
N,N′-Di-Boc-2H-Isoindole-2-carboxamidine—First Guanidine-Substituted Isoindole


Abstract
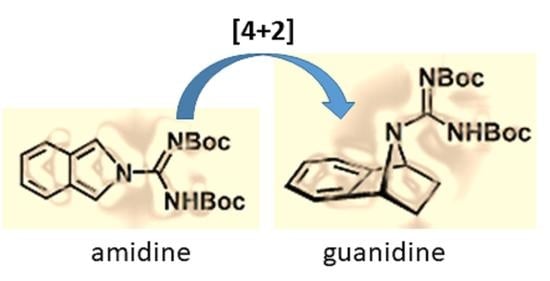
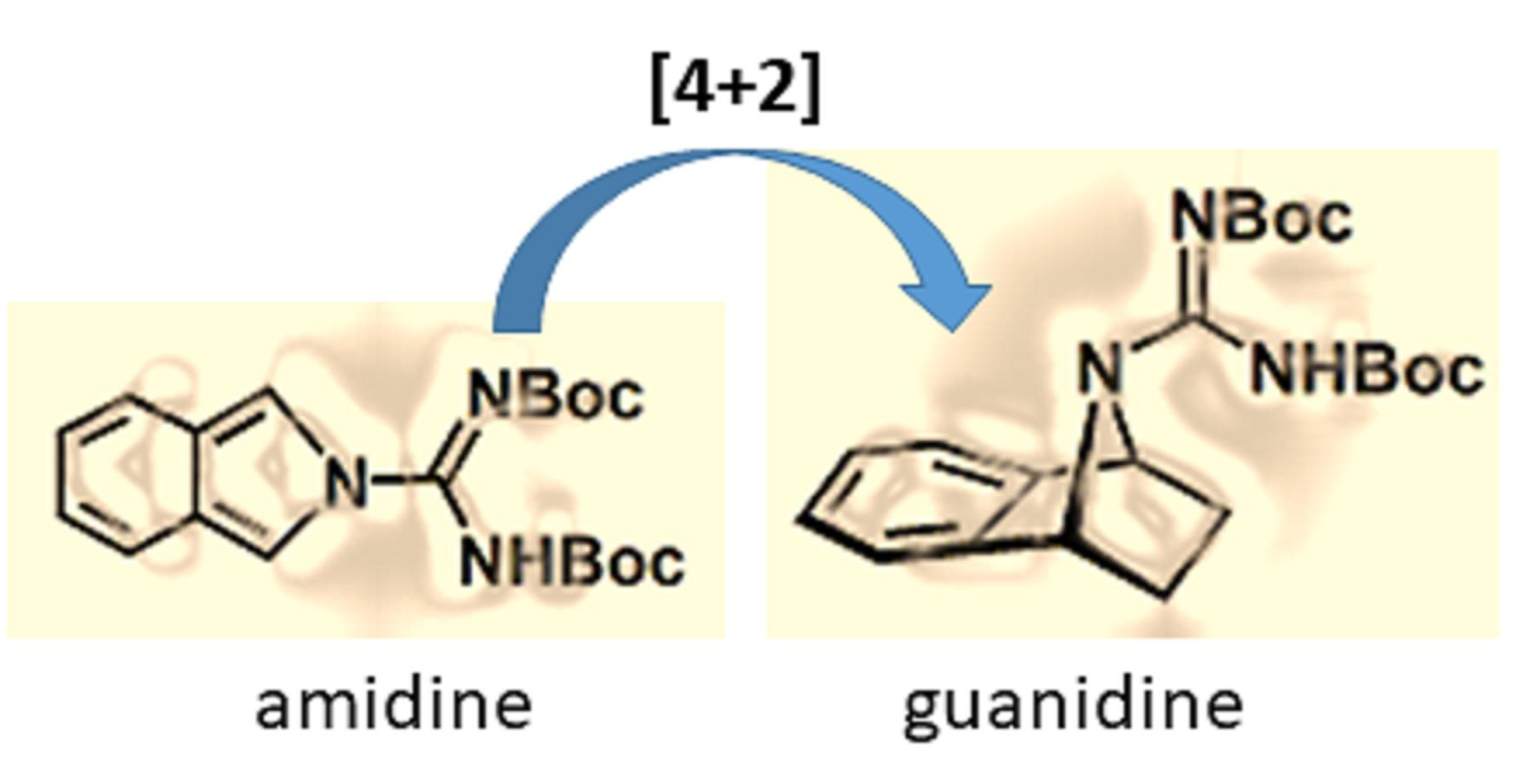
1. Introduction

{kind=link}
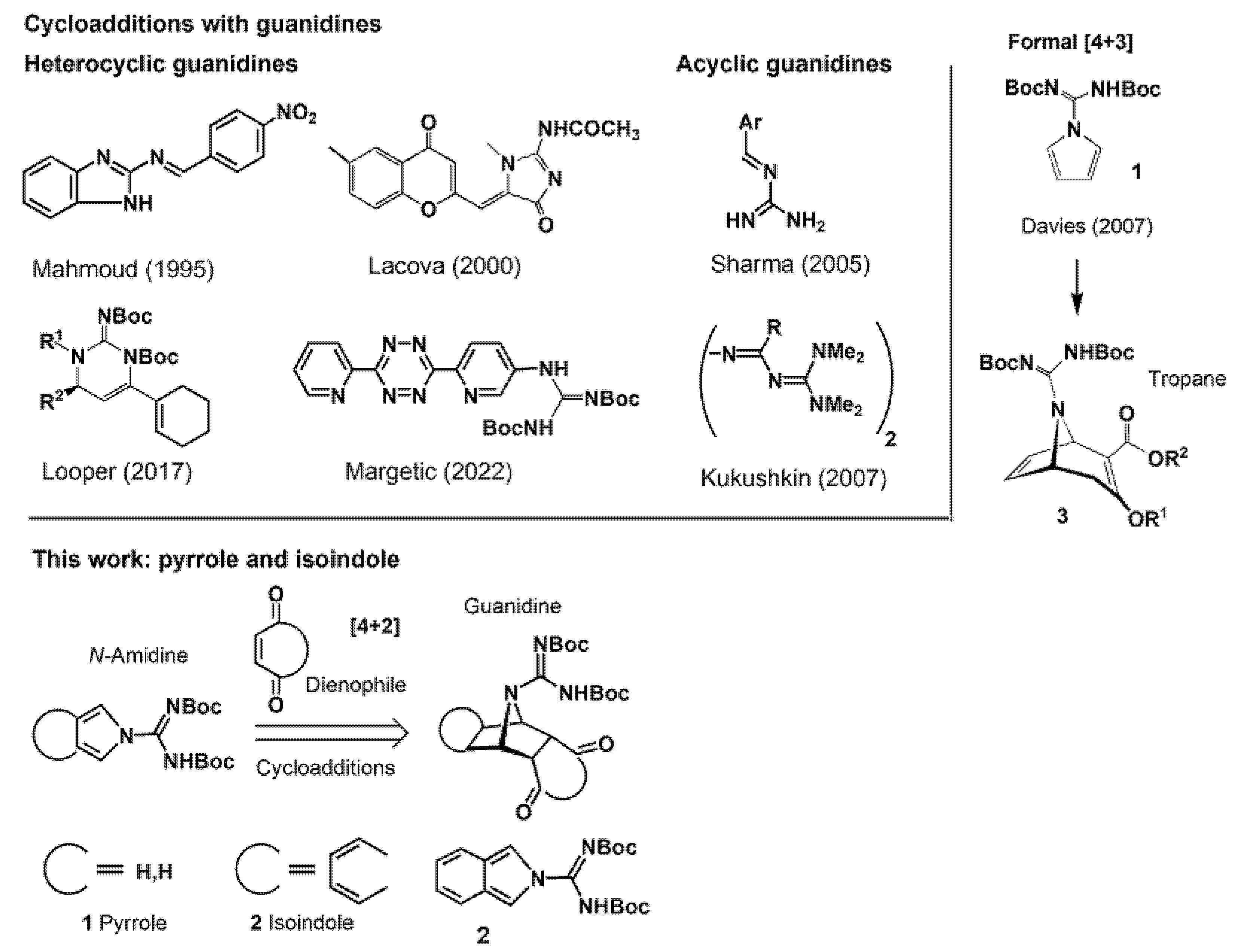{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Results and Discussion
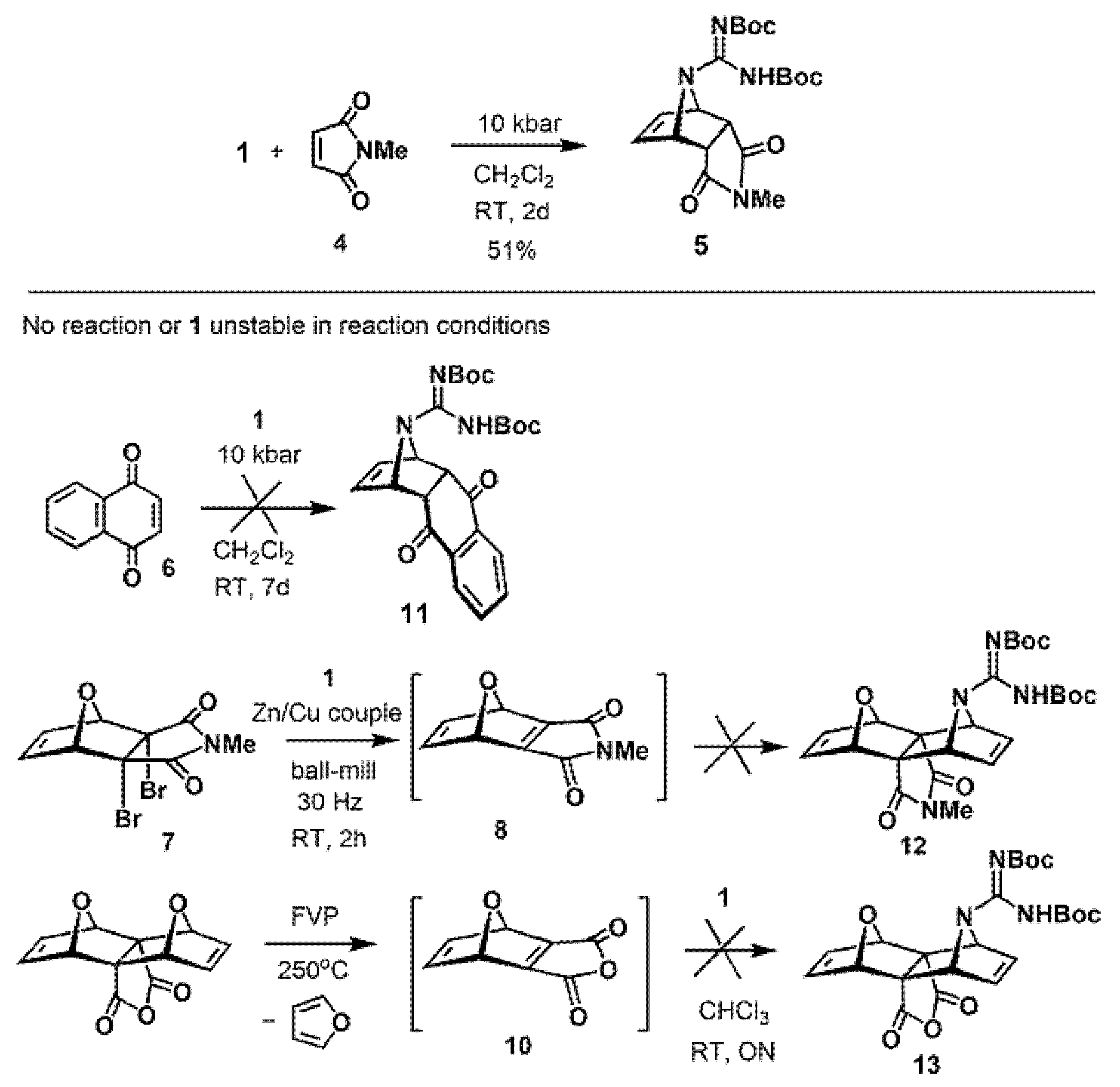
2.1. Cycloadditions of 1-Carboxamidine Pyrrole
2.2. Synthesis of Isoindoles and Their Reactivity
3. Materials and Methods
3.1. General
3.2. Synthesis of Cycloadduct 5
3.3. Synthesis of 20
3.4. Synthesis of Cycloadduct 23
3.5. Synthesis of Cycloadduct 26
3.6. Synthesis of Cycloadduct 28
3.7. Synthesis of Cycloadduct 29
3.8. Synthesis of 37
3.9. Synthesis of Cycloadduct 39
3.10. Synthesis of 42
3.11. Synthesis of 43
3.12. Synthesis of Guanidine 44
3.13. Synthesis of Cycloadduct 46
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Margetić, D. Physico-Chemical Properties of Organosuperbases. In Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; Ishikawa, T., Ed.; Wiley: Chichester, UK, 2009; Chapter 2; pp. 9–48. [Google Scholar] [CrossRef]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of novel uncharged organic superbases: Merging basicity and functionality. Acc. Chem. Res. 2021, 54, 3108–3123. [Google Scholar] [CrossRef] [PubMed]
- Saczewski, F.; Balewski, L. Biological activities of guanidine compounds. Expert Opin. Ther. Pat. 2009, 19, 1417–1448. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Urabe, D.; Isobe, M. An Efficient Total Synthesis of Optically Active Tetrodotoxin. Angew. Chem. Int. Ed. 2004, 43, 4782–4785. [Google Scholar] [CrossRef] [PubMed]
