

Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors
 , , , ,
, , , , 
Abstract
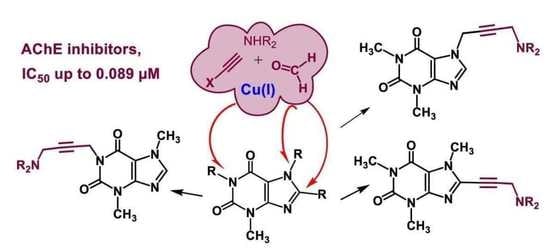
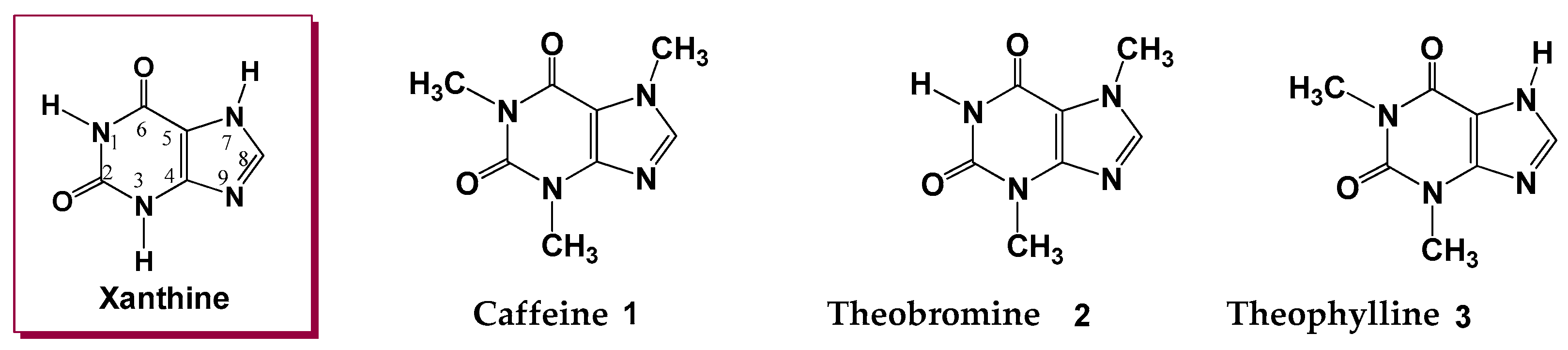
1. Introduction
2. Results and Discussion
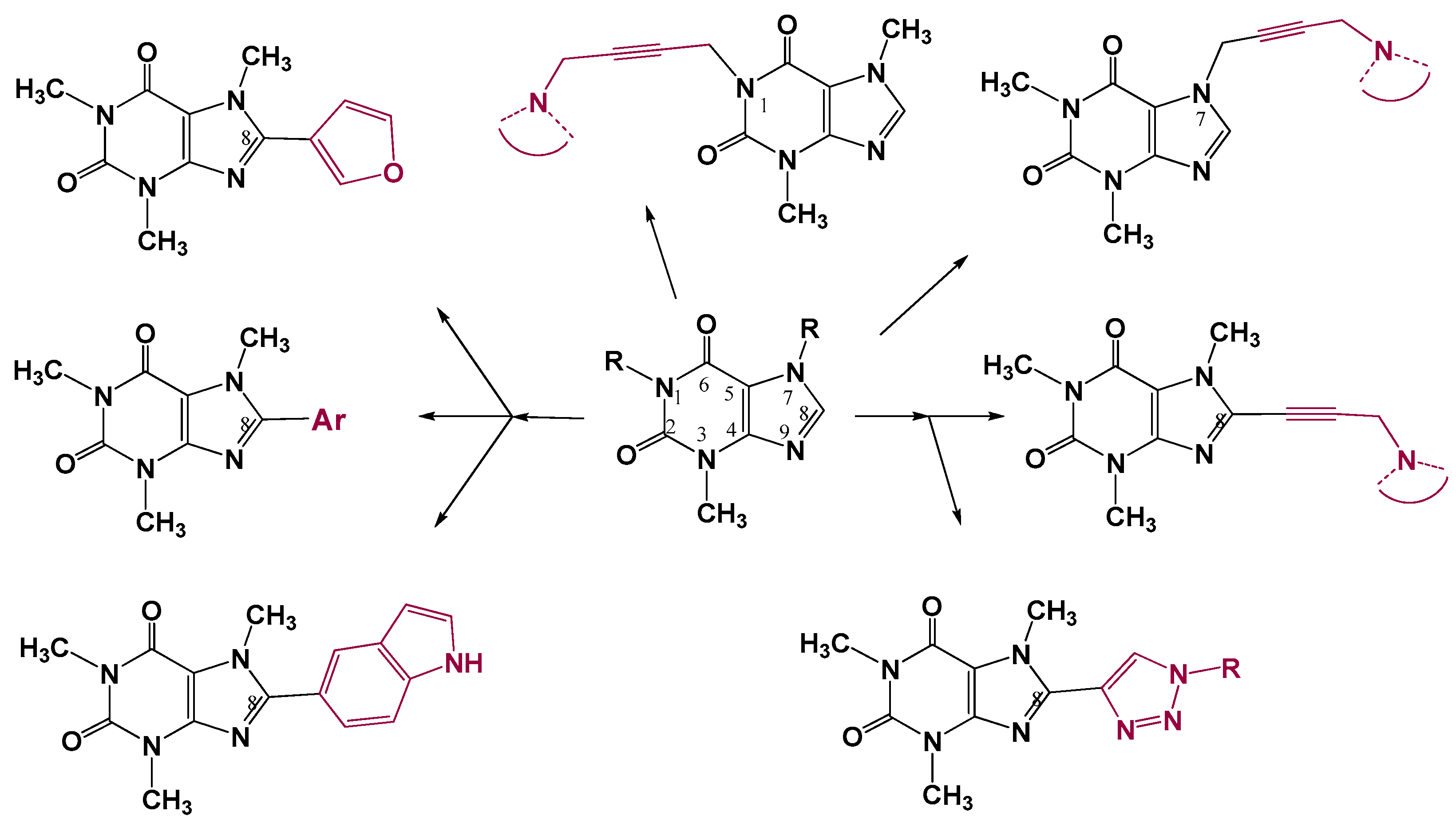
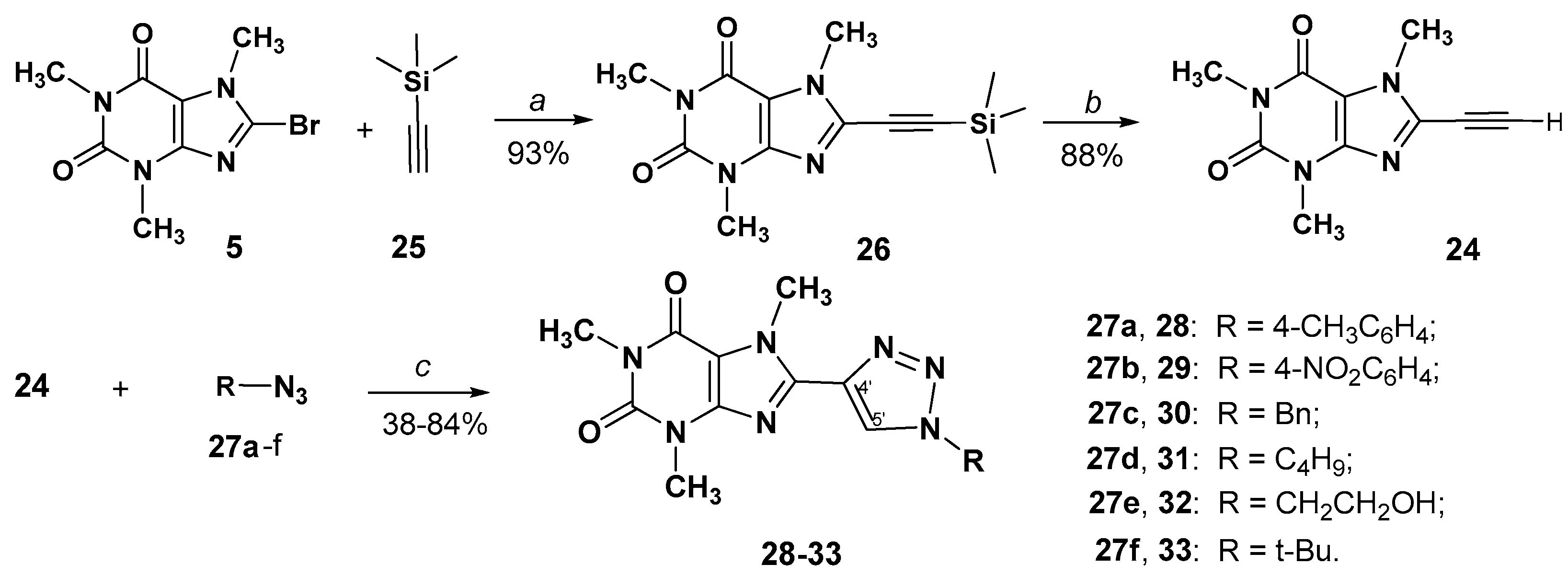
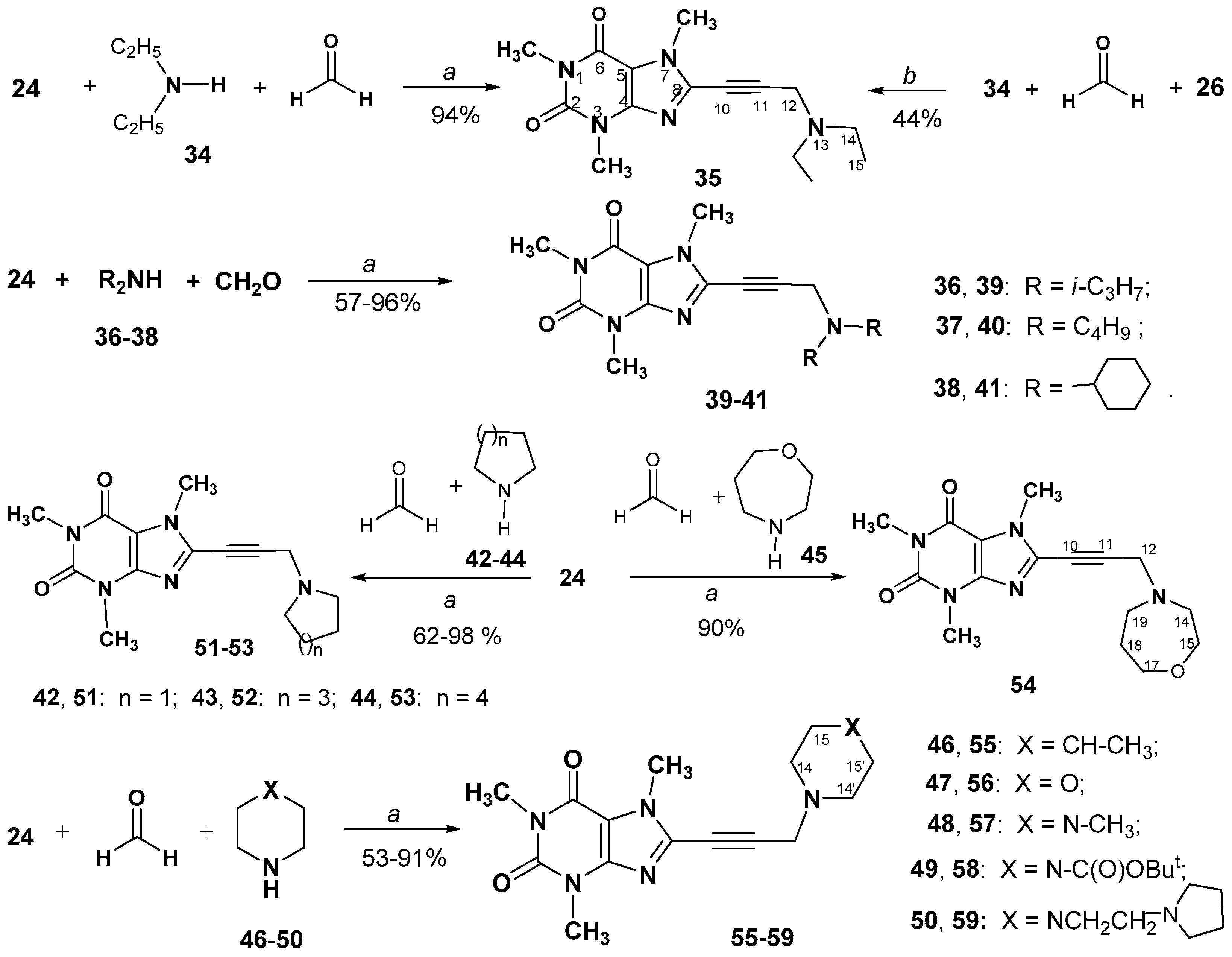
2.1. Chemistry
2.2. Enzyme Inhibition
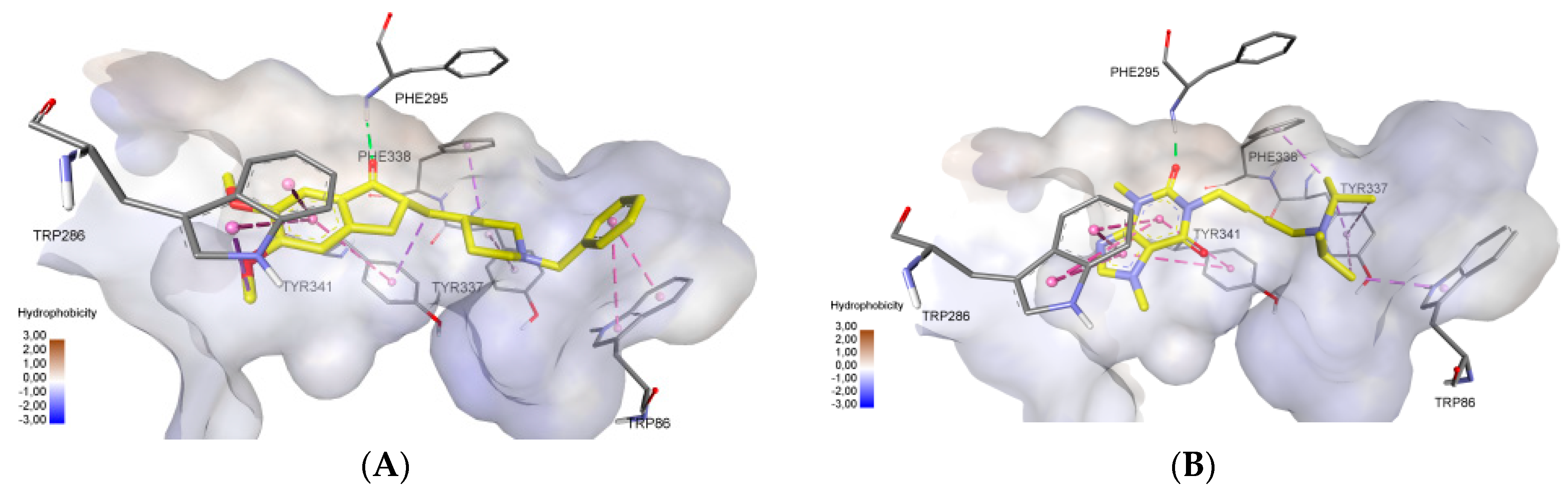
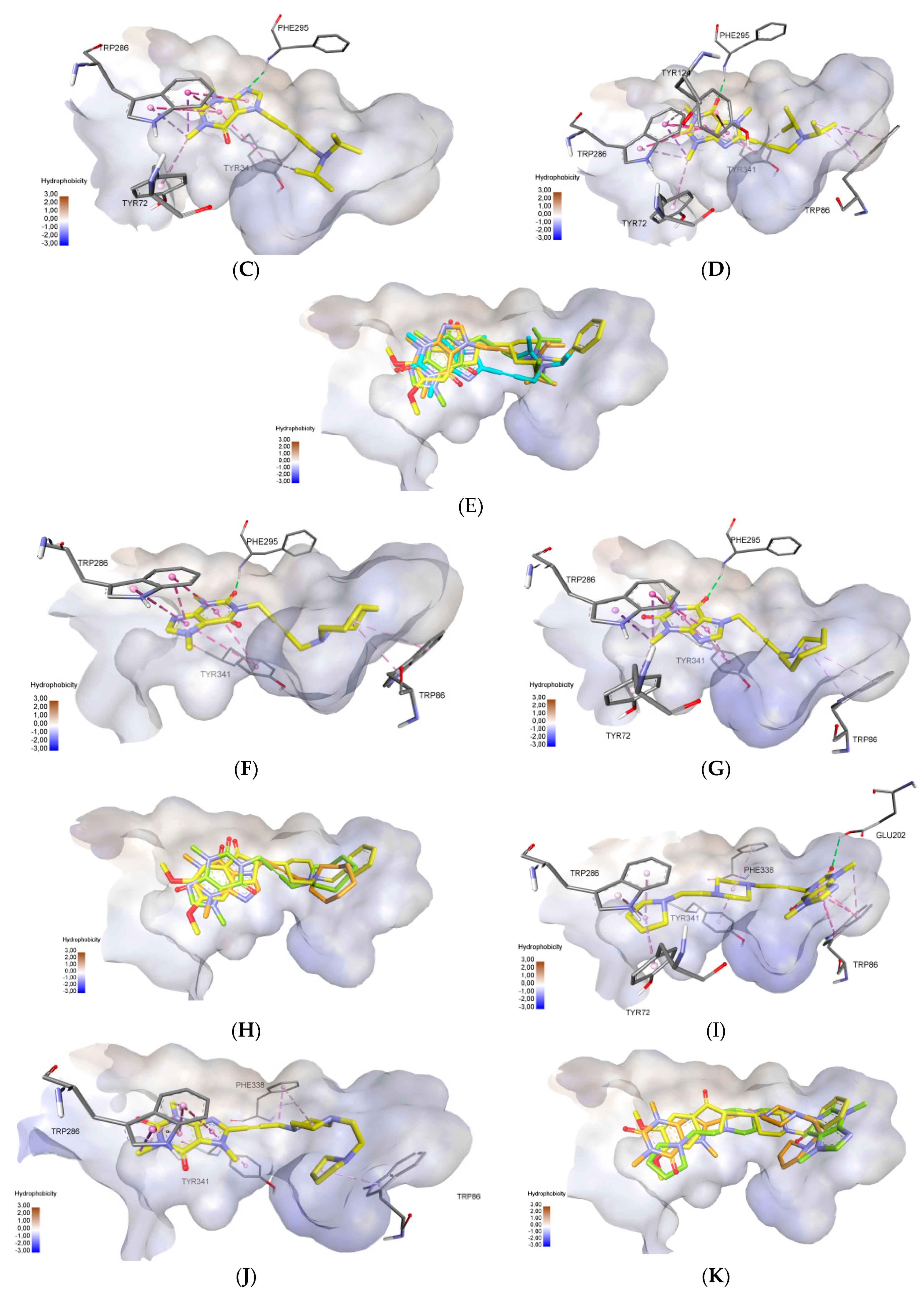
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
General Information
3.2. Synthesis and Spectral Data
3.2.1. General Method for the Preparation of 8-Aryl(hetaryl)-1,3,7-trimethyl- 3,7-dihydro-1H-purine-2,6-diones (7, 14–19, 22, 23)
1,3,7-Trimethyl-8-phenyl-3,7-dihydro-1H-purine-2,6-dione (7)
1,3,7-Trimethyl-8-o-tolyl-3,7-dihydro-1H-purine-2,6-dione (14)
8-(2-Aminophenyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (15)
8-(3-Methoxyphenyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (16)
8-(4-Methoxyphenyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (17)
8-(2,3-Dimethoxyphenyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (18)
8-(3,4,5-Trimethoxyphenyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (19)
8-(Furan-3-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (22)
8-(1H-Indol-5-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (23)
3.2.2. 1,3,7-Trimethyl-8-((trimethylsilyl)ethynyl)-3,7-dihydro-1H-purine-2,6-dione {8-[(trimethylsilyl)ethynyl]-caffeine} (26)
3.2.3. 8-Ethynyl-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione {8-Ethynylcaffeine} (24)
3.2.4. General Method for the Preparation of 8-(1,2,3-Triazol-4-yl)-3,7-дигидрo-1H-purine-2,6-diones (28–33)
1,3,7-Trimethyl-8-(1-(p-tolyl)-1H-1,2,3-triazol-4-yl)-3,7-dihydro-1H-purine-2,6-dione (28)
1,3,7-Trimethyl-8-(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)-3,7-dihydro-1H-purine- 2,6-dione (29)
8-(1-Benzyl-1H-1,2,3-triazol-4-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (30)
8-(1-Butyl-1H-1,2,3-triazol-4-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (31)
8-(1-(2-Hydroxyethyl)-1H-1,2,3-triazol-4-yl)-1,3,7-trimethyl-3,7-dihydro-1H-puri- ne-2,6-dione (32)
8-(1-(tert-Butyl)-1H-1,2,3-triazol-4-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (33)
3.2.5. Preparation of 8-(Piperidinyl)- or 8-(piperazinyl)-1,3,7-trimethyl-3,7-dihydro-1H-purine- 2,6-diones (60,61)
1,3,7-Trimethyl-8-(4-methylpiperidin-1-yl)-3,7-dihydro-1H-purine-2,6-dione (60)
tert-Butyl 4-(1,3,7-trimethyl-2,6-dioxo-3,7-dihydro-1H-purin-8-yl)piperazine-1- carboxylate (61)
