Zhang, Y.; Zhang, L.; Zhang, H.; Lu, C. Bioactive Compounds from Herbal Medicines to Manage Dyslipidemia. Biomed. Pharmacother. 2019, 118, 109338. [Google Scholar] [CrossRef]
- Sasikumar, P.; Aswathy, M.; Prem, P.T.; Radhakrishnan, K.V.; Chakrapani, P.S.B. Plant Derived Bioactive Compounds and Their Potential to Enhance Adult Neurogenesis. Phytomed. Plus 2022, 2, 100191. [Google Scholar]
- El Harrad, L.; Bourais, I.; Mohammadi, H.; Amine, A. Recent Advances in Electrochemical Biosensors Based on Enzyme Inhibition for Clinical and Pharmaceutical Applications. Sensors 2018, 18, 164. [Google Scholar] [CrossRef]
- Tlili, N.; Sarikurkcu, C. Bioactive Compounds Profile, Enzyme Inhibitory and Antioxidant Activities of Water Extracts from Five Selected Medicinal Plants. Ind. Crops Prod. 2020, 151, 112448. [Google Scholar] [CrossRef]
- Bellakhdar, J.; Claisse, R.; Fleurentin, J.; Younos, C. Repertory of Standard Herbal Drugs in the Moroccan Pharmacopoea. J. Ethnopharmacol. 1991, 35, 123–143. [Google Scholar] [CrossRef]
- Brown, D. Valerian: Clinical Overview-Phytotherapy Review & Commentary. Townsend Lett. Dr. 1995, 119, 1005–1012. [Google Scholar]
- Yeung, H.C. Handbook of Chinese Herbs and Formulas Institute of Chinese Medicine, Los Angeles. Am. Soc. Nutr. Sci. J. Nutr. 2004, 134, 1105–1159. [Google Scholar]
- Bouasla, A.; Bouasla, I. Ethnobotanical Survey of Medicinal Plants in Northeastern of Algeria. Phytomedicine 2017, 36, 68–81. [Google Scholar] [CrossRef]
- Petran, M.; Dragos, D.; Gilca, M. Historical Ethnobotanical Review of Medicinal Plants Used to Treat Children Diseases in Romania (1860s–1970s). J. Ethnobiol. Ethnomed. 2020, 16, 1–33. [Google Scholar] [CrossRef]
- Mardaninejad, S.; Janghorban, M.; Vazirpour, M. Collection and Identification of Medicinal Plants Used by the Indigenous People of Mobarakeh (Isfahan), Southwestern Iran. J. Med. Herbs 2013, 4, 23–32. [Google Scholar]
- Ziyyat, A.; Legssyer, A.; Mekhfi, H.; Dassouli, A.; Serhrouchni, M.; Benjelloun, W. Phytotherapy of Hypertension and Diabetes in Oriental Morocco. J. Ethnopharmacol. 1997, 58, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Güzel, Y.; Güzelşemme, M.; Miski, M. Ethnobotany of Medicinal Plants Used in Antakya: A Multicultural District in Hatay Province of Turkey. J. Ethnopharmacol. 2015, 174, 118–152. [Google Scholar] [CrossRef]
- Nasab, F.K.; Khosravi, A.R. Ethnobotanical Study of Medicinal Plants of Sirjan in Kerman Province, Iran. J. Ethnopharmacol. 2014, 154, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Sadgrove, N.J. The New Paradigm for Androgenetic Alopecia and Plant-Based Folk Remedies: 5α-Reductase Inhibition, Reversal of Secondary Microinflammation and Improving Insulin Resistance. J. Ethnopharmacol. 2018, 227, 206–236. [Google Scholar] [CrossRef]
- Jamila, F.; Mostafa, E. Ethnobotanical Survey of Medicinal Plants Used by People in Oriental Morocco to Manage Various Ailments. J. Ethnopharmacol. 2014, 154, 76–87. [Google Scholar] [CrossRef]