- Konrad, D.B.; Rühmann, K.-P.; Ando, H.; Hetzler, B.E.; Strassner, N.; Houk, K.N.; Matsuursa, B.S.; Trauner, D. A concise synthesis of tetrodotoxin. Science 2022, 377, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Margetić, D. Cycloadditions of guanidines. In Cycloaddition Reactions: Advances in Research and Applications; Margetić, D., Ed.; Nova Science Publishers: New York, NY, USA, 2019; Chapter 7; pp. 243–280. [Google Scholar]
- Briš, A.; Murata, Y.; Hashikawa, Y.; Margetić, D. Utilization of sym-Tetrazines as Guanidine Cycloaddition Delivery Reagents. An Experimental and Computational Study. J. Mol. Struct. 2023, 1272, 134207. [Google Scholar] [CrossRef]
- Margetić, D.; Russell, R.A.; Warrener, R.N. Cycloadditions Reagents for Rigidly Attaching the 1,4-Dimethoxynaphthalene Chromophore to Scaffold Alkenes. Org. Lett. 2000, 2, 4003–4006. [Google Scholar] [CrossRef]
- Warrener, R.N.; Schultz, A.C.; Houghton, M.A.; Butler, D.N. Rigid molecular racks featuring the 1,10-phenanthroline ligand especially those co-functionalised with redox-active groups or other bidentate ligands. Tetrahedron 1997, 53, 3991–4012. [Google Scholar] [CrossRef]
- Malpass, J.R.; Sun, G.; Fawcett, J.; Warrener, R.N. Novel ‘windscreen wiper’ cavity structures formed by the cycloaddition of N-substituted isoindoles onto molrac bis-alkenes. Tetrahedron Lett. 1998, 39, 3083–3086. [Google Scholar] [CrossRef]