3.2.6. Synthesis and Spectral Data of 8-(3-Aminoprop-1-ynyl)-1,3,7-trimethyl- 3,7-dihydro-1H-purine-2,6-diones (35, 39–41, 51–59)
8-(3-(Diethylamino)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (35)
8-(3-(Diisopropylamino)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine- 2,6-dione (39)
8-(3-(Dibutylamino)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (40)
8-(3-(Dicyclohexylamino)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine- 2,6-dione (41)
1,3,7-Trimethyl-8-(3-(pyrrolidin-1-yl)prop-1-yn-1-yl)-3,7-dihydro-1H-purine-2,6-dione (51)
8-(3-(Azepan-1-yl)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (52)
8-(3-(Azocan-1-yl)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione (53)
8-(3-(1,4-Oxazepan-4-yl)prop-1-yn-1-yl)-1,3,7-trimethyl-3,7-dihydro-1H-purine- 2,6-dione (54)
1,3,7-Trimethyl-8-(3-(4-methylpiperidin-1-yl)prop-1-yn-1-yl)-3,7-dihydro-1H- purine-2,6-dione (55)
1,3,7-Trimethyl-8-(3-morpholinoprop-1-yn-1-yl)-3,7-dihydro-1H-purine-2,6-dione (56)
1,3,7-Trimethyl-8-(3-(4-methylpiperazin-1-yl)prop-1-yn-1-yl)-3,7-dihydro-1H- purine-2,6-dione (57)
1,3,7-Trimethyl-8-(3-(4-(2-(pyrrolidin-1-yl)ethyl)piperazin-1-yl)prop-1-yn-1-yl) -3,7-dihydro-1H-purine-2,6-dione hydrate (59)
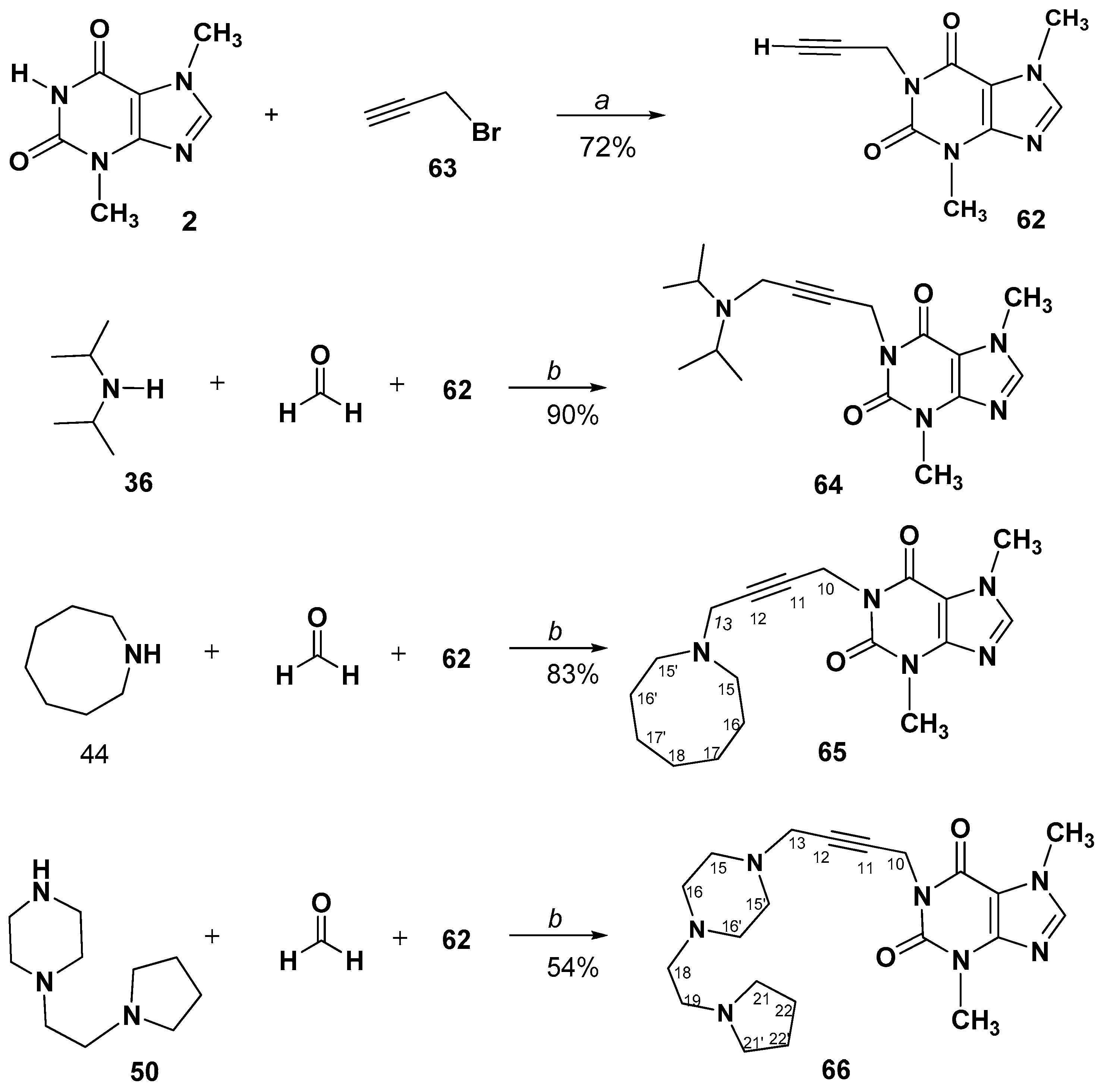
3.2.7. Propargylation of Dimethylxanthines (2,3)
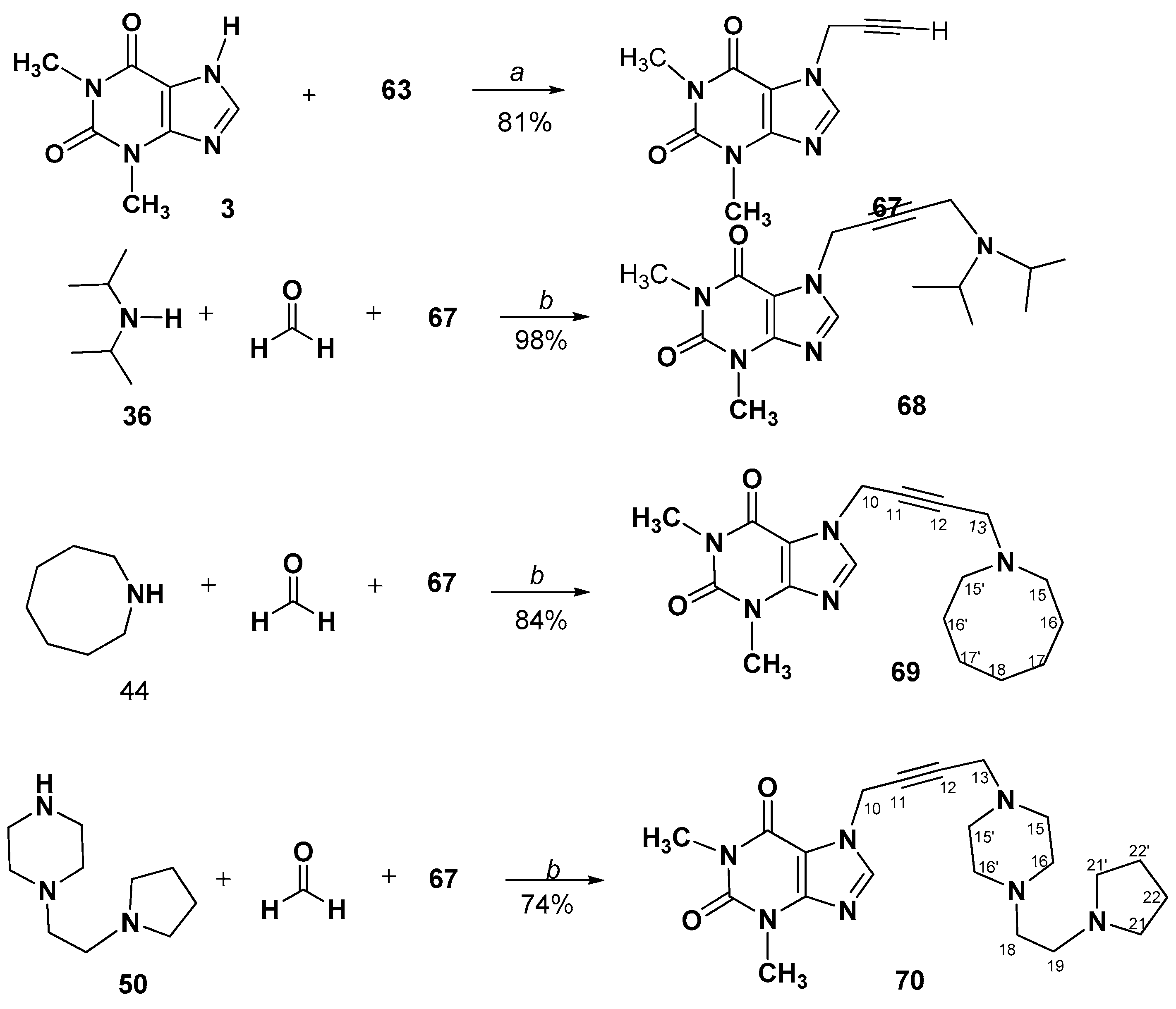
3.2.8. A3-Coupling of 1-Prop-2-ynyl-, or 7-Prop-2-ynyl- methylxanthines. Synthesis and Spectral Data of 1-(4-Aminobut-2-yn-1-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-diones (64–66) or 7-(4-Aminobut-2-yn-1-yl)-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-diones (68–70)
1-(4-(Diisopropylamino)but-2-yn-1-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (64)
1-(4-(Azocan-1-yl)but-2-yn-1-yl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione (65)
3,7-Dimethyl-1-(4-(4-(2-(pyrrolidin-1-yl)ethyl)piperazin-1-yl)but-2-yn-1-yl)-3,7- dihydro-1H-purine-2,6-dione hydrate (66)
7-(4-(Diisopropylamino)but-2-yn-1-yl)-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (68)
7-(4-(Azocan-1-yl)but-2-yn-1-yl)-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (69)
1,3-Dimethyl-7-(4-(4-(2-(pyrrolidin-1-yl)ethyl)piperazin-1-yl)but-2-yn-1-yl)-3,7- dihydro-1H-purine-2,6-dione (70)
3.3. Biochemical Method
3.4. Molecular Modelling and Molecular Dynamic Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Faudone, G.; Arifi, S.; Merk, D. The medicinal chemistry of caffeine. J. Med. Chem. 2021, 64, 7156–7178. [Google Scholar] [CrossRef] [PubMed]