- Jaradat, N.A.; Ayesh, O.I.; Anderson, C. Ethnopharmacological Survey about Medicinal Plants Utilized by Herbalists and Traditional Practitioner Healers for Treatments of Diarrhea in the West Bank/Palestine. J. Ethnopharmacol. 2016, 182, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Najem, M.; Harouak, H.; Ibijbijen, J.; Nassiri, L. Oral Disorders and Ethnobotanical Treatments: A Field Study in the Central Middle Atlas (Morocco). Heliyon 2020, 6, e04707. [Google Scholar] [CrossRef]
- El-Hilaly, J.; Hmammouchi, M.; Lyoussi, B. Ethnobotanical Studies and Economic Evaluation of Medicinal Plants in Taounate Province (Northern Morocco). J. Ethnopharmacol. 2003, 86, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Teixidor-Toneu, I.; Martin, G.J.; Ouhammou, A.; Puri, R.K.; Hawkins, J.A. An Ethnomedicinal Survey of a Tashelhit-Speaking Community in the High Atlas, Morocco. J. Ethnopharmacol. 2016, 188, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Zougagh, S.; Belghiti, A.; Rochd, T.; Zerdani, I.; Mouslim, J. Medicinal and Aromatic Plants Used in Traditional Treatment of the Oral Pathology: The Ethnobotanical Survey in the Economic Capital Casablanca, Morocco (North Africa). Nat. Prod. Bioprospect. 2019, 9, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Arranz, S.; Pérez-Jiménez, J.; Saura-Calixto, F. Antioxidant Capacity of Walnut (Juglans regia L.): Contribution of Oil and Defatted Matter. Eur. Food Res. Technol. 2008, 227, 425–431. [Google Scholar] [CrossRef]
- Erdemoglu, N.; Küpeli, E.; Yeşilada, E. Anti-Inflammatory and Antinociceptive Activity Assessment of Plants Used as Remedy in Turkish Folk Medicine. J. Ethnopharmacol. 2003, 89, 123–129. [Google Scholar] [CrossRef]
- Vieira, V.; Pereira, C.; Pires, T.C.; Calhelha, R.C.; Alves, M.J.; Ferreira, O.; Barros, L.; Ferreira, I.C. Phenolic Profile, Antioxidant and Antibacterial Properties of Juglans regia L. (Walnut) Leaves from the Northeast of Portugal. Ind. Crops Prod. 2019, 134, 347–355. [Google Scholar] [CrossRef]
- Pereira, J.A.; Oliveira, I.; Sousa, A.; Valentão, P.; Andrade, P.B.; Ferreira, I.C.; Ferreres, F.; Bento, A.; Seabra, R.; Estevinho, L. Walnut (Juglans regia L.) Leaves: Phenolic Compounds, Antibacterial Activity and Antioxidant Potential of Different Cultivars. Food Chem. Toxicol. 2007, 45, 2287–2295. [Google Scholar] [CrossRef]
- Zhang, Z.; Liao, L.; Moore, J.; Wu, T.; Wang, Z. Antioxidant Phenolic Compounds from Walnut Kernels (Juglans regia L.). Food Chem. 2009, 113, 160–165. [Google Scholar] [CrossRef]
- Ghasemi, M.; Arzani, K.; Hasani, D.; Ghasemi, S. Fatty Acids Composition of Some Selected Walnut (Juglans regia L.) Genotypes in Markazi Province. J. Food Sci. Technol. 2010, 7, 31–37. [Google Scholar]
- Bourais, I.; Elmarrkechy, S.; Taha, D.; Mourabit, Y.; Bouyahya, A.; El Yadini, M.; Machich, O.; El Hajjaji, S.; El Boury, H.; Dakka, N. A Review on Medicinal Uses, Nutritional Value, and Antimicrobial, Antioxidant, Anti-Inflammatory, Antidiabetic, and Anticancer Potential Related to Bioactive Compounds of J. regia. Food Rev. Int. 2022, 1–51. [Google Scholar] [CrossRef]