- Warrener, R.N.; Margetic, D.; Sun, G.; Russell, R.A. Position-Addressable Nano-Scaffolds. I. The Preparation of N,O-, N,C- and N,N-Bridged Sesquinorbornadiene Succinimides as Compact, Highly Functionalized Addressable Building Blocks. Aust. J. Chem. 2003, 56, 263–267. [Google Scholar] [CrossRef]
- Donohoe, T.J. Product class 14: 1H- and 2H-isoindoles. Sci. Synth. 2001, 10, 653–692. [Google Scholar] [CrossRef]
- Reddy, R.P.; Davies, H.M.L. Asymmetric Synthesis of Tropanes by Rhodium-Catalyzed [4+3] Cycloaddition. J. Am. Chem. Soc. 2007, 129, 10312–10313. [Google Scholar] [CrossRef]
- Antol, I.; Barešić, L.; Glasovac, Z.; Margetić, D. Computational Study of Electronic Influence of Guanidine Substitution on Diels-Alder Reactions of Heterocyclic Dienes. Croat. Chem. Acta 2019, 92, 279–286. [Google Scholar] [CrossRef]
- Parr, B.T.; Economou, C.; Herzon, S.B. A concise synthesis of (+)-batzelladine B from simple pyrrole-based starting materials. Nature 2015, 525, 507–510. [Google Scholar] [CrossRef]
- Economou, C.; Romaire, J.P.; Scott, T.Z.; Parr, B.T.; Herzon, S.B. A convergent approach to batzelladine alkaloids. Total syntheses of (+)-batzelladine E, (−)-dehydrobatzelladine C, and (+)-batzelladine K. Tetrahedron 2018, 74, 3188–3197. [Google Scholar] [CrossRef]
- Margetić, D. High Pressure Organic Synthesis; Verlag Walter de Gruyter: Berlin, Germany, 2019; ISBN 978-3-11-055602-5. [Google Scholar] [CrossRef]