- Janitschke, D.; Lauer, A.A.; Bachmann, C.M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Methylxanthines and neurodegenerative diseases: An update. Nutrients 2021, 13, 803. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W. Caffeine analogs: Biomedical impact. Cell. Mol. Life Sci. 2007, 64, 2153–2169. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.P.; Alves, M.G.; Oliveira, P.F.; Silva, B.M. Structure-bioactivity relationships of methylxanthines: Trying to make sense of all the promises and the drawbacks. Molecules 2016, 21, 974. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Shreshtha, A.K.; Thakur, M.S.; Patra, S. Xanthine scaffold: Scope and potential in drug development. Heliyon 2018, 4, e00829. [Google Scholar] [CrossRef]
- Bhat, J.A.; Gupta, S.; Kumar, M. Neuroprotective effects of theobromine in transient global cerebral ischemia-reperfusion rat model. Biochem. Biophys. Res. Commun. 2021, 571, 74–80. [Google Scholar] [CrossRef]
- Liu, W.; Meissner, G. Structure-activity relationship of xanthines and skeletal muscle ryanodine receptor/Ca2+ release channel. Pharmacology 1997, 54, 135–143. [Google Scholar] [CrossRef]
- Schepici, G.; Silvestro, S.; Bramanti, P.; Mazzon, E. Caffeine: An over-view of its beneficial effects in experimental models and clinical trials of Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 4766. [Google Scholar] [CrossRef]
- Oñatibia-Astibia, A.; Franco, R.; Martínez-Pinilla, E. Health benefits of methylxanthines in neurodegenerative diseases. Mol. Nutr. Food Res. 2017, 61, 1600670. [Google Scholar] [CrossRef]
- Karadsheh, N.; Kussie, P.; Linthicum, D.S. Inhibition of acetylcholinesterase by caffeine, anabasine, methyl pyrrolidine and their derivatives. Toxicol. Lett. 1991, 55, 335–342. [Google Scholar] [CrossRef]
- Okello, E.J.; Leylabi, R.; McDougall, G.J. Inhibition of acetylcholinesterase by green and white tea and their simulated intestinal metabolites. Food Funct. 2012, 3, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Dobes, P. Caffeine inhibits acetylcholinesterase, but not butyrylcholinesterase. Int. J. Mol. Sci. 2013, 14, 9873–9882. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. The effects of caffeine on the cholinergic system. Mini Rev. Med. Chem. 2014, 14, 543–549. [Google Scholar] [CrossRef]
- Pohanka, M. Role of Caffeine in the Age-related Neurodegenerative Diseases: A Review. Mini Rev. Med. Chem. 2022, 22, 2726–2735. [Google Scholar] [CrossRef]