- Papoutsi, Z.; Kassi, E.; Chinou, I.; Halabalaki, M.; Skaltsounis, L.A.; Moutsatsou, P. Walnut Extract (Juglans regia L.) and Its Component Ellagic Acid Exhibit Anti-Inflammatory Activity in Human Aorta Endothelial Cells and Osteoblastic Activity in the Cell Line KS483. Br. J. Nutr. 2008, 99, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Qamar, W.; Sultana, S. Kernel Modulate Cigarette Smoke Extract Induced Acute Inflammation, Oxidative Stress and Lung Injury in Wistar Rats. Hum. Exp. Toxicol. 2011, 30, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Nasiry, D.; Ahmadvand, H.; Talebpour Amiri, F.; Akbari, E. Protective Effects of Methanolic Extract of Juglans regia L. Leaf on Streptozotocin-Induced Diabetic Peripheral Neuropathy in Rats. BMC Complement. Altern. Med. 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, J.; Saadipour, K.; Delaviz, H.; Mohammadi, B. Anti-Diabetic Effects of an Alcoholic Extract of Juglans regia in an Animal Model. Turk. J. Med. Sci. 2011, 41, 685–691. [Google Scholar] [CrossRef]
- Mollica, A.; Zengin, G.; Locatelli, M.; Stefanucci, A.; Macedonio, G.; Bellagamba, G.; Onaolapo, O.; Onaolapo, A.; Azeez, F.; Ayileka, A. An Assessment of the Nutraceutical Potential of Juglans regia L. Leaf Powder in Diabetic Rats. Food Chem. Toxicol. 2017, 107, 554–564. [Google Scholar] [CrossRef]
- Gutfinger, T. Polyphenols in Olive Oils. J. Am. Oil Chem. Soc. 1981, 58, 966–968. [Google Scholar] [CrossRef]
- Brighente, I.M.C.; Dias, M.; Verdi, L.G.; Pizzolatti, M.G. Antioxidant Activity and Total Phenolic Content of Some Brazilian Species. Pharm. Biol. 2007, 45, 156–161. [Google Scholar] [CrossRef]
- Julkunen-Tiitto, R.; Sorsa, S. Testing the Effects of Drying Methods on Willow Flavonoids, Tannins, and Salicylates. J. Chem. Ecol. 2001, 27, 779–789. [Google Scholar] [CrossRef]
- Pallauf, K.; Rivas-Gonzalo, J.C.; Del Castillo, M.D.; Cano, M.P.; de Pascual-Teresa, S. Characterization of the Antioxidant Composition of Strawberry Tree (Arbutus unedo L.) Fruits. J. Food Compos. Anal. 2008, 21, 273–281. [Google Scholar] [CrossRef]
- Mraihi, F.; Fadhil, H.; Trabelsi-Ayadi, M.; Chérif, J.-K. Chemical Characterization by HPLC-DAD-ESI/MS of Flavonoids from Hawthorn Fruits and Their Inhibition of Human Tumor Growth. J. New Sci. 2015, 3, 840–846. [Google Scholar]
- Yamasaki, K.; Hashimoto, A.; Kokusenya, Y.; Miyamoto, T.; Sato, T. Electrochemical Method for Estimating the Antioxidative Effects of Methanol Extracts of Crude Drugs. Chem. Pharm. Bull. 1994, 42, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Applying an Improved ABTS Radical Cation Decolorization Assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, S.A.; Dzoyem, J.P.; Shai, L.J.; Eloff, J.N. The Anti-Inflammatory and Antioxidant Activity of 25 Plant Species Used Traditionally to Treat Pain in Southern African. BMC Complement. Altern. Med. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-H.; Chen, Q.-X.; Wang, Q.; Song, K.-K.; Wang, J.; Sha, L.; Guan, X. Inhibition of the Activity of Mushroom Tyrosinase by Alkylbenzoic Acids. Food Chem. 2006, 94, 1–6. [Google Scholar] [CrossRef]