- Štrbac, P.; Margetić, D. Complementarity of solution and solid state mechanochemical reaction conditions demonstrated by 1,2-debromination of tricyclic imides. Beilstein J. Org. Chem. 2022, 18, 746–753. [Google Scholar] [CrossRef]
- Butler, D.N.; Margetić, D.; O’Neill, P.J.C.; Warrener, R.N. Parity Reversal: A New Diels-Alder Strategy for the Synthesis of Sesquinorbornadienes, Including Those with Heterobridges and Those of Unusual Stereochemistry. Synlett 2000, 1, 98–100. [Google Scholar] [CrossRef]
- Juršić, B.S. AM1 semiempirical study of benzopyrroles as dienes for Diels-Alder reaction. Can. J. Chem. 1996, 74, 114–120. [Google Scholar] [CrossRef]
- Warrener, R.N.; Butler, D.N.; Margetić, D. Preparation of the First Isobenzofuran Containing Two Ring Nitrogens: A New Diels-Alder Diene for the Synthesis of Molecular Scaffolds Containing one or more End-Fused 3,6-di(2-pyridyl)pyridazine Ligands. Aust. J. Chem. 2003, 56, 811–817. [Google Scholar] [CrossRef]
- Warrener, R.N. Isolation of isobenzofuran, a stable but highly reactive molecule. J. Am. Chem. Soc. 1971, 93, 2346–2348. [Google Scholar] [CrossRef]
- Priestley, G.M.; Warrener, R.N. A new route to isoindole (benzo[c]indole) and its derivatives. Tetrahedron Lett. 1972, 42, 4295–4298. [Google Scholar] [CrossRef]
- Kreher, R.P.; Use, G. Untersuchungen zur Chemie von Isoindolen und Isoindoleninen, XXIX. Reaktionen des 2H-Isoindols mit Maleinimiden: Ein einfaches Herstellungsverfahren für 7-Azabicyclo[2.2.1]heptene. Eur. J. Inorg. Chem. 1988, 121, 927–934. [Google Scholar] [CrossRef]