- Fabiani, K.; Murray, A.P.; Corradi, J.; Antollini, S.S. A novel pharmacological activity of caffeine in the cholinergic system. Neuropharmacology 2018, 135, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Yelanchezian, Y.M.M.; Waldfogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Neuroprotective effect of caffeine in Alzheimer’s disease. Molecules 2022, 27, 3737. [Google Scholar] [CrossRef]
- Lambertucci, C.; Marucci, G.; Catarzi, D.; Colotta, V.; Francucci, B.E.; Spinaci, A.; Varano, F.; Volpini, R. A2A Adenosine receptor antagonists and their potential in neurological disorders. Curr. Med. Chem. 2022, 29, 4780–4795. [Google Scholar] [CrossRef]
- Müller, C.E.; Baqi, Y.; Hinz, S.; Namasivayam, V. Medicinal chemistry of A2B adenosine receptors. In The Adenosine Receptors; Borea, P.A., Varani, K., Gessi, S., Merighi, S., Vincenzi, F., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Volume 34, pp. 137–168. [Google Scholar]
- Rodriguez-Franco, M.I.; Fernandez-Bachiller, M.I. 1-Benzyl-4-chloromethylpiperidine: A Building block in the synthesis of compounds of pharmacological interest. Synthesis 2002, 2002, 911–915. [Google Scholar] [CrossRef]
- Rodriguez-Franco, M.I.; Fernandez-Bachiller, M.I.; Perez, C.; Castro, A.; Martinez, A. Design and synthesis of N- benzylpiperidine-purine derivatives as new dual inhibitors of acetyl- and butyrylcholinesterase. Bioorg. Med. Chem. 2005, 13, 6795–6802. [Google Scholar] [CrossRef]
- Mohamed, T.; Osman, W.; Tin, G.; Rao, P.P.N. Selective inhibition of human acetylcholinesterase by xanthine derivatives: In vitro inhibition and molecular modeling investigations. Bioorg. Med. Chem. Lett. 2013, 23, 4336–4342. [Google Scholar] [CrossRef]
- Fabiani, C.; Biscussi, B.; Munafó, J.P.; Murray, A.P.; Corradi, J.; Antollini, S.S. New synthetic caffeine analogs as modulators of the cholinergic system. Mol. Pharmacol. 2022, 101, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Bolea, I.; Gella, A.; Unzeta, M. Propargylamine-derived multitarget-directed ligands: Fighting Alzheimer’s disease with monoamine oxidase inhibitors. J. Neural Transm. 2013, 120, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Samadi, A.; Chioua, M.; Nikolic, K.; Filipic, S.; Agbaba, D.; Soriano, E.; de Andrés, L.; Rodríguez- Franco, M.I.; Alcaro, S.; et al. N-Methyl-N-((1-methyl-5-(3-(1-(2-methyl benzyl)piperidin-4-yl)propoxy)-1H-indol-2-yl)methyl)prop-2-yn-1-amine, a new cholinesterase and monoamine oxidase dual inhibitor. J. Med. Chem. 2014, 57, 10455–10463. [Google Scholar] [CrossRef] [PubMed]