- Kusano, R.; Ogawa, S.; Matsuo, Y.; Tanaka, T.; Yazaki, Y.; Kouno, I. α-Amylase and Lipase Inhibitory Activity and Structural Characterization of Acacia Bark Proanthocyanidins. J. Nat. Prod. 2011, 74, 119–128. [Google Scholar] [CrossRef]
- Li, C.-W.; Chu, Y.-C.; Huang, C.-Y.; Fu, S.-L.; Chen, J.-J. Evaluation of Antioxidant and Anti-α-Glucosidase Activities of Various Solvent Extracts and Major Bioactive Components from the Seeds of Myristica Fragrans. Molecules 2020, 25, 5198. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Appendino, G.; Ottino, M.; Marquez, N.; Bianchi, F.; Giana, A.; Ballero, M.; Sterner, O.; Fiebich, B.L.; Munoz, E. Arzanol, an Anti-Inflammatory and Anti-HIV-1 Phloroglucinol α-Pyrone from Helichrysum italicum ssp. Microphyllum. J. Nat. Prod. 2007, 70, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Ieri, F.; Martini, S.; Innocenti, M.; Mulinacci, N. Phenolic Distribution in Liquid Preparations of Vaccinium myrtillus L. and Vaccinium vitis Idaea L. Phytochem. Anal. 2013, 24, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Vagiri, M.; Ekholm, A.; Andersson, S.C.; Johansson, E.; Rumpunen, K. An Optimized Method for Analysis of Phenolic Compounds in Buds, Leaves, and Fruits of Black Currant (Ribes nigrum L.). J. Agric. Food Chem. 2012, 60, 10501–10510. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Theodoridis, G.A.; Plumb, R.S.; Wilson, I.D. Current Practice of Liquid Chromatography–Mass Spectrometry in Metabolomics and Metabonomics. J. Pharm. Biomed. Anal. 2014, 87, 12–25. [Google Scholar] [CrossRef]
- Li, Y.; Kong, D.; Fu, Y.; Sussman, M.R.; Wu, H. The Effect of Developmental and Environmental Factors on Secondary Metabolites in Medicinal Plants. Plant Physiol. Biochem. 2020, 148, 80–89. [Google Scholar] [CrossRef]
- Bhatia, K.; Rahman, S.; Ali, M.; Raisuddin, S. In Vitro Antioxidant Activity of Juglans regia L. Bark Extract and Its Protective Effect on Cyclophosphamide-Induced Urotoxicity in Mice. Redox Rep. 2006, 11, 273–279. [Google Scholar] [CrossRef]
- Noumi, E.; Snoussi, M.; Noumi, I.; Valentin, E.; Aouni, M.; Al-sieni, A. Comparative Study on the Antifungal and Antioxydant Properties of Natural and Colored Juglans regia L. Barks: A High Activity against Vaginal Candida Strains. Life Sci. J. 2014, 11, 327–335. [Google Scholar]
- Jabli, M.; Sebeia, N.; Boulares, M.; Faidi, K. Chemical Analysis of the Characteristics of Tunisian Juglans regia L. Fractions: Antibacterial Potential, Gas Chromatography–Mass Spectroscopy and a Full Investigation of Their Dyeing Properties. Ind. Crops Prod. 2017, 108, 690–699. [Google Scholar] [CrossRef]
- Wisastra, R.; Dekker, F.J. Inflammation, Cancer and Oxidative Lipoxygenase Activity Are Intimately Linked. Cancers 2014, 6, 1500–1521. [Google Scholar] [CrossRef]