- Ohwada, T.; Ishikawa, S.; Mine, Y.; Inami, K.; Yanagimoto, T.; Karaki, F.; Kabasawa, Y.; Otani, Y.; Mochizuki, M. 7-Azabicyclo [2.2.1]heptane as a structural motif to block mutagenicity of nitrosamines. Bioorganic Med. Chem. 2011, 19, 2726–2741. [Google Scholar] [CrossRef] [PubMed]
- Kreher, R.P.; Seubert, J.; Kohl, N. Investigations on the Chemistry of Isoindoles and Isoindolenines. Part 26. Simple Methods for the Preparation of 2H-Isoindole. Chem. Ztg. 1987, 111, 349–356. [Google Scholar] [CrossRef]
- Kreher, R.P.; Kohl, N. A Rational Synthetic Method for 2H-Isoindoles. Angew. Chem. Int. Ed. 1984, 23, 517–518. [Google Scholar] [CrossRef]
- Davies, J.W.; Malpass, J.R.; Moss, R.E. Barriers to inversion at nitrogen in bicyclic amines and hydrazines. Tetrahedron Lett. 1985, 26, 4533–4536. [Google Scholar] [CrossRef]
- Đud, M.; Glasovac, Z.; Margetić, D. The utilization of ball-milling in synthesis of aryl guanidines through guanidinylation and N-Boc-deprotection sequence. Tetrahedron 2019, 75, 109–115. [Google Scholar] [CrossRef]
- Hewson, M.J.C.; Schmutzler, R. Phosphorus-fluorine chemistry part XLIII. Pyrrole-substituted fluorophosphoranes. Phosphorus Sulfur Relat. Elem. 1980, 8, 9–26. [Google Scholar] [CrossRef]
- Davies, J.W.; Durrant, M.L.; Walker, M.P.; Belkacemi, D.; Malpass, J.R. Preparation and spectroscopic studies of the 1,4-dihydro- 1,4-iminonaphthalene (7-azabenzonorbornadiene) ring system. Tetrahedron 1992, 48, 861–884. [Google Scholar] [CrossRef]
- Wojciechowski, K. Synthesis of 4-nitro-2H-isoindole derivatives. Liebigs Ann. Chem. 1991, 1991, 831–832. [Google Scholar] [CrossRef]