- Zindo, F.T.; Joubert, J.; Malan, S.F. Propargylamine as functional moiety in the design of multifunctional drugs for neurodegenerative disorders: MAO inhibition and beyond. Future Med. Chem. 2015, 7, 609–629. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Di Pietro, N.O.; Alencar, G.; Esteban, E.; Viayna, N.; Szałaj, J.; Vázquez, J.; Juárez-Jiménez, I.; Sola, B.; Pérez, M.; Solé, M.; et al. Design, synthesis and biological evaluation of N-methyl-N-[(1,2,3-triazol-4- yl)alkyl]propargylamines as novel monoamine oxidase B inhibitors. Bioorg. Med. Chem. 2016, 24, 4835–4854. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Tipton, K.F. Assessment of enzyme inhibition: A review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 2017, 22, 1192. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, V.; Prashar, V.; Saini, S.; Dwivedi, A.R.; Bajaj, B.; Mehta, D.; Parkash, J.; Kumar, V. Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase. Eur. J. Med. Chem. 2019, 177, 221–234. [Google Scholar] [CrossRef]
- Mesiti, F.; Chavarria, D.; Gaspar, A.; Alcaro, S.; Borges, F. The chemistry toolbox of multitarget-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 181, 111572. [Google Scholar] [CrossRef]
- Carreiras, M.D.; Ismaili, L.; Marco-Contelles, J. Propargylamine-derived multi-target directed ligands for Alzheimer’s disease therapy. Bioorg. Med. Chem. Lett. 2020, 30, 126880. [Google Scholar] [CrossRef]
- Krátky, M.; Vu, Q.A.; Štepánková, S.; Maruca, A.; Silva, T.B.; Ambroz, M.; Pflégr, V.; Rocca, R.; Svrčcková, K.; Alcaro, S.; et al. Novel propargylamine-based inhibitors of cholinesterases and monoamine oxidases: Synthesis, biological evaluation and docking study. Bioorg. Chem. 2021, 116, 105301. [Google Scholar] [CrossRef]
- Kumar, B.; Dwivedi, A.R.; Arora, T.; Raj, K.; Prashar, V.; Kumar, V.; Singh, S.; Prakash, J.; Kumar, V. Design, synthesis, and pharmacological evaluation of N-propargylated diphenylpyrimidines as multitarget directed ligands for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2022, 13, 2122–2139. [Google Scholar] [CrossRef] [PubMed]
- Bolea, I.; Juárez-Jiménez, J.; de los Ríos, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]- N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [PubMed]
- Bautista-Aguilera, O.M.; Esteban, G.; Bolea, I.; Nikolic, K.; Agbaba, D.; Moraleda, I.; Iriepa, I.; Samadi, A.; Soriano, E.; Unzeta, M.; et al. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.H.; Romero, A.; et al. Donepezil+propargylamine+8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martínez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, as a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front. Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef]