- Sun, Y.; Li, S.; Zeng, F.; Qi, J.; Qin, W.; Tan, C.; Luo, Q.; Wu, D.; Zhang, Q.; Lin, D. Functional Components, Antioxidant Activity and Hypoglycemic Ability Following Simulated Gastro-Intestinal Digestion of Pigments from Walnut Brown Shell and Green Husk. Antioxidants 2019, 8, 573. [Google Scholar] [CrossRef]
- Berkoz, M. Antioxidant and Anti-Lipoxygenase Activities of Cydonia oblonga. Medicine 2020, 9, 251–254. [Google Scholar] [CrossRef]
- Alam, F.; Ashraf, M. Gaultheria Trichophylla (Royle): A Source of Minerals and Biologically Active Molecules, Its Antioxidant and Anti-Lipoxygenase Activities. BMC Complement. Altern. Med. 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Lončarić, M.; Strelec, I.; Moslavac, T.; Šubarić, D.; Pavić, V.; Molnar, M. Lipoxygenase Inhibition by Plant Extracts. Biomolecules 2021, 11, 152. [Google Scholar] [CrossRef]
- Russell, W.R.; Scobbie, L.; Duthie, G.G.; Chesson, A. Inhibition of 15-Lipoxygenase-Catalysed Oxygenation of Arachidonic Acid by Substituted Benzoic Acids. Bioorg. Med. Chem. 2008, 16, 4589–4593. [Google Scholar] [CrossRef]
- Jenner, P.M.; Hagan, E.C.; Taylor, J.M.; Cook, E.L.; Fitzhugh, O.G. Food Flavourings and Compounds of Related Structure I. Acute Oral Toxicity. Food Cosmet. Toxicol. 1964, 2, 327–343. [Google Scholar] [CrossRef]
- Loke, W.M.; Proudfoot, J.M.; Stewart, S.; McKinley, A.J.; Needs, P.W.; Kroon, P.A.; Hodgson, J.M.; Croft, K.D. Metabolic Transformation Has a Profound Effect on Anti-Inflammatory Activity of Flavonoids Such as Quercetin: Lack of Association between Antioxidant and Lipoxygenase Inhibitory Activity. Biochem. Pharmacol. 2008, 75, 1045–1053. [Google Scholar] [CrossRef]
- Ha, T.J.; Shimizu, K.; Ogura, T.; Kubo, I. Inhibition Mode of Soybean Lipoxygenase-1 by Quercetin. Chem. Biodivers. 2010, 7, 1893–1903. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chem. Eur. J. 2018, 24, 47–55. [Google Scholar] [CrossRef]
- Rusu, M.E.; Gheldiu, A.-M.; Mocan, A.; Moldovan, C.; Popa, D.-S.; Tomuta, I.; Vlase, L. Process Optimization for Improved Phenolic Compounds Recovery from Walnut (Juglans regia L.) Septum: Phytochemical Profile and Biological Activities. Molecules 2018, 23, 2814. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A Comprehensive Review on Tyrosinase Inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Akin, M.; Arabaci, G.; Saki, N. Total Phenols, Antioxidant Potential and Tyrosinase Inhibitory Activity of Walnut (Juglans regia L.) Leaf, Husk and Seed. Asian J. Chem. 2013, 25, 9337. [Google Scholar] [CrossRef]
- Besrour, N.; Oludemi, T.; Mandim, F.; Pereira, C.; Dias, M.I.; Soković, M.; Stojković, D.; Ferreira, O.; Ferreira, I.C.; Barros, L. Valorization of Juglans regia Leaves as Cosmeceutical Ingredients: Bioactivity Evaluation and Final Formulation Development. Antioxidants 2022, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Zengin, G.; Aktumsek, A.; Karatas, S. Chemical and Biological Approaches on Nine Fruit Tree Leaves Collected from the Mediterranean Region of Turkey. J. Funct. Foods 2016, 22, 518–532. [Google Scholar] [CrossRef]