- Murashima, T.; Tamai, R.; Nishi, K.; Nomura, K.; Fujita, K.; Uno, H.; Ono, N. Synthesis and X-ray structure of stable 2H-isoindoles. J. Chem. Soc. Perkin Trans. 2000, 6, 995–998. [Google Scholar] [CrossRef]
- Use, G.; Kreher, R. Studies of the chemistry of isoindoles and isoindolenines 18. 2-tert-butyl-5-nitro-2H-isoindole. Preparation and reactions. Chem. Ztg. 1982, 106, 143–144. [Google Scholar] [CrossRef]
- Lin, C.; Zhen, L.; Cheng, Y.; Du, H.-J.; Zhao, H.; Wen, X.; Kong, L.-Y.; Xu, Q.-L.; Sun, H. Visible-Light Induced Isoindoles Formation To Trigger Intermolecular Diels-Alder Reactions in the Presence of Air. Org. Lett. 2015, 17, 2684–2687. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štrbac, P.; Briš, A.; Margetić, D. N,N′-Di-Boc-2H-Isoindole-2-carboxamidine—First Guanidine-Substituted Isoindole. Molecules 2022, 27, 8954. https://doi.org/10.3390/molecules27248954
Štrbac P, Briš A, Margetić D. N,N′-Di-Boc-2H-Isoindole-2-carboxamidine—First Guanidine-Substituted Isoindole. Molecules. 2022; 27(24):8954. https://doi.org/10.3390/molecules27248954
Chicago/Turabian StyleŠtrbac, Petar, Anamarija Briš, and Davor Margetić. 2022. "N,N′-Di-Boc-2H-Isoindole-2-carboxamidine—First Guanidine-Substituted Isoindole" Molecules 27, no. 24: 8954. https://doi.org/10.3390/molecules27248954
APA StyleŠtrbac, P., Briš, A., & Margetić, D. (2022). N,N′-Di-Boc-2H-Isoindole-2-carboxamidine—First Guanidine-Substituted Isoindole. Molecules, 27(24), 8954. https://doi.org/10.3390/molecules27248954