- Guieu, B.; Lecoutey, C.; Legay, R.; Davis, A.; Sopkova de Oliveira Santos, J.; Altomare, C.D.; Catto, M.; Rochais, C.; Dallemagne, P. First synthesis of racemic trans propargylamino donepezil, a pleiotrope agent able to both inhibit AChE and MAOB, with potential interest against Alzheimer’s disease. Molecules 2021, 26, 80. [Google Scholar] [CrossRef]
- Rad, M.N.S.; Behrouz, S.; Nekoei, A.-R. 8-Bromocaffeine (8-BC): A New versatile reagent for conversion of aldoximes into nitriles. Synlett 2012, 23, 1191–1198. [Google Scholar]
- Reshetnikov, D.V.; Patrushev, S.S.; Shults, E.E. Synthetic transformations of sesquiterpene lactones. 11. Conjugates based on caffeine and eudesmanolides with N-containing linkers. Chem. Nat. Compd. 2020, 56, 855–860. [Google Scholar] [CrossRef]
- Vollmann, K.; Müller, C.E. Synthesis of 8-Substituted Xanthine Derivatives by Suzuki Cross-Coupling Reaction. Heterocycles 2002, 57, 871–879. [Google Scholar] [CrossRef]
- Reetz, M.T.; Westermann, E. Phosphane-free palladium-catalyzed coupling reactions: The decisive role of Pd nanoparticles. Angew. Chem. Int. Ed. 2000, 39, 165–168. [Google Scholar] [CrossRef]
- Albuquerque, H.M.T.; Pinto, D.C.G.A.; Silva, A.M.S. Microwave irradiation: Alternative heating process for the synthesis of biologically applicable chromones, quinolones, and their precursors. Molecules 2021, 26, 6293. [Google Scholar] [CrossRef] [PubMed]
- Rybár, A.; Pfleiderer, W. 7-Alkyl-8-ethinyltheophylline. Collect. Czeh. Chem. Commun. 1987, 52, 2730–2734. [Google Scholar] [CrossRef]
- Thompson, R.; Beauglehole, A.; Wang, G. Xanthine-Substituted Alkynyl Carbamates/Reverse Carbamates as A2B Antagonists. Publication Number WO/2016/164838, 13 October 2016. International Application No. PCT/US2016/026807. [Google Scholar]
- Arsenjans, P.; Vasiljeva, J.; Domracheva, I.; Shestakova, I.; Gulbe, A.; Kanepe-Lapsa, I.; Kauss, V.; Kalvins, I. Ethynylxanthines, preparation and use as calcium ion channel modulators. Publication Number WO 2016159747A, 10 June 2016. Application PCT/LV2015/000003 events. [Google Scholar]
- Arsenjans, P.; Vasiljeva, J.; Domracheva, I.; Kanepe-Lapsa, I. 8-Ethynylxanthines as promising antiproliferative agents, angiogenesis inhibitors, and calcium channel activity modulators. Chem. Heterocyclic Compd. 2020, 56, 776–785. [Google Scholar]