- Opperman, L.; De Kock, M.; Klaasen, J.; Rahiman, F. Tyrosinase and Melanogenesis Inhibition by Indigenous African Plants: A Review. Cosmetics 2020, 7, 60. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, P.; Meena, A.; Luqman, S. Acacetin, a Flavone with Diverse Therapeutic Potential in Cancer, Inflammation, Infections and Other Metabolic Disorders. Food Chem. Toxicol. 2020, 145, 111708. [Google Scholar] [CrossRef]
- Wang, W.; Yue, R.-F.; Jin, Z.; He, L.-M.; Shen, R.; Du, D.; Tang, Y.-Z. Efficiency Comparison of Apigenin-7-O-Glucoside and Trolox in Antioxidative Stress and Anti-Inflammatory Properties. J. Pharm. Pharmacol. 2020, 72, 1645–1656. [Google Scholar] [CrossRef]
- Rahimzadeh, M.; Jahanshahi, S.; Moein, S.; Moein, M.R. Evaluation of Alpha-Amylase Inhibition by Urtica Dioica and Juglans regia Extracts. Iran. J. Basic Med. Sci. 2014, 17, 465. [Google Scholar]
- Boulfia, M.; Lamchouri, F.; Toufik, H. Mineral Analysis, in Vitro Evaluation of Alpha-Amylase, Alpha-Glucosidase, and Beta-Galactosidase Inhibition, and Antibacterial Activities of Juglans regia L. Bark Extracts. BioMed Res. Int. 2021, 2021, 1585692. [Google Scholar] [CrossRef]
- Sun, Q.-Y.; Zhou, H.-H.; Mao, X.-Y. Emerging Roles of 5-Lipoxygenase Phosphorylation in Inflammation and Cell Death. Oxid. Med. Cell. Longev. 2019, 2019, 2749173. [Google Scholar] [CrossRef]
- Yaghobian, F.; Dehghan, F.; Azarbayjani, M.A. Forecast of Ameliorating Effect of Dietary Flavonol Consumption in White Tea with or without Aerobic Training on Type 2 Diabetes (T2D) in Females. Clin. Nutr. ESPEN 2021, 45, 134–140. [Google Scholar]
- Spínola, V.; Castilho, P.C. Evaluation of Asteraceae Herbal Extracts in the Management of Diabetes and Obesity. Contribution of Caffeoylquinic Acids on the Inhibition of Digestive Enzymes Activity and Formation of Advanced Glycation End-Products (in Vitro). Phytochemistry 2017, 143, 29–35. [Google Scholar] [CrossRef]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A Review of Alpha-Glucosidase Inhibitors from Plants as Potential Candidates for the Treatment of Type-2 Diabetes. Phytochem. Rev. 2021, 21, 1049–1079. [Google Scholar] [CrossRef]
- Nasiry, D.; Khalatbary, A.R.; Ahmadvand, H.; Talebpour Amiri, F.B. Effects of Juglans regia L. Leaf Extract Supplementation on Testicular Functions in Diabetic Rats. Biotech. Histochem. 2021, 96, 41–47. [Google Scholar] [CrossRef]
- Hosseini, S.; Jamshidi, L.; Mehrzadi, S.; Mohammad, K.; Najmizadeh, A.R.; Alimoradi, H.; Huseini, H.F. Effects of Juglans regia L. Leaf Extract on Hyperglycemia and Lipid Profiles in Type Two Diabetic Patients: A Randomized Double-Blind, Placebo-Controlled Clinical Trial. J. Ethnopharmacol. 2014, 152, 451–456. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPC | TFC | TTC | |
|---|---|---|---|