- Peshkov, V.A.; Pereshivko, O.P.; Van der Eycken, E.V. A walk around the A3-coupling. Chem. Soc. Rev. 2012, 41, 3790–3807. [Google Scholar] [CrossRef] [PubMed]
- Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Synthesis and Reactivity of Propargylamines in Organic Chemistry. Chem. Rev. 2017, 117, 14091–14200. [Google Scholar] [CrossRef]
- Allen, F.H.; Kenard, O.; Watson, D.G.; Bramer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Seale, T.W.; Abla, K.A.; Shamim, M.T.; Carney, J.M.; Daly, J.W. 3,7-Dimethyl-1-propargylxanthine: A potent and selective in vivo antagonist of adenosine analogs. Life Sci. 1988, 43, 1671–1684. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Program for Area Detector Adsorption Correction; Institute for Inorganic Chemistry, University of Göttingen: Goettingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXS-97 and SHELXL-97—Program for Crystal Structure Solution and Refinement; University of Göttingen: Goettingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Guo, Q.; Cheng, Y.; Lan, J.; You, J. Palladium-catalyzed desulfitative C-H arylation of heteroarenes with sodium sulfinates. Chem. -A Eur. J. 2011, 17, 13415–13419. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W.; Padgel, W.L.; Shamim, M.T. Analogs of caffeine and theophylline: Effect of structural alterations on affinity at adenosine receptors. J. Med. Chem. 1986, 29, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger Small Molecule Drug Discovery Suite; Schrödinger, LLC: New York, NY, USA, 2016.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Gerlits, O.; Ho, K.Y.; Cheng, X.; Blumenthal, D.; Taylor, P.; Kovalevsky, A.; Radić, Z. A new crystal form of human acetylcholinesterase for exploratory room-temperature crystallography studies. Chem.-Biol. Interact. 2019, 309, 108698. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Biovia, D.S.; Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Richmond, T.J. Dassault Systèmes BIOVIA, Discovery Studio Visualizer, v.17.2; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
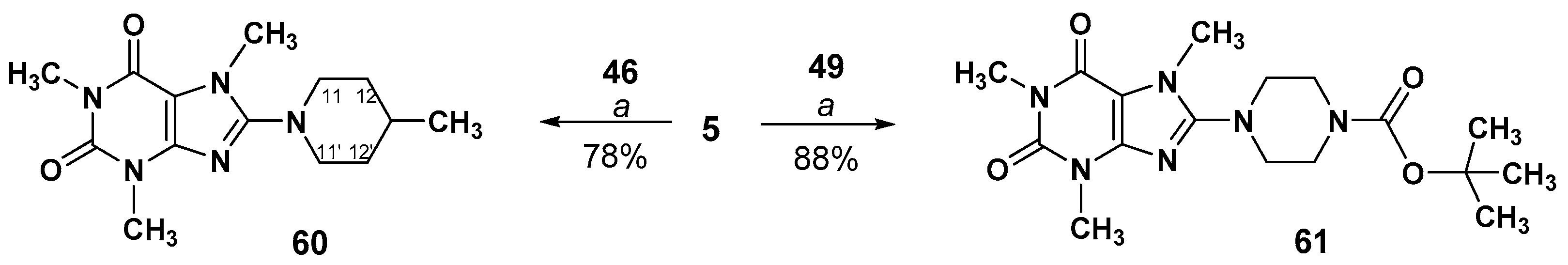{kind=link}
{kind=link}
{kind=link}
| Compound Type | Code | IC50, μM | Compound Type | Code | IC50, μM |
|---|---|---|---|---|---|
8-Aryl(hetaryl)caffeines | 7 | 510.4 ± 9.1 | 8-(3-R-amino)prop-1-yn-1-yl)-caffeines | 35 | 4.7 ± 0.1 |
| 14 | 201.4 ± 5.4 | 39 | 11.7 ± 0.1 | ||
| 15 | 94.5 ± 18.9 | 40 | 50.1 ± 4.5 | ||
| 16 | 105.0 ± 2.0 | 41 | 26.3 ± 0.5 | ||
| 17 | 146.0 ± 1.0 | 51 | 43.7 ± 3.6 | ||
| 18 | 142.0 ± 2.0 | 52 | 12.28 ± 0.31 | ||