| EqGA mg/g | EqRU mg/g | EqCat mg/g | |
| MWL | 389.40 ± 2.74 | 306.36 ± 9.73 | – |
| MWH | 306.36 ± 4.74 | 66.07 ± 2.68 | – |
| MWK | 406.95 ± 7.60 | 18.44 ± 4.75 a | – |
| MWB | 413.71 ± 9.73 | 395.71 ± 39.44 | 5.44 ± 1.07 |
| No | Rt (min) | MS/MS (m/z) | Fragment | Compounds | Chemical Class |
|---|---|---|---|---|---|
| 1 | 4.63 | 439 | 169/183/341 | Caffeoyl Hexoside | Caffeic acid derivatives |
| 2 | 10.47 | 435 | 177/183/195/331/435 | Quercetin Pentoside | Flavonols |
| 3 | 17.85 | 369 | 183/195/233/331/369 | Fraxetin-8-O-glucoside | Hydroxycoumarin |
| 4 | 18.74 | 615 | 233/255/331/447/615 | Quercetin Galloyl-glucoside | Flavonols |
| 5 | 19.61 | 545 | 233/255/435/447/545 | O-methyl-epicatechin-sulfate-O-glucoside | Flavanols |
| 6 | 20.30 | 447 | 299/447 | Kaempferol Glucoside | Flavonols |
| 7 | 21.81 | 331 | 285/331 | Gallic Acid glucoside | Phenolic acid |
| 8 | 32.26 | 487 | 375/457/487 | Caffeoyl hexose deoxyhexose | Caffeic acid derivatives |
| 9 | 37.32 | 893 | 297/397/527 | Di-caffeic acid derivatives | Caffeic acid derivatives |
| No | Rt (min) | MS/MS (m/z) | Fragment | Compounds | Chemical Class |
|---|---|---|---|---|---|
| 1 | 4.65 | 369 | 269/293/315/369 | Fraxetin-8-O-glucoside | Hydroxycoumarin |
| 2 | 10.68 | 565 | 255/345/483/565 | Myricetin malonyl-glucoside | Flavonols |
| 3 | 11.50 | 665 | 113/335/407/665 | Gallotannin | Phenolic acid |
| 4 | 20.11 | 691 | 195/435/545/691 | Quercetin galloyl-glycoside | Flavonols |
| 5 | 27.94 | 593 | 117/253/425/593 | Kaempferol glucosyl-rhamnoside | Flavonols |
| 6 | 32.20 | 563 | 239/365/427/563 | Apigenin pentosyl glucoside | Flavones |
| 7 | 37.25 | 737 | 311/409/435/737 | Quercetin pentoside | Flavonols |
| 8 | 42.21 | 445 | 291/309/345/445 | Apigenin-O-glucuronide | Flavones |
| 9 | 43.25 | 701 | 311/409/663/701 | Tiliroside | Flavonols |
| 10 | 50.90 | 677 | 295/313/452/677 | Dicaffeoylquinic acid glycoside | Phenolic |
| 11 | 51.38 | 505 | 141/387/411/505 | Quercetin Malonyl Hexoside 1 | Flavonols |
| 12 | 54.10 | 563 | 255/281/563 | Apigenin Peptosyl glucoside | Flavones |
| 13 | 54.66 | 715 | 141/279/309/715 | Acacetin-7-O-rutinoside | Flavone |
| 14 | 58.04 | 903 | 283/667/751/903 | Tetragalloyl glucose | Glucide |
| No | Rt (min) | MS/MS (m/z) | Fragment | Compounds | Chemical Class |
|---|---|---|---|---|---|
| 1 | 4.72 | 197 | 147/183/197 | Dihydroxybenzoic acid derivative | Phenolic acid |
| 2 | 10.20 | 283 | 255/283 | Acacetin aglycone | Flavone |
| 3 | 11.64 | 339 | 177/197/339 | Caffeoyl-D-glucose | Caffeic acid derivatives |
| 4 | 17.41 | 507 | 191/207/435/507 | Quercetin O hexoside 1 | Flavonols |
| 5 | 18.97 | 431 | 207/385/417/431 | Apigenin-7-O-glucoside | Flavones |
| 6 | 27.11 | 427 | 297/339/373/427 | Caffeoyl derivative | Caffeic acid derivatives |
| 7 | 32.20 | 529 | 141/293/313/441/529 | p-Coumaroyl derivative | Coumaric acid |
| 8 | 36.38 | 487 | 157/171/267/409/433/487 | Caffeoyl hexose-deoxyhexoside | Caffeic acid derivatives |