| 19 | >1000 | 53 | 0.25 ± 0.001 | ||
| R = Furan-3-yl | 22 | 21.8 ± 0.2 | 54 | 124.0 ± 9.0 | |
| R = Indol-5-yl | 23 | >1000 | 55 | 2.5 ± 0.01 | |
8-Triazolylcaffeines | 28 | 531.0 ± 7.5 | 56 | 224.0 ± 5.0 | |
| 29 | 279.0 ± 6.0 | 57 | 60.0 ± 1.0 | ||
| 30 | 242.0 ± 2.0 | 58 | 1.20 ± 0.01 | ||
| 31 | 21.3 ± 0.4 | 59 | 0.552 ± 0.004 | ||
| 32 | 35.3 ± 0.9 |  | 60 | 113.0 ± 2.0 | |
| 33 | 42.4 ± 1.0 | 61 | 42.0 ± 1.0 | ||
 | 24 | 101.0 ± 1.7 |  | 68 | 14.9 ± 0.4 |
| 26 | 102.0 ± 4.3 | 69 | 0.121 ± 0.001 | ||
 | 64 | 2.4 ± 0.1 | 70 | 1.8 ± 0.1 | |
| 65 | 0.089 ± 0.001 | Caffeine | 1 | 30.0 ± 2.4 | |
| 66 | 0.746 ± 0.021 | Galantamine | 4.9 ± 0.2 |
| Entry | Compound | Ligand Efficiency, kcal/mol | Binding Energy a, kcal/mol |
|---|---|---|---|
| 1 | 64 | −0.634 | −15.207 |
| 2 | Donepezil | −0.529 | −14.817 |
| 3 | 65 | −0.590 | −14.742 |
| 4 | 68 | −0.614 | −14.733 |
| 5 | 66 | −0.486 | −14.590 |
| 6 | 69 | −0.554 | −13.840 |
| 7 | 59 | −0.429 | −12.860 |
| 8 | 55 | −0.529 | −12.693 |
| 9 | 58 | −0.518 | −12.432 |
| 10 | 39 | −0.493 | −11.833 |
| 11 | 53 | −0.417 | −10.691 |
| 12 | 70 | −0.356 | −10.542 |
| 12 | 41 | −0.351 | −9.562 |
| 13 | 52 | −0.382 | −9.561 |
| 14 | 22 | −0.351 | −7.931 |
| 15 | 32 | −0.319 | −7.712 |
| 16 | 31 | −0.235 | −7.053 |
| 17 | 24 | −0.407 | −6.512 |
| 18 | 1 | −0.449 | −6.284 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reshetnikov, D.V.; Ivanov, I.D.; Baev, D.S.; Rybalova, T.V.; Mozhaitsev, E.S.; Patrushev, S.S.; Vavilin, V.A.; Tolstikova, T.G.; Shults, E.E. Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors. Molecules 2022, 27, 8787. https://doi.org/10.3390/molecules27248787
Reshetnikov DV, Ivanov ID, Baev DS, Rybalova TV, Mozhaitsev ES, Patrushev SS, Vavilin VA, Tolstikova TG, Shults EE. Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors. Molecules. 2022; 27(24):8787. https://doi.org/10.3390/molecules27248787
Chicago/Turabian StyleReshetnikov, Danila V., Igor D. Ivanov, Dmitry S. Baev, Tatyana V. Rybalova, Evgenii S. Mozhaitsev, Sergey S. Patrushev, Valentin A. Vavilin, Tatyana G. Tolstikova, and Elvira E. Shults. 2022. "Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors" Molecules 27, no. 24: 8787. https://doi.org/10.3390/molecules27248787
APA StyleReshetnikov, D. V., Ivanov, I. D., Baev, D. S., Rybalova, T. V., Mozhaitsev, E. S., Patrushev, S. S., Vavilin, V. A., Tolstikova, T. G., & Shults, E. E. (2022). Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors. Molecules, 27(24), 8787. https://doi.org/10.3390/molecules27248787