| 9 | 37.25 | 435 | 359/409/417/435 | Quercetin pentoside | Flavonols |
| No | Rt (min) | MS/MS (m/z) | Fragment | Compounds | Chemical Class |
|---|---|---|---|---|---|
| 1 | 4.28 | 437 | 145/171/249/437 | p-Coumaroyl derivative | Coumaric acid |
| 2 | 11.60 | 453 | 159/177/339/383/437/453 | p-Coumaroyl derivative | Coumaric acid |
| 3 | 16.65 | 531 | 317/ 417/ 447 /483/531 | Caffeoyl derivative | Caffeic acid derivatives |
| 4 | 18.80 | 533 | 447/ 463/515/533 | Kaempferol-malonylglucoside | Flavonols |
| 5 | 19.92 | 565 | 395/435/533/565 | Myricetin malonyl-glucoside | Flavonols |
| 6 | 20.47 | 631 | 171/263/459/533/631 | Quercetin galloyl-glycoside | Flavonols |
| 7 | 20.93 | 583 | 183/201/485/583 | Myricetin acetylglycoside | Flavonols |
| 8 | 21.61 | 711 | 183/255/389/485/711 | Quercetin-7-O-hexoside-3-O- (malonyl) hexoside | Flavonols |
| 9 | 22.04 | 633 | 325/469/595/633 | Trigalloylglucose | Glucide |
| 10 | 32.28 | 483 | 313/339/483 | Digalloylglucose | Glucide |
| 11 | 36.43 | 487 | 313/343/355/487 | Caffeoyl hexose-deoxyhexoside | Caffeic acid derivatives |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourais, I.; Elmarrkechy, S.; Taha, D.; Badaoui, B.; Mourabit, Y.; Salhi, N.; Alshahrani, M.M.; Al Awadh, A.A.; Bouyahya, A.; Goh, K.W.; et al. Comparative Investigation of Chemical Constituents of Kernels, Leaves, Husk, and Bark of Juglans regia L., Using HPLC-DAD-ESI-MS/MS Analysis and Evaluation of Their Antioxidant, Antidiabetic, and Anti-Inflammatory Activities. Molecules 2022, 27, 8989. https://doi.org/10.3390/molecules27248989
Bourais I, Elmarrkechy S, Taha D, Badaoui B, Mourabit Y, Salhi N, Alshahrani MM, Al Awadh AA, Bouyahya A, Goh KW, et al. Comparative Investigation of Chemical Constituents of Kernels, Leaves, Husk, and Bark of Juglans regia L., Using HPLC-DAD-ESI-MS/MS Analysis and Evaluation of Their Antioxidant, Antidiabetic, and Anti-Inflammatory Activities. Molecules. 2022; 27(24):8989. https://doi.org/10.3390/molecules27248989
Chicago/Turabian StyleBourais, Ilhame, Salma Elmarrkechy, Douae Taha, Bouabid Badaoui, Yassine Mourabit, Najoua Salhi, Mohammed Merae Alshahrani, Ahmed Abdullah Al Awadh, Abdelhakim Bouyahya, Khang Wen Goh, and et al. 2022. "Comparative Investigation of Chemical Constituents of Kernels, Leaves, Husk, and Bark of Juglans regia L., Using HPLC-DAD-ESI-MS/MS Analysis and Evaluation of Their Antioxidant, Antidiabetic, and Anti-Inflammatory Activities" Molecules 27, no. 24: 8989. https://doi.org/10.3390/molecules27248989
APA StyleBourais, I., Elmarrkechy, S., Taha, D., Badaoui, B., Mourabit, Y., Salhi, N., Alshahrani, M. M., Al Awadh, A. A., Bouyahya, A., Goh, K. W., Tan, C. S., El Hajjaji, S., Dakka, N., & Iba, N. (2022). Comparative Investigation of Chemical Constituents of Kernels, Leaves, Husk, and Bark of Juglans regia L., Using HPLC-DAD-ESI-MS/MS Analysis and Evaluation of Their Antioxidant, Antidiabetic, and Anti-Inflammatory Activities. Molecules, 27(24), 8989. https://doi.org/10.3390/molecules27248989